

Summary
Matrix metalloproteases (MMPs) play a role in remodeling the extracellular matrix during brain development, and have been implicated in synaptic plasticity. Here we report that a member of the neuronal pentraxin (NP) family, neuronal pentraxin receptor (NPR), undergoes regulated cleavage by the MMP tumor necrosis factor-α converting enzyme (TACE). NPR is enriched at excitatory synapses, where it associates with AMPA-type glutamate receptors (AMPAR) and enhances synaptogenesis (Sia et al., 2007). However, in response to activation of group 1 mGluRs (mGluR1/5), TACE cleaves NPR and releases the pentraxin domain from its N-terminal transmembrane domain. Cleaved NPR rapidly accumulates in endosomes where it co-localizes with AMPAR. This process is necessary for mGluR1/5-dependent LTD in hippocampal and cerebellar synapses. These observations suggest that cleaved NPR functions to “capture” AMPAR for endocytosis and reveal a bifunctional role of NPs in both synapse strengthening and weakening.
Introduction
The neuronal pentraxins (NPs) NP1 and Neuronal Activity Regulated Pentraxin (Narp) are secreted, Ca2+ dependent lectins that are present at excitatory synapses, and are part of the extracellular synaptic scaffolding protein complex that contributes to synaptogenesis (Goodman et al., 1996; O'Brien et al., 1999; Tsui et al., 1996; Xu et al., 2003; Sia et al., 2007). Narp is regulated as an immediate early gene, and both its mRNA and protein are rapidly and transiently upregulated following patterned synaptic activity or seizure (Tsui et al., 1996). In assays of synaptogenesis, Narp overexpression increases excitatory synapse formation (O'Brien et al., 2002; O'Brien et al., 1999; Xu et al., 2003), while dominant-negative Narp decreases excitatory synapse formation (O'Brien et al., 2002). In similar assays, NP1 is less effective than Narp but markedly enhances the synaptogenic activity of submaximal levels of Narp. Studies of the molecular basis of their synaptogenic activity identified interactions that mediate assembly of NPs into highly structured multimers that are covalently linked. The N-terminal 200 amino acids of Narp and NP1 include coiled-coil repeats and three cysteines that form specific disulfide bonds between different NP molecules. Clusters that form on the surface of heterologous cells that express NP1 alone are relatively small, but co-expression of Narp results in disulfide linked NP1-Narp mixed multimers that form large clusters similar to Narp alone. This ability of Narp to enhance the clustering activity of NP1 may underlie their cooperative action in synaptogenesis (Xu et al., 2003).
Analysis of the ability of NPs to bind and co-cluster AMPARs revealed a critical role of the C-terminal pentraxin domain (Xu et al., 2003). The pentraxin domain is structurally similar to certain plant lectins, including wheat germ agglutinin (Emsley et al., 1994), and is a distant member of the LNS domain that is present in other synaptic proteins including agrin and neurexin (Beckmann et al., 1998; Rudenko et al., 2001; Rudenko et al., 1999). The pentraxin domain of Narp and NP1 appear identical in their affinity for binding AMPAR (Xu et al., 2003). Accordingly, NPs can be understood to be bimodular proteins; the N-terminus mediates self-association and clustering, while the C-terminus mediates co-clustering of AMPAR.
Neuronal Pentraxin Receptor (NPR) is a third member of the NP family that is primarily expressed in the central nervous system, and physically associates with Narp and NP1 (Dodds et al., 1997; Kirkpatrick et al., 2000). Like Narp and NP1, NPR binds AMPAR and contributes to synapse formation (Sia et al., 2007). Unlike Narp and NP1, NPR possesses an N-terminal transmembrane domain and a short (∼7 amino acid) intracellular sequence. In the present study, we report that NPR is cleaved by the extracellular protease TACE to release a soluble form of NPR. TACE is an MMP that is thought to play a central role in ectodomain shedding, a process by which regulated cleavage of membrane proteins can release functional domains (Black 2002; Blobel, 2000; Prenzel et al., 1999). MMPs have been implicated in migration of neuronal precursor cells, axonal guidance, neural cell adhesion, and activity-dependent remodeling of neuronal connections (Del Bigio et al., 1999; Domeniconi et al., 2005; Galko and Tessier-Lavigne, 2000; McFarlane, 2003; Ethell and Ethell, 2007). MMPs have recently been implicated in models of learning and memory (Tomimatsu et al., 2002, Nagy et al., 2006). However, the mechanisms of MMP involvement in synaptic plasticity, as well as their targets, are largely unknown. In exploring the role of NPR proteolytic processing, we found that TACE is activated by group 1 metabotropic glutamate receptors, mGluR1 and mGluR5 (mGluR1/5), in neurons. Using a combination of biochemical and cell biological techniques, we show that TACE-dependent NPR cleavage is an essential, early event required for mGluR1/5-dependent internalization of AMPAR, and for mGluR1/5-dependent long-term depression (mGluR1/5-dependent LTD) in both the hippocampus and the cerebellum. These studies support of model in which cleavage of NPR releases NPs from their transmembrane tether and allows them to be trafficked in endosomes with AMPAR, and thereby contribute to the down regulation of synaptic AMPARs.
Results
NPR associates with Narp, NP1, and AMPARs, and is present at excitatory synapses
The domain structure of NPR (Figure 1A) is notable for a predicted N-terminal transmembrane domain (Dodds et al., 1997; Kirkpatrick et al., 2000), coiled-coil domains and cysteines that are predicted to mediate homophilic interactions (Xu et al., 2003), and a C-terminal pentraxin domain. As anticipated from sequence conservation, NPR and Narp form heteromultimer complexes when co-expressed in HEK293T cells, and their association is dependent on specific cysteine residues within the N-terminus of Narp (See Xu et al., 2003; Supplemental Figure 1). To examine NP complexes in brain, lysates from WT mouse were prepared and IPed with rabbit antibody specific to NP1 or Narp. Lysates from NP1 KO, Narp KO and combined NP1/Narp/NPR KO (NP TKO) were also utilized as controls (Bjartmar et al., 2006). NPR (Ab 4999) specifically co-IPed with Narp and NP1 (Figure 1B). Consistent with the notion that NPs physically interact, there is overlap of NP expression in specific regions of the hippocampus and cerebellum (Supplemental Figure 2).
Figure 1. NPR associates with Narp, NP1, and AMPAR at excitatory synapses, and is proteolytically processed.

(A) Schematic of NPR structure. NPR contains 2 coiled-coil domains and a C-terminal pentraxin domain. Cysteines predicted to form disulfide linkages with other NPR subunits as well as other NPs are labeled. Regions of NPR used to generate Ab LFC-NPR, Ab 4999, and Ab 4450 are identified. Cleavage Sites A and B are identified and occur just before L36 and D176, respectively. Cleavage produces three NPR fragments, Long Form Cleaved (LFC-NPR), N-terminal Fragment (NTF-NPR), and C-terminal Fragment (CTF-NPR).
(B) Co-IP analysis of interactions between native NPR, Narp, and NP1 using lysates from WT, NP TKO, NP1 KO, and Narp KO mouse forebrain. The ∼62 kDa band detected by NPR antibody (Ab 4999) corresponds to the molecular weight predicted for full-length NPR. The ∼20 kDa band corresponds to NTF-NPR.
(C) GluR1-HA, but not GluR6-HA, co-IPs with NPR from transfected HEK293T cells.
(D) GluR1 co-IPs with NPR from forebrain extracts derived from WT mice. NPT KO and NPR KO are provided as control.
(E) NPR is localized to excitatory synapses in cultured hippocampal neurons (DIV 14; Ab 4450 at 1:200 labeled live). Scale bar = 10 μm.
(F) Multiple fragments of NPR are present in culture medium of NPR transfected HEK293T cells. Western blot analysis was performed on lysates and medium from transfected cells using Ab 4450 and Ab 4999.
(G) Identification of NPR cleavage sites.
(H) Western blot analysis of NPR protein expression in extracts of adult hippocampus, cerebellum, and cortex using Ab 4450 and Ab 4999.
The pentraxin domain of Narp and NP1 is required for interaction with AMPAR (Xu et al., 2003). The pentraxin domains of the NPs are highly conserved and, therefore, we predicted that NPR would interact with AMPAR. To test this, AMPAR subunit HA tagged GluR1 (GluR1-HA) was co-transfected with myc tagged Narp (Narp-myc) or NPR (NPR-myc) into HEK293T cells and lysates were assayed for co-IP. As previously described (O'Brien et al., 1999), GluR1-HA co-IPed with Narp-myc while kainate receptor subunit GluR6-HA did not (Figure 1C). Similarly, GluR1-HA but not GluR6-HA co-IPed with NPR-myc (Figure 1C). Additionally, GluR1 co-IPed with NPR from forebrain extracts derived from WT mice suggesting that GluR1 and NPR form associations in vivo (Figure 1D), although interactions may be indirect.
We examined the subcellular distribution of NPR in primary hippocampal cultures. NPR antibody (Ab 4450) detected large clusters that co-localized with PSD-95, a marker for excitatory synapses (Figure 1E), but not glutamic acid decarboxylase, a marker for inhibitory axons (not shown). These observations indicate that NPR is like Narp and NP1 in that it forms physical associations with other neuronal pentraxins and AMPARs, and localizes to excitatory synapses.
NPR is proteolytically processed in heterologous cells and in brain
We noted that when NPR was expressed in HEK293T cells, Ab 4450 detected ∼62 kDa and ∼45 kDa proteins in the medium (Figure 1F, left blot). Their abundance in the medium suggested that NPR may be proteolytically processed. To examine this, we affinity purified the ∼62 kDa and ∼45 kDa bands from the medium and performed N-terminal Edman sequencing. The ∼62-kDa species was identified to be a fragment of NPR that begins at Site A at amino acid 36L, just C-terminal to the predicted transmembrane domain (Figure 1A and 1G). The ∼45 kDa species was identified as an NPR fragment that begins at Site B, at amino acid 176D. Cleavage at Site B was predicted to produce a corresponding N-terminal fragment ending at amino acid 175A (Figure 1A and 1G). Consistent with this prediction, Ab 4999 (see Figure 1A) detected a ∼62 kDa band in both the lysates and medium (Figure 1F, right blot), and an additional band of ∼20 kDa that was present in the lysates, but not in the medium. The 20 kDa fragment corresponds to the predicted molecular weight of a membrane associated N-terminal fragment produced by cleavage at Site B.
Cleavage at Site A (L36) produces a secreted product that we designate long form cleaved NPR (LFC-NPR) that lacks only the transmembrane domain and the short cytosolic region. LFC-NPR migrates on SDS-PAGE with nearly the same apparent molecular mass as full-length NPR (FL-NPR) at ∼62 kDa, although on optimal gels they are resolved as a doublet (Supplemental Figure 5C). An antibody generated against the cytosolic N-terminus was not specific and we were not able to detect the predicted residual N-terminus. Cleavage at Site B (D176) produces two cleavage products, which we designate N-terminal fragment (NTF-NPR) and C-terminal fragment (CTF-NPR), which migrate at ∼20 kDa and ∼45 kDa, respectively. It is noteworthy that we were able to detect CTF-NPR in the medium, but NTF-NPR was present only in lysates of transfected cells (Figure 1F). This suggests that cleavage at Site B produces NTF-NPR that remains on the cell surface. Taken together, these data suggest that NPR is cleaved at either Site A or Site B, but not both, since a truncated NTF-NPR fragment is not detected in the medium. The possibility that these products might arise from alternative splice forms of NPR can be excluded since the indicated fragments were isolated from transgene expression of cloned NPR cDNA.
We sought to detect NPR cleavage products in rat brain. Extracts from adult rat hippocampus, cerebellum, and cortex were analyzed by Western blot. Ab 4450 and Ab 4999 detected bands that run at the expected ∼62 kDa after treatment with reducing reagent (Figure 1H). Both antibodies detected lower molecular weight species from the brain extracts: Ab 4450 detected a band of ∼45 kDa that corresponds to CTF-NPR, and Ab 4999 detected a band of ∼20 kDa that corresponds to NTF-NPR (Figure 1H). All immunoreactivity for NPR using Ab 4450 or Ab 4999 was absent in brain extracts from mice that lack NPR (Supplemental Figure 3 and Figure 1B, respectively).
We considered the possibility that the observed fragments of NPR might result from intracellular processing of an unconventional signal sequence and subsequent secretion, rather than proteolytic cleavage of membrane bound FL-NPR. However, FL-NPR is the predominant form of NPR on the surface of neurons (Supplemental Figure 6). Additionally, NPR was originally purified from the membrane fraction of neurons derived from rat brain, and Edman sequencing confirmed its identity as full length NPR (Dodds et al., 1997). Finally, drugs that block cleavage of NPR (see below) are not known to regulate signal sequence processing or secretion. Therefore, the cleaved fragments of NPR appear to be products of surface membrane bound, full length NPR, and not the result of secretion of a signal-sequence processed form of NPR.
The transmembrane domain of NPR inhibits its ability to form surface clusters
Cleavage at Site A releases LFC-NPR from its transmembrane domain. In assays comparing the cell biological properties of NPR and Narp, we noted that FL-NPR is uniformly distributed on the cell surface while Narp forms large clusters (Figure 2A). The retention of secreted Narp on the cell surface requires oligomerization, but its physical basis remains to be defined (Xu et al., 2003). Nevertheless, the marked difference in the properties of Narp and NPR on the cell surface suggested that the transmembrane domain of NPR might restrict its ability to form clusters. To examine this possibility, we generated chimeric mutants (CM) of Narp and NPR in which the N-terminal and C-terminal ends were swapped (Figure 2B). CM-1 and CM-2, which include various lengths of Narp N-terminus that replace the NPR transmembrane domain, formed clusters on the surface of transfected COS-7 cells stained live with anti-myc. By contrast, CM-4 and CM-5, which maintain one or both coiled-coil domains of Narp coupled to the NPR transmembrane domain, did not (Figure 2A). Moreover, CM-3, which replaces the transmembrane domain of NPR with the signal peptide of Narp, and CM-6, which replaces the signal peptide of Narp with the transmembrane domain of NPR demonstrated that the transmembrane domain of NPR is necessary (CM-3) and sufficient (CM-6) to prevent pentraxin cluster formation. These observations suggest that cleavage at Site A could dramatically alter the cellular properties of NPR.
Figure 2. Transmembrane domain of NPR is necessary and sufficient to inhibit cluster formation.
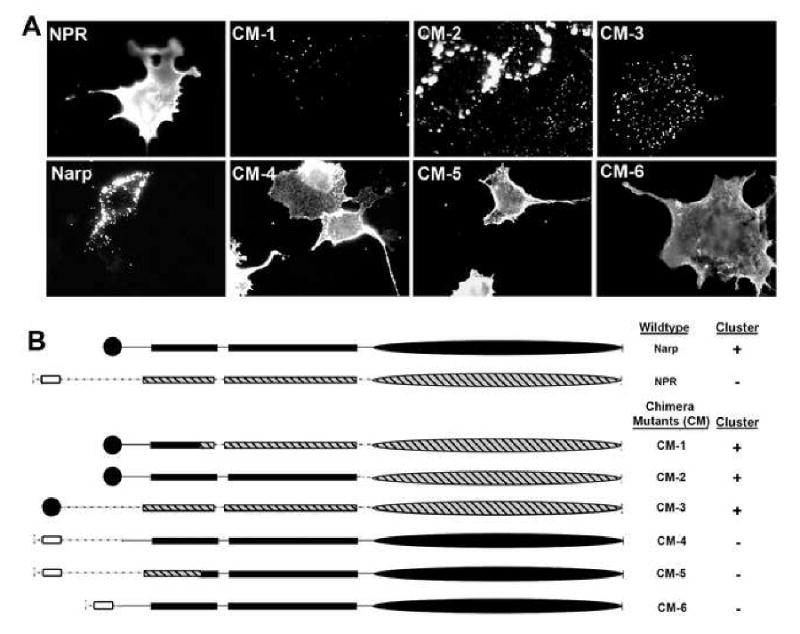
(A) Live cell surface staining of Narp, NPR, and Narp/NPR chimera mutants (CM) in transfected COS-7 cells.
(B) Schematic of the Narp, NPR, and Narp/NPR chimera constructs used. Narp signal peptide (closed circle) and NPR transmembrane domain (open box) are illustrated. All constructs have a C-terminal myc tag.
NPR is cleaved by TACE
We sought to identify mechanisms that could generate cleaved fragments of NPR. Type I and II transmembrane proteins can be cleaved by regulated matrix metalloproteases (MMPs) (Black, 2002; Blobel, 2000). Accordingly, we tested whether cleavage of NPR could be induced by signaling pathways that activate MMPs. Cultures transfected with NPR were treated with phorbol myristate acetate (PMA; 150 nM), which is known to activate MMPs (Arribas et al., 1996; Buxbaum 1998), and medium from the cultures was sampled at subsequent time points (Figure 3A). PMA increased the accumulation of NPR in the medium which corresponds to LFC-NPR (Figure 3A). The ∼45 kDa CTF-NPR was also present in the medium, but its stimulated accumulation was less abundant than LFC-NPR and varied between experiments.
Figure 3. TACE cleaves NPR and promotes clustering with AMPAR.

(A) – (B) HEK293T cells were transfected with NPR-myc transgene. Two days after transfection, medium was replaced with drug and vehicle. At the indicated time points, samples of medium were removed. At the end of the time course, cells were lysed. Lysates and medium were then analyzed by Western blot analysis using Ab 4450.
(A) PMA (150 nM) stimulates generation of LFC- and CTF-NPR in the medium of transfected HEK293T cells. Generation of LFC- and CTF-NPR is inhibited by TAPI-2 (50 μM).
(B) Co-transfection of HEK293T cells with siRNA targeting human TACE (TACE (h) siRNA) reduces PMA (150 nM) stimulated generation of LFC-NPR and CTF-NPR. NPR cleaved products are restored by co-transfection of mouse TACE (TACE (m)) cDNA. Lysates were probed with anti-TACE to confirm knockdown and expression of TACE as well as anti-β-actin as loading control.
(C) Representative images of live anti-myc stained NPR-myc transfected COS-7 cells 90 min after treatment with PMA (150 nM) or vehicle. Scale bar = 5 μm.
(D) Representative images of live Ab 4450 and anti-HA stained COS-7 cells co-transfected with wtNPR-myc and GluR2-HA 90 min after treatment with PMA (150 nM) or vehicle. Magnified regions of PMA treated cells (boxed) are provided in the right inset. Scale bars = 5 μm.
Based on these observations, we hypothesized that cleavage at Site A might be mediated by TACE, since TACE is reported to mediate PKC-stimulated cleavage of Type I and II proteins such as TNFα, TGFα, and APP in multiple cell types (Black, 2002; Blobel, 2000). Consistent with this hypothesis, PMA-stimulated generation of LFC-NPR was completely prevented by the drug TAPI-2 (50 μM), which inhibits TACE activity (Arribas et al., 1996; Moss et al., 2007) (Figure 3A). Stimulated generation of LFC-NPR was also inhibited by the general MMP inhibitor GM6001 (25 μM; Supplemental Figure 4). To further examine the role of TACE, we transfected HEK293 cells with siRNA specific for human TACE and confirmed that it reduced expression of native TACE (Figure 3B, right blot). siRNA markedly reduced PMA-stimulated generation of LFC-NPR (Figure 3B, left blot). Co-transfection of mouse TACE restored the ability of PMA to generate LFC-NPR. Together, these observations implicate TACE in regulated cleavage of NPR at Site A. The comparatively modest generation of CTF-NPR by PMA was also blocked by Tapi-2 and TACE siRNA suggesting that cleavage at Site B may also mediated by TACE (Figure 3A and 3B). Note, however, that GM6001, which is less selective than Tapi-2, blocks Site A, but not Site B cleavage (Supplemental Figure 4). This does not exclude a role for TACE in Site B cleavage since inhibitors of MMPs are reported to be differentially effective for different substrates (Hooper et al., 1997).
PMA induces NPR clusters on the surface of transfected cells
We examined the prediction that regulated cleavage of NPR could result in its redistribution to clusters on the cell surface. COS-7 cells expressing NPR-myc were stained live with anti-myc antibody after treatment with PMA (150 nM) or vehicle. Cells treated with vehicle exhibited a uniform NPR distribution (Figure 3C). When cells were treated with PMA (90 min), clusters of NPR were detected (Figure 3C).
We next asked if induction of NPR clusters by PMA results in co-clusters with AMPAR. This is based on precedent that Narp induces GluR1/2 to form co-clusters on heterologous cells (O'Brien et al., 1999). As anticipated, COS-7 cells co-transfected with NPR-myc and GluR2-HA without PMA treatment exhibited diffuse NPR and GluR2 staining (Figure 3D). By contrast, COS-7 cells treated with PMA exhibited co-clusters of NPR and GluR2-HA (Figure 3D).
LFC-NPR co-clusters with AMPAR in cultured hippocampal neurons, both on the cell surface, and internally, following DHPG treatment
To evaluate the localization of endogenous LFC-NPR as well as the regulation of its production by Site A cleavage in neurons, we generated a cleavage site selective antibody against LFC-NPR (Ab LFC-NPR) using a synthetic peptide that mimics the new epitope at the N-terminus of LFC-NPR (See Methods and Figure 1A). When Ab LFC-NPR was used to blot C-terminal tagged NPR-myc expressed in HEK293T cells, it selectively detected a ∼60 kDa protein that was enriched in the medium relative to lysates, while the anti-myc detected NPR equally in both fractions (Supplemental Figure 5A). Ab LFC-NPR detected a protein that migrates at ∼60 kDa that superimposes with LFC-NPR detected by Ab 4450 in brain lysates from WT mouse forebrain (Supplemental Figure 5C), and that was absent from brain lysates prepared from NPR KO mouse (Supplemental Figure 5B). These observations confirm that Ab LFC-NPR selectively detects LFC-NPR.
We examined the possibility that LFC-NPR associates with AMPAR on the surface of neurons. Cultured hippocampal neurons were transfected with GluR1 tagged with N-terminal extracellular α-bungarotoxin (BTX)-binding site (BBS) (Sekine-Aizawa and Huganir, 2004). Surface GluR1-BBS and native LFC-NPR were then visualized by live application of rhodamine-linked BTX and Ab LFC-NPR. Labeling was performed at 10°C to limit internalization. Live staining revealed co-clusters of LFC-NPR and GluR1-BBS on the surface of cultured hippocampal neurons (Figure 4A). These findings indicate that LFC-NPR co-localizes with surface AMPAR. The absence of LFC-NPR in medium of neuronal cultures, and labeling on the cell surface indicates that it remains adherent to the neuronal cell surface, like Narp and NP1 (Xu et al., 2003).
Figure 4. LFC-NPR co-localizes with internalized AMPAR at synapses.
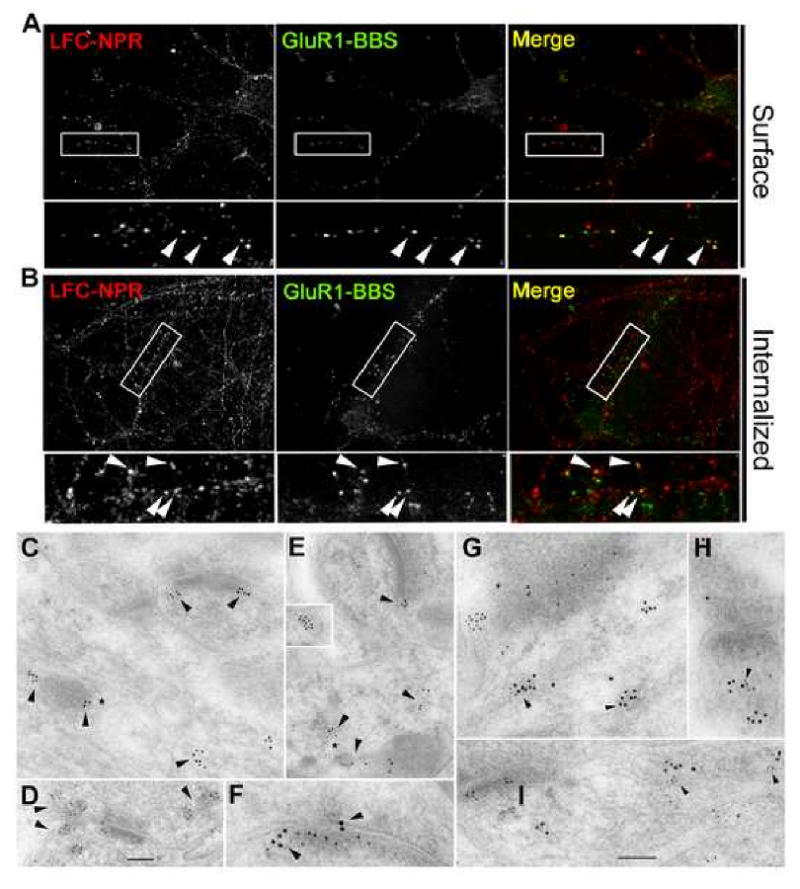
(A) GluR1-BBS transgene co-localizes with endogenous LFC-NPR clusters in cultured hippocampal neurons (DIV14).
(B) Internalized GluR1-BBS and endogenous LFC-NPR co-localize in cultured hippocampal neurons (DIV 14) 45 min after DHPG (50 μM) treatment. Scale bar = 10 μm.
(C-E) Sections of hippocampus (C, CA1 stratum radiatum; D, CA3 stratum lucidum) or cerebellar molecular layer (E) labeled with Ab LFC-NPR (10 nm gold), single-labeled (C-E). Gold labeling (arrowheads) is in various tubulovesicular structures in the presynaptic terminal (D) and postsynaptic spine (C and D). Labeling for LFC-NPR is also associated with the endosomal complex found in the dendrite shaft near the base of a spine (* in C and E). Inset in E shows labeling in a vesicle (appears to be partly clathrin-coated) that probably is newly endocytosed from the adjacent cell membrane (an unidentified process in contact with the side of an axon terminal in the cerebellum).
(F-I) Sections of the hippocampus CA1 stratum radiatum (F, G, and H) and CA3 stratum lucidum (I) synapses double-labeled for LFC-NPR (Ab LFC-NPR; 10 nm gold) and either GluR2/3 (F, G, and H) or GluR2 (I; 5 nm gold), illustrating co-localization (arrowheads) of labeling in postsynaptic tubulovesicular organelles including some distinctive endosomes (* in G and I). Labeling for LFC-NPR co-localizes with AMPAR labeling associated with postsynaptic membrane and adjacent cytoplasm as well as synaptic cleft. The synapse in G is an interneuron dendrite shaft synapse with an oblique synaptic cleft, and those in F, H, and I are synaptic spines. Scale bar (D) is 100 nm in C and D, 125 nm in E and inset, and 50 nm in F. Scale bar (I) is 100 nm in G-I.
We next assessed whether LFC-NPR co-localizes with AMPAR after endocytosis using an acid strip immunocytochemical staining protocol (Carroll et al. 1999; see Supplemental Experimental Procedures). Consistent with published results (Snyder et al. 2001), transient application of DHPG, a selective agonist for mGluR1/5 receptors, evoked endocytosis of GluR1-BBS (Figure 4B). Internalized puncta of LFC-NPR and GluR1-BBS co-localized in discrete puncta within dendrites (Figure 4B) supporting the notion that LFC-NPR and AMPAR co-localize in endosomes. In support of this hypothesis, NPR co-localized with EEA1, a marker for early endosomes (Supplemental Figure 7A). Finally, internal LFC-NPR punctae often co-localized with PSD-95 (Supplemental Figure 7B), suggesting that the vesicles play a role in trafficking to or from the synapse.
LFC-NPR co-localizes with AMPAR in vesicles at synapses in brain
We performed double immunogold labeling with Ab LFC-NPR and antibodies to AMPAR to assess if these proteins co-localize in postsynaptic endosomes in vivo. We were unable to use a monoclonal antibody to GluR1 for double labeling and, therefore, used antibodies to GluR2. Ab LFC-NPR immunoEM exhibited gold labeling in tubulovesicular and vesicular structures in presynaptic terminals and postsynaptic spines in the CA1 and CA3 regions of the hippocampus (Figure 4C, D, F, and G-I) and in the Purkinje spines of the cerebellar cortex (Figure 4E). Labeling was also associated with distinctive endosomal complexes in the dendrite adjacent to the spine synapse (Figure 4C, E, G, and I), and in other endosomal structures (4E, inset). In double-labeling for LFC-NPR and AMPAR, co-localization was evident in the postsynaptic membrane and endosomal structures (Figure 4F-I).
Cleavage of NPR is regulated by mGluR1/5 in cultured neurons
We next used Ab LFC-NPR to examine the possibility that mGluR1/5 might regulate cleavage of NPR. mGluR1/5 activates PKC via phospholipase C and the production of diacylglycerol (DAG) and inositol triphosphate (IP3) that releases intracellular Ca2+. Cultured cortical neurons (∼DIV 14) were treated with DHPG for 5 minutes then returned to control medium for intervals of 0 to 90 min. Ab LFC-NPR detected a ∼60kDa band from lysates that increased over time after stimulation with DHPG (Figure 5A). 90 minutes after DHPG, levels of LFC-NPR increased by 60.7% ± 2.5% (Figure 5A). No specific band was detected in the medium by Ab LFC-NPR or other NPR Abs (not shown). Pretreatment of cultured neurons with TAPI-2 blocked the DHPG-stimulated increase in LFC-NPR (Supplemental Figure 8). These results support the hypothesis that NPR undergoes TACE cleavage in neurons that can be regulated by mGluR1/5. They also establish that there is a low but detectable basal level of LFC-NPR on the surface of cultured neurons (Supplemental Figure 6).
Figure 5. mGluR1/5-dependent LTD in neuronal culture requires NPs and TACE-like activity.
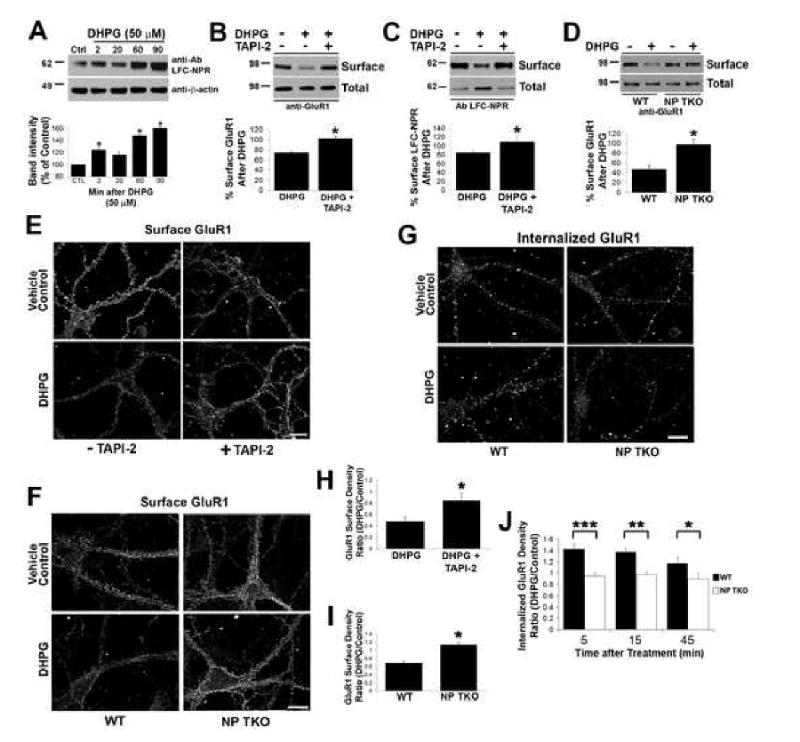
(A) Representative Western blot using Ab LFC-NPR of lysates from primary cortical cultures at indicated time points after treatment with DHPG (50 μM). Quantification of band intensity at indicated time points compared to control (Mean ± SD, n=3 paired animals, *p < 0.05).
(B) Representative Western blot of biotinylated surface GluR1 from primary cortical cultures probed with C-terminal GluR1C antibody 45 min after 5 min treatment with DHPG (50 μM) +/- TAPI-2 (50 μM treated 20 min before DHPG stimulation). Quantification of band intensity ratio of surface GluR1 after DHPG treatment to vehicle control (Mean ± SD, n= 3, *p < 0.05).
(C) Representative western blot of biotinylated surface LFC-NPR from primary cortical cultures 45 min after 5 min treatment with DHPG (50 μM) +/- TAPI-2 (50 μM). Quantification of band intensity ratio of treated surface to control surface LFC-NPR (Mean ± SD, n=3, *p < 0.05).
(D) Representative Western blot of biotinylated surface GluR1 from WT and NP TKO primary cortical neurons 45 min after treatment with DHPG (50 μM). Quantification of ratio of surface GluR1 after DHPG treatment to vehicle control (Mean ± SD, n=4, *p < 0.05).
(E) DHPG (50 μM) stimulated surface GluR1 loss in cultured hippocampal neurons (DIV1 14) is blocked by treatment with TAPI-2 (50 μM). See Figure 5H for quantification. Scale bar = 10 μm.
(F) DHPG (50 μM) stimulated surface GluR1 loss is impaired in neuronal cultures derived from NP TKO. See Figure 5I for quantification. Scale bar = 10 μm.
(G) DHPG induced GluR1 endocytosis is impaired in TKO hippocampal neurons compared to WT. Internalized GluR1 was measured +/- DHPG (50 μM) at 5 min (not shown), 15 min (not shown), and 45 min using acid strip protocol to visualize internalized labeled GluR1. See Figure 5J for quantification.
(H) Quantification of surface GluR1 puncta density after vehicle control or DHPG stimulation +/- TAPI-2. The mean surface GluR1 puncta density per unit length of dendrite was calculated for each treatment and expressed as a ratio (DHPG/vehicle control). These ratios are compared +/- TAPI-2 (mean ± SEM; n> 14; *p < 0.0001).
(I) Quantification of density of surface GluR1 per unit length after vehicle control or DHPG stimulation cultured neurons from WT and NP TKO mice. Ratio of DHPG/vehicle control was calculated as described above (mean ± SEM; n > 66; *p < 0.001).
(J) Quantification of internalized GluR1 density per unit length after vehicle control or DHPG stimulation in cultured neurons from WT and NP TKO mice at indicated time points (mean ± SEM; 5 min: n>110; 15 min n>160; 45 min n>36; ***p = 0.002; **p = 0.004; *p = 0.04).
mGluR1/5-dependent AMPAR endocytosis in cultured neurons requires TACE and NPs
Activation of mGluR1/5 using DHPG in neurons produces a rapid and sustained reduction of surface AMPAR that represents a cellular model of LTD (Huber et al., 2001; Snyder et al., 2001; Xiao et al., 2001). Since NPR processing is induced by mGluR1/5 stimulation, and LFC-NPR co-localizes with internalized AMPAR, we examined the role that NPR processing might play in mGluR1/5-dependent trafficking of AMPAR. We assayed cell surface expression of GluR1 in cultured cortical neurons 45 min after DHPG treatment using the soluble biotinylating reagent to enrich surface proteins (Figure 5B). As reported previously (Snyder et al., 2001), surface GluR1 was reduced 45 min after DHPG treatment compared to vehicle control treated cultured cortical neurons (74.1 ± 2.2% of vehicle control, Figure 5B). The effect of DHPG on surface GluR1 was blocked by pretreatment with TAPI-2 (102.4 ± 4.9% of vehicle control, Figure 5B) suggesting that TACE activity is necessary for DHPG mediated reduction of cell surface GluR1. Using the same technique, we next examined DHPG mediated trafficking of LFC-NPR. Similar to GluR1, surface LFC-NPR was reduced after DHPG treatment (84.3 ± 7.2% of vehicle control, Figure 5C). These changes in LFC-NPR were blocked by pretreatment of cultures with TAPI-2 (108.9 ± 16.0% of vehicle control, Figure 5C).
To examine the role of NPs in mGluR1/5 mediated AMPAR surface expression, we compared surface GluR1 in neurons cultured from WT mouse and NP TKO mouse cortex after DHPG treatment. WT neurons exhibited a significant reduction in surface GluR1 45 min after DHPG treatment (46.3 ± 8.3% of vehicle control, Figure 5D). By contrast, there was no change in surface GluR1 in NP TKO after treatment with DHPG (98.4 ± 9.7% of vehicle control).
An immunocytochemical approach was employed to validate the biochemical findings. Cultured hippocampal neurons (DIV 14) were treated with DHPG for 5 min. In parallel studies, we found that addition of an mGluR5 allosteric potentiator 3, 3′-diflourobenzaldazine (DFB; 50 μM; (O'Brien et al., 2003)) 5 min before DHPG stimulation increased the consistency of the DHPG-evoked response. Neurons were live labeled with GluR1 N-terminal specific antibody 45 min after DHPG treatment. Consistent with our results using biochemistry, surface GluR1 puncta density was reduced in DHPG treated neurons (47.6 ± 8.5% of vehicle control, Figure 5E and 5H). There was little change in surface GluR1 puncta density after DHPG treatment if neurons were pretreated with TAPI-2 (84.7 ± 12.6% of vehicle control, Figure 5E and 5H). Surface GluR1 puncta density was also examined in cultured neurons from NP TKO and WT mice. Again, surface GluR1 puncta density was reduced in DHPG treated neurons derived from WT mice (68.5 ± 6.0% of vehicle control, Figure 5F and 5I) while surface GluR1 puncta density was unchanged by DHPG treatment in neurons cultured from NP TKO mice (113.4 ± 6.3% of vehicle control, Figure 5F and 5I). These immunocytochemistry experiments confirm the requirement of TACE activity and NP expression in mGluR1/5 mediated AMPAR cell surface reduction.
We examined internalization of AMPAR in neurons derived from NP TKO and WT mice using an acid-strip immunocytochemical staining protocol (Carroll et al. 1999). Consistent with previous results (Snyder et al., 2001), DHPG treatment evoked an increase in internalized GluR1 in neurons derived from WT mice (Figure 5G and 5J). Internalized GluR1 was maximal within 5 min after addition of DHPG. This decreased at later time points (15 min and 45 min) presumably due to cycling of labeled GluR1 internalized through the internalized pool and reinsertion into the membrane. By contrast, DHPG treatment did not increase GluR1 internalization in neurons from NP TKO mice (Figure 5G and 5J). The largest difference in DHPG induced GluR1 internalization density between WT and NP TKO neurons was observed at the earliest time point (5 min). This suggests that NPs are required for mGluR1/5-dependent rapid endocytosis of GluR1.
mGluR1/5 dependent hippocampal LTD requires TACE and NPs
To examine the hypothesis that regulated cleavage of NPR plays a role in synaptic AMPAR trafficking, we examined the Schaffer collateral-CA1 synapse since it exhibits reliable mGluR1/5-dependent LTD that is mediated by postsynaptic AMPAR internalization (Camodeca et al., 1999; Fitzjohn et al., 1999; Huber et al., 2000; Kemp and Bashir, 1999; Palmer et al., 1997; Snyder et al., 2001). Using field potential recording, we examined a chemical form of mGluR1/5-dependent LTD in the CA1 region of WT mouse (3 - 4 week old) hippocampal slices. Application of the mGluR1/5 agonist DHPG (50 μM for 5 min) elicited a biphasic effect: a large depression of evoked field excitatory postsynaptic potential (fEPSP) slope during DHPG exposure, followed by a smaller depression of fEPSP slope that remained after washout (72 ± 3.0% of baseline at t = 76 min, n = 11; Figure 6A). This is similar to previous reports (Camodeca et al., 1999; Fitzjohn et al., 1999; Huber et al., 2001; Palmer et al., 1997).
Figure 6. mGluR1/5-dependent LTD at the Schaffer-CA1 synapse requires NPR and TACE-like activity.

(A) DHPG induced mGluR1/5-dependent LTD is blocked by general MMP inhibitor GM6001 (4 μM) and TACE inhibitor TAPI-2 (50 μM). Single representative traces from before (-10 min) and after (+76 min) DHPG are shown in the corresponding insets. All scale bars = 0.5 mV, 10 ms. All points represent the mean ± SEM of the corresponding group.
(B) DHPG induced mGluR1/5-dependent LTD is absent in NPR KO and NP TKO. Representative single traces from before (-15 min) and after (+75 min) DHPG are shown in the inset.
(C) PP-1Hz induced mGluR1/5-dependent LTD is blocked by TAPI-2 (50 μM) and is absent in NP TKO and NPR KO mice. Representative single traces from before (-10 min) and after (+50 min) are shown in the inset.
To assess the role of TACE in mGluR1/5-dependent LTD, two different inhibitors were used: GM6001 and TAPI-2. Treatment of slices with GM6001 or inactive GM6001 control compound (4 μM for 15 min starting 10 min before onset of DHPG) and TAPI-2 (50 μM for 20 min, starting 15 min before onset of DHPG) did not alter basal fEPSP slope or probability of release as indexed by paired-pulse ratio (PPR) (Supplemental Figure 9A), and had no effect on the initial, phasic EPSP depression during DHPG treatment. However, GM6001 completely blocked DHPG-induced LTD measured at t = 76 min (102 ± 5.6%, n = 6; p<0.01; Figure 6A) while application of the inactive GM6001 control compound (4 μM) failed to do so (71 ± 4.6% of baseline, n = 4, Figure 6A). Additionally, TAPI-2, an inhibitor of TACE, effectively blocked DHPG-induced LTD measured at t = 76 min (95 ± 1.5% of baseline, n = 6; p<0.01; Figure 6A). These results suggest that TACE activity is necessary for mGluR1/5-dependent hippocampal LTD.
We next examined chemical mGluR1/5-dependent LTD in the hippocampal slices prepared from WT, NPR KO, and NP TKO mice. Presynaptic function and basal synaptic transmission in hippocampal slices prepared from NPR KO and NP TKO at the Schaffer collateral-CA1 synapse exhibited no differences compared to WT as indexed by PPR and fiber volley/fEPSP slope functions, respectively (Supplemental Figures 9B and 9C). Moreover, the initial DHPG-induced depression of fEPSP slope was identical in WT, NPR KO, and NP TKO slices (Figure 6B). However, DHPG-induced LTD was significantly impaired in slices prepared from both NPR KO and NP TKO mice (96 ± 2.5% n = 7; p<0.01; 86 ± 4.1% n = 9; p<0.01, respectively) when compared to wild-type mice (60 ± 3.7 % n = 13, Figure 6B).
We next examined a synaptically induced form of mGluR1/5-dependent LTD at the Schaffer collateral-CA1 synapse. Paired-pulse stimulation repeated at 1 Hz for 15 min (PP-1Hz) at the Schaffer collateral-CA1 synapses produced an mGluR1/5 dependent form of LTD (Huber et al., 2000; Kemp and Bashir, 1999). Using field recordings in WT hippocampal slices, PP-1Hz induced LTD, consistent with previous studies (80 ± 2.2% of baseline at t = 50 min; n = 10; Figure 6C). Pretreatment of WT slices with TAPI-2 (50 μM) blocked PP-1Hz induced LTD (102 ± 5.6% of baseline at t = 50 min; n = 4; p<0.01, Figure 6C). Finally, PP-1Hz induced LTD was absent in NPR KO slices (92 ± 4.4% of baseline at t = 50 min; n = 7) and NP TKO slices (96 ± 1.0% of baseline at t = 50 min; n = 6; p<0.01).
mGluR1-dependent LTD in cultured cerebellar Purkinje cells is TACE and NP dependent
Previous work has shown that LTD in Purkinje cells cultured from embryonic mouse cerebellum requires activation of mGluR1, the only mGluR1/5 expressed in Purkinje cells, and LTD is expressed postsynaptically as a reduction in surface AMPA receptors (Aiba et al., 1994; Chung et al., 2003; Leitges et al., 2004; Linden, 2001; Matsuda et al., 2000; Steinberg et al., 2006; Wang and Linden, 2000). Here, we have examined LTD using a purely postsynaptic model in which iontophoretic test pulses of glutamate are applied to voltage-clamped Purkinje cells in culture. Following a baseline recording period, LTD was induced by pairing six 3 sec long depolarizing steps to 0 mV with six glutamate test pulses. When test pulses were resumed after pairing, LTD of the glutamate-evoked inward current was evident (52 ± 9.0% of baseline at t = 40 min, n = 7 cells, Figure 7A), as has been previously reported. When these experiments were repeated in cultures derived from NPR KO mice (98 ± 8.6% of baseline, n = 6) or NP TKO mice (108 ± 9.3% of baseline, n = 8), LTD was absent (Figure 7A). As a test of the specificity of the NPR KO interruption of LTD, a rescue experiment was attempted. NPR KO Purkinje cells were subject to biolistic transfection with a plasmid driving strong expression of wtNPR. This manipulation succeeded in restoring LTD to near-WT levels (61 ± 7.6% of baseline, n = 6, Figure 7A).
Figure 7. mGluR1-dependent LTD in cultured cerebellar Purkjine neurons requires NPR and TACE.
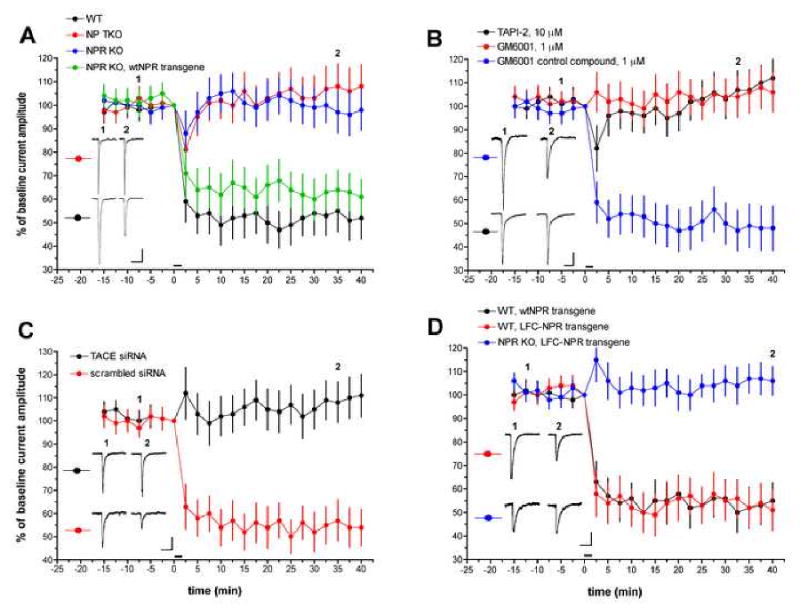
(A) LTD experiments in Purkinje cells derived from mice harboring mutations in NP genes. Test stimuli are iontophoretic pulses of glutamate. Following a baseline recording period, LTD was induced by six, 3 sec long depolarizing steps to 0 mV, each paired with a glutamate test pulse. This is indicated by the horizontal bar at t = 0 min. Exemplar current traces are single, unaveraged records corresponding to the points indicated on the time-course graph. Scale bars = 50 pA, 1 sec. WT (n = 7 cells); NP TKO (n = 8 cells); NPR KO (n = 6 cells); NPR KO, wtNPR transgene (n = 6 cells).
(B) LTD experiments in WT Purkinje cells treated with TACE inhibitors. Scale bars = 30 pA, 1 sec. TAPI-2 (n = 7 cells); GM6001 (n = 7 cells); GM6001 control compound (n = 7 cells).
(C) LTD experiments in WT Purkinje cells treated with siRNA directed against mouse TACE. Scale bars = 100 pA, 1 sec. TACE (m) siRNA (n = 8 cells); scrambled siRNA (n = 6 cells).
(D) LTD experiments in Purkinje cells transfected with either full-length NPR or LFC-NPR. Scale bars = 100 pA. 1 sec. WT, NPR transgene (n = 5 cells); WT, LFC-NPR transgene (n = 7 cells); NPR KO, LFC-NPR transgene (n = 8 cells).
To assess the role of TACE in cerebellar LTD, TAPI-2 (10 μM) and GM6001 (1 μM) were used. Both of these treatments produced a complete blockade of LTD induction (112 ± 8.6% and 106 ± 8.4% of baseline at t = 40 min, respectively, n = 7 for both groups, Figure 7B). However, application of the inactive GM6001 control compound (1 μM) failed to do so (48 ± 9.8% of baseline, n = 7, Figure 7B). This finding was confirmed by a TACE knockdown strategy using biolistic transfection of siRNA directed against mouse TACE (> 36 hours before recording; see Supplemental Figure 10 for siRNA confirmation). Mouse TACE siRNA-treated Purkinje cells showed no LTD (Figure 7C; 111 ± 9.3% of baseline at t = 40 min, n = 8) while Purkinje cells treated with a control scrambled siRNA showed robust LTD (54 ± 7.9% of baseline, n = 6), comparable to that seen in untreated cells.
If TACE cleavage of NPR is required for LTD induction then perhaps the TACE cleavage product, LFC-NPR, could rescue LTD induction when overexpressed in NPR KO Purkinje cells. This was not the case: LTD induction failed in this configuration (106 ± 6.2% of baseline at t = 40 min, n = 8; Figure 7D). In control experiments, neither LFC-NPR nor full-length NPR altered LTD induction when expressed in WT Purkinje cells (51 ± 8.7%, n = 7 and 55 ± 7.7%, n = 5, respectively; Figure 7D). This suggests that the action of NPR cleavage by TACE, not merely the LFC-NPR product, is required for LTD.
Efforts to identify point mutants of NPR that are resistant to TACE cleavage were not successful. Precedent indicates that MMP cleavage is regulated by interactions outside the site of cleavage. For instance, ADAM10 cleaves Ephrin-A5 only when Ephrin-A5 is bound to EphA3, presumably by interaction of ADAM10 with the ligand binding domain of EphA3 (Janes et al., 2005). Our model incorporates the failure of LFC-NPR to rescue mGluR1-dependent LTD by proposing that the regulated cleavage must occur at local sites of mGluR1/5 signaling (see Figure 8).
Figure 8. NPs exert bifunctional control of synaptic AMPAR via the regulated action of TACE.

Consistent with previous models, NPs can act as synaptogenic agents by recruiting AMPAR to synapses (Xu et al., 2003; Sia et al., 2007). However, upon activation of TACE, consequent to mGluR1/5 or other signaling events, TACE cleaves NPR, and enables NPR with associated NPs to cluster and co-cluster AMPAR at the site of TACE activity, and thereby increase the rate of AMPAR endocytosis. This process is essential for durable LTD.
Finally, none of these genetic manipulations of NPs or TACE appeared to affect basal synaptic strength as indexed by mEPSC amplitude and kinetics (Supplemental Table 1). Drugs and genetic manipulations can sometimes impact cerebellar LTD induction through their side effects on either mGluR1 function or voltage-sensitive Ca2+ channel function. None of the manipulations herein affected either depolarization-evoked or DHPG-evoked dendritic Ca2+ transients (Supplemental Table 1). Furthermore, when LTD was induced chemically, by bath application of the PKC activator phorbol-12,13-diacetate (PDA, 200 nM), a process which bypasses the initial signals for LTD induction, the same pattern of LTD blockade and rescue was seen with NP and TACE manipulations (Supplemental Figure 11). Taken together, these findings indicate that induction side effects cannot explain the observed blockade and rescue of LTD.
Discussion
A Model of Regulated NPR Cleavage in LTD
The present findings demonstrate that NPR, and its regulated cleavage by TACE, are essential for group 1 mGluR1/5-dependent LTD (Figure 8). Several steps in the process are experimentally defined. mGluR1/5 induces the cleavage of NPR, and this is blocked by TACE inhibitors. Cleaved NPR undergoes rapid internalization, and co-localizes with AMPAR in postsynaptic vesicular structures that are confirmed by immunoEM. mGluR1/5 stimulation results in accelerated AMPAR endocytosis and a reduction of steady state AMPAR levels, and both of these processes are blocked by TACE inhibitors, and are absent in NP TKO neurons. Finally, mGluR1/5-dependent LTD is blocked by TACE inhibitors, and is absent or dramatically reduced in neurons derived from mice lacking NPR. Our model envisions that the transmembrane domain of NPR prevents its being incorporated into endosomes. Cleavage of NPR allows it, together with linked NPs and their associated pool of AMPAR, to enter endosomes, and thereby increases the efficacy of AMPAR endocytosis. This mechanism appears to be broadly relevant at excitatory synapses since similar responses are evident at both the hippocampal Schaffer collatoral-CA1 synapse and in Purkinje cells in primary culture.
MMPs and synaptic plasticity
Our model implicates the MMP TACE in mGluR1/5-dependent LTD, a cellular model for synaptic plasticity that is thought to underlie learning and memory. Our data suggest that TACE is necessary and sufficient to cleave NPR. The possibility that other members of the ADAM family of MMPs may play a direct or indirect role in cleavage of NPR can not be excluded. Other ADAM candidates include ADAM10, which is localized at synapses by interaction with synapse-associated protein-97 (SAP97), an AMPAR associated protein that is involved in trafficking of AMPAR (Marcello et al., 2007; Mauceri et al., 2004). Additionally, other ADAMs may play a role in the proper targeting, localization, and/or complex formation of NPR, mGluR1/5, and AMPARs at specific sites of the synapses. For example, ADAM22, a catalytically inactive ADAM, associates with the postsynaptic complex of PSD-95 and stargazin, a complex which has also been described to regulate AMPAR trafficking (Fukata et al., 2006; Nicoll et al., 2006).
MMPs, in addition to TACE, have been implicated in synaptic plasticity. MMP-9 KO mice exhibit impairments in LTP in hippocampal slices that can be rescued by addition of recombinant MMP-9 (Nagy et al., 2006). Additionally, MMP-9 KO mice exhibit behavioral impairments in associative learning (Nagy et al., 2006). MMP protein expression is reported to be regulated by activity, and protein localization may be regulated in part by activity-dependent localization of MMP mRNA (Meighan et al., 2006; Nagy et al., 2007; Konopacki et al., 2007). The specific upstream pathways that activate MMPs and downstream cleavage targets that mediate expression of synaptic plasticity are not well understood. The observations that mGluR1/5 activated TACE cleavage of NPR is required for expression of mGluR1/5 dependent LTD provide an important precedent.
mGluR1/5 and AMPAR Trafficking
The molecular mechanisms governing mGluR1/5-dependent LTD are perhaps best understood in Purkinje cells. mGluR1 receptors activate a Ca2+ signaling cascade that activates PKC (Bear, 2000; Crepel and Krupa, 1988; Ito, 2002; Khodakhah and Armstrong, 1997; Linden and Connor, 1991). Of the various isoforms of PKC, the classical PKCα has been implicated in mGluR1-dependent LTD by KO and RNAi studies, as well as by a transgenic mouse model expressing a PKC inhibitor (isoform non-specific) selectively in Purkinje cells (De Zeeuw et al., 1998; Goossens et al., 2001; Leitges et al., 2004). One of the actions of PKCα is to phosphorylate GluR2 at serine 880, and this has the effect of reducing its binding to the scaffolding protein GRIP (Chung et al., 2003). By contrast, the binding of the BAR domain containing protein PICK1 is not altered by phosphorylation and consequently, there results an increase in PICK1 binding to GluR2(Jin et al., 2006). The physiological correlate of enhanced PICK1 binding is an increase in GluR2 endocytosis rate and a reduction in steady state level of synaptic AMPAR (Kim et al., 2001; Steinberg et al. 2006).
In the hippocampus, mGluR1/5-dependent LTD of the Schaffer collateral-CA1 synapse shares several mechanistic similarities to mGluR1-dependent LTD in the Purkinje cells. Schaffer collatoral/CA1 synapse LTD induced by mGluR1/5 is mediated by a postsynaptic increase in the rate of AMPAR endocytosis (Snyder et al., 2001). However, hippocampal mGluR1/5-dependent LTD is insensitive to PKC specific inhibitors and is sensitive to phosphatase inhibitors (Schnabel et al., 1999, 2001). Our current data indicating a role for TACE and NPR in mGluR1/5-dependent LTD of the Schaffer collateral-CA1 synapse beg the question of how TACE might be activated if, as reported, PKC inhibitors do not block LTD. There is substantial diversity of PKC isoforms and perhaps the critical kinase is resistant to inhibitors that have been used including chelerythrine, Ro 31-8220, Go6976 (inhibits PKC α, β, and μ isoforms) and Go6983 (inhibits PKC α, β, γ, δ, and ζ isoforms) (Schnabel et al.,2001). We also note that mGluR1/5-dependent LTD in the medial perforant path of the dentate gyrus is blocked by PKC inhibitors (Huang et al.,1999).
Another striking feature of mGluR1/5-dependent LTD at the Schaffer collateral-CA1 synapse is that its maintenance, after as brief a time as 10 min, is dependent on de novo protein translation of preexisting mRNA (Snyder et al., 2001, Huber 2000). The identity of the translated proteins that are required for this process is currently unknown. However, Arc/Arg3.1 is a candidate for such a protein. Its mRNA is robustly induced and transported to dendrites where is may be locally translated at synaptic sites (Lyford et al.,1995; Steward et al.,1998). Additionally, Arc/Arg3.1 interacts with proteins involved in endocytotic machinery and modulates trafficking of AMPAR (Chowdhury et al.,2006; Shepherd et al., 2006). Currently, the role of Arc/Arg3.1 in mGluR1/5-dependent LTD is not known but may involve maintenance of mGluR1/5-dependent LTD expression which has been shown to be protein synthesis dependent (Snyder et al., 2001). It is possible that TACE dependent cleavage of NPR may mediate the early phase of mGluR1/5-dependent LTD and Arc/Arg3.1 may mediate the maintenance of LTD expression.
Neuronal Pentraxins and Synaptic Plasticity: Emerging Insights
Narp and NP1 have previously been implicated in synapse formation (O'Brien et al., 2002; O'Brien et al., 1999; Xu et al., 2003; Sia et al., 2007). Like Narp and NP1, NPR has been shown to be synaptogenic in an in-vitro system that mimics synapse formation where NPR clusters transfected GluR4 in cultured glia (Sia et al., 2007). Moreover, the number of GluR4 puncta are reduced in interneurons derived from NP TKO mice compared to WT. The requirement of NPR for clustering GluR4 may be limited to interneurons by associations with interneuron specific proteins or by other mechanisms. In contrast, we did not observe any differences in basal synaptic strength or AMPAR number between NP KOs (single and TKO) and WT preparations examining excitatory neurons or excitatory neurotransmission (Supplemental Figure 9 and Supplemental Table 1). These results are consistent with a previous report that GluR1 clusters in NP single KO or TKO hippocampal neurons are identical to WT (Bjartmar et al., 2006). Glutamatergic synapse function was reduced in cultured retinal ganglion cells derived from Narp/NP1 double KO compared to WT, but normal in more mature cultures. Accordingly, the contribution of NPs to synaptogenesis appears to be developmentally regulated and cell type specific.
While the physiology of synapse formation and synaptic depression, as revealed in the present study, appear overtly reciprocal, the ability of NPs to bind and cluster AMPAR may be central to both. In synapse formation, NPs may capture and cluster AMPAR at synapses, while in synaptic depression, NPs appear to capture AMPAR at sites of regulated endocytosis. It is notable that the present model of NPs in LTD is dependent on NPR, which shows a marked developmental increase during the 2nd and 3rd postnatal weeks in the hippocampus (Supplemental Figure 12). Early in development, NPR may function primarily to anchor secreted Narp and NP1 at sites of emerging synapses. In more mature synapses, NPR may play a central role in forming a signaling complex that is required for mGluR1/5-dependent LTD. Our model proposes that the transmembrane domain is required for proper targeting of NPR or for the formation of a signaling complex that is present at mature synapses where rapid, mGluR1/5-dependent cleavage by TACE is required for mGluR1/5-dependent LTD.
Several questions remain to be addressed to fully integrate the present findings into our current understanding of synaptic plasticity. The signaling complex that serves as a molecular link to NPR, mGluR1/5, and TACE, or TACE-like proteases is unknown, and the absence or presence of this complex may confer the bifunctional role of NPs in emerging versus mature synapses. Moreover, physiological interactions as well as epistatic analysis of NPs with Arc/Arg.1, PICK1 and proteins that mediate clathrin-dependent endosome formation remain to be defined. NPs are present in many compartments of the spine including the presynaptic compartment (Xu et al., 2003; and see Figure 4) suggesting that they may contribute to forms of plasticity in addition to those described here, such as synapse elimination (Bjartmar et al., 2006), maintenance of pre- to post- synaptic ratio, or bidirectional signaling at synapses.
Experimental Procedures
DNA constructs, antibodies, cell culture, transfection, Western blot analysis, co-IP analysis, identification of cleavage sites, immunoelectron microscopy, immunohistochemistry, immunocytochemistry, cleavage assay, knockdown of TACE using siRNA, and statistical analysis are included in Supplemental Experimental Procedures.
Image acquisition, analysis, and quantification
Images of stained heterologous cells were obtained using a Nikon E800 epifluorescence microscope. Images of stained neurons were obtained using a UltraVIEW spinning disk confocal microscope fitted with a Nikon Eclipse TE 200 microscope. Image analysis was performed using Metamorph imaging software (Universal Imaging, Downingtown, PA). See Supplemental Experimental Procedures for details.
Electrophysiology
fEPSPs were recorded from the stratum radiatum of acute rat hippocampal slices (P21-P28) in response to stimulation of the Schaffer collateral-commissural pathway. LTD was induced by application of mGluR1/5-selective agonist 3,5-dihydroxyphenylglycine (DHPG; 50 μM, 5min) or by a paired pulse low frequency stimulation protocol (PP-1Hz: 2 pulses with 50 ms interstimulus interval at 1 Hz for 15 min) with D-AP5 (50 μM). Strength of synaptic response was measured as fEPSPs slope. See Supplemental Experimental Procedures for details.
Cultures of embryonic mouse cerebellum were prepared as previously described and recordings of voltage-clamped Purkinje cells were performed according to our previously published method (Leitges 2004). LTD was induced by LTD-inducing pairing stimuli consisting of six, 3-s-long depolarizations to 0mV, each delivered together with a test pulse of glutamate. See Supplemental Experimental Procedures for more details and detailed description of methods for Ca2+ imaging and gene delivery.
Supplementary Material
Acknowledgments
We thank Michael Chang, Sung Jin Park, and Jason Shepherd for helpful discussion on the manuscript as well as technical assistance. We thank Maurice D. Hinson for technical assistance with NPR mutant constructs, Ya-Xian Wang for help with the immunogold studies, and Roland Bock for cerebellum cultures. We also thank Yoko Sekine-Aizawa and Richard Huganir for providing BBS tagged GluR1 and helpful discussion. This study was supported by National Institutes of Health grants 5R01NS039156-08 (P.F.W), R37MM51106 (D.J.L), and in part by the National Eye Institute's Visual Neuroscience Training Program (R.W.C.) and the NIDCD Intramural Program (R.S.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- Arribas J, Coodly L, Vollmer P, Kishimoto TK, Rose-John S, Massague J. Diverse cell surface protein ectodomains are shed by a system sensitive to metalloprotease inhibitors. J Biol Chem. 1996;271:11376–11382. doi: 10.1074/jbc.271.19.11376. [DOI] [PubMed] [Google Scholar]
- Bear MF, Linden DJ. The mechanisms and meaning of long-term synaptic depression in the mammalian brain. Baltimore: Johns Hopkins University Press; 2000. [Google Scholar]
- Beckmann G, Hanke J, Bork P, Reich JG. Merging extracellular domains: fold prediction for laminin G-like and amino-terminal thrombospondin-like modules based on homology to pentraxins. J Mol Biol. 1998;275:725–730. doi: 10.1006/jmbi.1997.1510. [DOI] [PubMed] [Google Scholar]
- Bjartmar L, Huberman AD, Ullian EM, Renteria RC, Liu X, Xu W, Prezioso J, Susman MW, Stellwagen D, Stokes CC, et al. Neuronal pentraxins mediate synaptic refinement in the developing visual system. Journal of Neuroscience. 2006 doi: 10.1523/JNEUROSCI.4212-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RA. Tumor necrosis factor-alpha converting enzyme. Int J Biochem Cell Biol. 2002;34:1–5. doi: 10.1016/s1357-2725(01)00097-8. [DOI] [PubMed] [Google Scholar]
- Blobel CP. Remarkable roles of proteolysis on and beyond the cell surface. Curr Opin Cell Biol. 2000;12:606–612. doi: 10.1016/s0955-0674(00)00139-3. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- Camodeca N, Breakwell NA, Rowan MJ, Anwyl R. Induction of LTD by activation of group I mGluR in the dentate gyrus in vitro. Neuropharmacology. 1999;38:1597–1606. doi: 10.1016/s0028-3908(99)00093-3. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, Xia H, Luscher C, Altschuler Y, Nicoll RA, Malenka RC, von Zastrow M. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Steinberg JP, Huganir RL, Linden DJ. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science. 2003;300:1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]
- Crepel F, Krupa M. Activation of protein kinase C induces a long-term depression of glutamate sensitivity of cerebellar Purkinje cells. An in vitro study. Brain Res. 1988;458:397–401. doi: 10.1016/0006-8993(88)90486-6. [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ, Oberdick J. Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron. 1998;20:495–508. doi: 10.1016/s0896-6273(00)80990-3. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR, Tchelingerian JL, Jacque CM. Expression of extracellular matrix degrading enzymes during migration of xenografted brain cells. Neuropathol Appl Neurobiol. 1999;25:54–62. doi: 10.1046/j.1365-2990.1999.00158.x. [DOI] [PubMed] [Google Scholar]
- Dodds DC, Omeis IA, Cushman SJ, Helms JA, Perin MS. Neuronal pentraxin receptor, a novel putative integral membrane pentraxin that interacts with neuronal pentraxin 1 and 2 and taipoxin- associated calcium-binding protein 49. J Biol Chem. 1997;272:21488–21494. doi: 10.1074/jbc.272.34.21488. [DOI] [PubMed] [Google Scholar]
- Domeniconi M, Zampieri N, Spencer T, Hilaire M, Mellado W, Chao MV, Filbin MT. MAG induces regulated intramembrane proteolysis of the p75 neurotrophin receptor to inhibit neurite outgrowth. Neuron. 2005;46:849–855. doi: 10.1016/j.neuron.2005.05.029. [DOI] [PubMed] [Google Scholar]
- Emsley J, White HE, O'Hara BP, Oliva G, Srinivasan N, Tickle IJ, Blundell TL, Pepys MB, Wood SP. Structure of pentameric human serum amyloid P component. Nature. 1994;367:338–345. doi: 10.1038/367338a0. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Ethell DW. Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J Neurosci Res. 2007;85:2813–2823. doi: 10.1002/jnr.21273. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Kingston AE, Lodge D, Collingridge GL. DHPG-induced LTD in area CA1 of juvenile rat hippocampus; characterisation and sensitivity to novel mGlu receptor antagonists. Neuropharmacology. 1999;38:1577–1583. doi: 10.1016/s0028-3908(99)00123-9. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- Galko MJ, Tessier-Lavigne M. Function of an axonal chemoattractant modulated by metalloprotease activity. Science. 2000;289:1365–1367. doi: 10.1126/science.289.5483.1365. [DOI] [PubMed] [Google Scholar]
- Goodman AR, Cardozo T, Abagyan R, Altmeyer A, Wisniewski HG, Vilcek J. Long pentraxins: an emerging group of proteins with diverse functions. Cytokine Growth Factor Rev. 1996;7:191–202. doi: 10.1016/1359-6101(96)00019-6. [DOI] [PubMed] [Google Scholar]
- Goossens J, Daniel H, Rancillac A, van der Steen J, Oberdick J, Crepel F, De Zeeuw CI, Frens MA. Expression of protein kinase C inhibitor blocks cerebellar long-term depression without affecting Purkinje cell excitability in alert mice. J Neurosci. 2001;21:5813–5823. doi: 10.1523/JNEUROSCI.21-15-05813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper NM, Karran EH, Turner AJ. Membrane protein secretases. Biochem J. 1997;321(Pt 2):265–279. doi: 10.1042/bj3210265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LQ, Rowan MJ, Anwyl R. Role of protein kinases A and C in the induction of mGluR-dependent long-term depression in the medial perforant path of the rat dentate gyrus in vitro. Neurosci Lett. 1999;274:71–74. doi: 10.1016/s0304-3940(99)00561-3. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis--dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- Ito M. The molecular organization of cerebellar long-term depression. Nat Rev Neurosci. 2002;3:896–902. doi: 10.1038/nrn962. [DOI] [PubMed] [Google Scholar]
- Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;123:291–304. doi: 10.1016/j.cell.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Jin W, Ge WP, Xu J, Cao M, Peng L, Yung W, Liao D, Duan S, Zhang M, Xia J. Lipid binding regulates synaptic targeting of PICK1, AMPA receptor trafficking, and synaptic plasticity. J Neurosci. 2006;26:2380–2390. doi: 10.1523/JNEUROSCI.3503-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp N, Bashir ZI. Induction of LTD in the adult hippocampus by the synaptic activation of AMPA/kainate and metabotropic glutamate receptors. Neuropharmacology. 1999;38:495–504. doi: 10.1016/s0028-3908(98)00222-6. [DOI] [PubMed] [Google Scholar]
- Khodakhah K, Armstrong CM. Induction of long-term depression and rebound potentiation by inositol trisphosphate in cerebellar Purkinje neurons. Proc Natl Acad Sci U S A. 1997;94:14009–14014. doi: 10.1073/pnas.94.25.14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci U S A. 2001;98:11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick LL, Matzuk MM, Dodds DC, Perin MS. Biochemical interactions of the neuronal pentraxins. Neuronal pentraxin (NP) receptor binds to taipoxin and taipoxin-associated calcium-binding protein 49 via NP1 and NP2. J Biol Chem. 2000;275:17786–17792. doi: 10.1074/jbc.M002254200. [DOI] [PubMed] [Google Scholar]
- Konopacki FA, Rylski M, Wilczek E, Amborska R, Detka D, Kaczmarek L, Wilczynski GM. Synaptic localization of seizure-induced matrix metalloproteinase-9 mRNA. Neuroscience. 2007 doi: 10.1016/j.neuroscience.2007.08.026. [DOI] [PubMed] [Google Scholar]
- Leitges M, Kovac J, Plomann M, Linden DJ. A unique PDZ ligand in PKCalpha confers induction of cerebellar long-term synaptic depression. Neuron. 2004;44:585–594. doi: 10.1016/j.neuron.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Linden DJ. The expression of cerebellar LTD in culture is not associated with changes in AMPA-receptor kinetics, agonist affinity, or unitary conductance. Proc Natl Acad Sci U S A. 2001;98:14066–14071. doi: 10.1073/pnas.241384598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, Gilbert DJ, Jenkins NA, Lanahan AA, Worley PF. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron. 1995;14:433–445. doi: 10.1016/0896-6273(95)90299-6. [DOI] [PubMed] [Google Scholar]
- Marcello E, Gardoni F, Mauceri D, Romorini S, Jeromin A, Epis R, Borroni B, Cattabeni F, Sala C, Padovani A, et al. Synapse-associated protein-97 mediates alpha-secretase ADAM10 trafficking and promotes its activity. J Neurosci. 2007;27:1682–1691. doi: 10.1523/JNEUROSCI.3439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Launey T, Mikawa S, Hirai H. Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. Embo J. 2000;19:2765–2774. doi: 10.1093/emboj/19.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauceri D, Cattabeni F, Di Luca M, Gardoni F. Calcium/calmodulin-dependent protein kinase II phosphorylation drives synapse-associated protein 97 into spines. J Biol Chem. 2004;279:23813–23821. doi: 10.1074/jbc.M402796200. [DOI] [PubMed] [Google Scholar]
- McFarlane S. Metalloproteases: carving out a role in axon guidance. Neuron. 2003;37:559–562. doi: 10.1016/s0896-6273(03)00089-8. [DOI] [PubMed] [Google Scholar]
- Meighan SE, Meighan PC, Choudhury P, Davis CJ, Olson ML, Zornes PA, Wright JW, Harding JW. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J Neurochem. 2006;96:1227–1241. doi: 10.1111/j.1471-4159.2005.03565.x. [DOI] [PubMed] [Google Scholar]
- Moss ML, Rasmussen FH. Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal Biochem. 2007;366:144–148. doi: 10.1016/j.ab.2007.04.043. [DOI] [PubMed] [Google Scholar]
- Nagy V, Bozdagi O, Huntley GW. The extracellular protease matrix metalloproteinase-9 is activated by inhibitory avoidance learning and required for long-term memory. Learn Mem. 2007;14:655–664. doi: 10.1101/lm.678307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy V, Bozdagi O, Matynia A, Balcerzyk M, Okulski P, Dzwonek J, Costa RM, Silva AJ, Kaczmarek L, Huntley GW. Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J Neurosci. 2006;26:1923–1934. doi: 10.1523/JNEUROSCI.4359-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311:1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- O'Brien JA, Lemaire W, Chen TB, Chang RS, Jacobson MA, Ha SN, Lindsley CW, Schaffhauser HJ, Sur C, Pettibone DJ, et al. A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol Pharmacol. 2003;64:731–740. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- O'Brien R, Xu D, Mi R, Tang X, Hopf C, Worley P. Synaptically targeted narp plays an essential role in the aggregation of AMPA receptors at excitatory synapses in cultured spinal neurons. J Neurosci. 2002;22:4487–4498. doi: 10.1523/JNEUROSCI.22-11-04487.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P. Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron. 1999;23:309–323. doi: 10.1016/s0896-6273(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997;36:1517–1532. doi: 10.1016/s0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- Rudenko G, Hohenester E, Muller YA. LG/LNS domains: multiple functions -- one business end? Trends Biochem Sci. 2001;26:363–368. doi: 10.1016/s0968-0004(01)01832-1. [DOI] [PubMed] [Google Scholar]
- Rudenko G, Nguyen T, Chelliah Y, Sudhof TC, Deisenhofer J. The structure of the ligand-binding domain of neurexin Ibeta: regulation of LNS domain function by alternative splicing. Cell. 1999;99:93–101. doi: 10.1016/s0092-8674(00)80065-3. [DOI] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick IC, Collingridge GL. An investigation into signal transduction mechanisms involved in DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology. 1999;38:1585–1596. doi: 10.1016/s0028-3908(99)00062-3. [DOI] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick IC, Collingridge GL. Protein phosphatase inhibitors facilitate DHPG-induced LTD in the CA1 region of the hippocampus. Br J Pharmacol. 2001;132:1095–1101. doi: 10.1038/sj.bjp.0703905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine-Aizawa Y, Huganir RL. Imaging of receptor trafficking by using alpha-bungarotoxin-binding-site-tagged receptors. Proc Natl Acad Sci U S A. 2004;101:17114–17119. doi: 10.1073/pnas.0407563101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia GM, Beique JC, Rumbaugh G, Cho R, Worley PF, Huganir RL. Interaction of the N-Terminal Domain of the AMPA Receptor GluR4 Subunit with the Neuronal Pentraxin NP1 Mediates GluR4 Synaptic Recruitment. Neuron. 2007;55:87–102. doi: 10.1016/j.neuron.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the immediate early gene Arc to localize selectively near activated postsynaptic sites on neuronal dendrites. Neuron. 1998;21:741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- Tomimatsu Y, Idemoto S, Moriguchi S, Watanabe S, Nakanishi H. Proteases involved in long-term potentiation. Life Sci. 2002;72:355–361. doi: 10.1016/s0024-3205(02)02285-3. [DOI] [PubMed] [Google Scholar]
- Tsui CC, Copeland NG, Gilbert DJ, Jenkins NA, Barnes C, Worley PF. Narp, a novel member of the pentraxin family, promotes neurite outgrowth and is dynamically regulated by neuronal activity. J Neurosci. 1996;16:2463–2478. doi: 10.1523/JNEUROSCI.16-08-02463.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Linden DJ. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron. 2000;25:635–647. doi: 10.1016/s0896-6273(00)81066-1. [DOI] [PubMed] [Google Scholar]
- Xiao MY, Zhou Q, Nicoll RA. Metabotropic glutamate receptor activation causes a rapid redistribution of AMPA receptors. Neuropharmacology. 2001;41:664–671. doi: 10.1016/s0028-3908(01)00134-4. [DOI] [PubMed] [Google Scholar]
- Xu D, Hopf C, Reddy R, Cho RW, Guo L, Lanahan A, Petralia RS, Wenthold RJ, O'Brien RJ, Worley P. Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron. 2003;39:513–528. doi: 10.1016/s0896-6273(03)00463-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.