

Cells respond to unfolded and misfolded proteins in the endoplasmic reticulum by activating the unfolded protein response, which arrests protein synthesis and removes the accumulated, aberrant protein load, so that normal function of the organelle can be restored as soon as possible. This response has been linked to a variety of diseases, including neurodegenerative conditions, immune response disorders, cancer, diabetes, and fatty liver disease (1). The transcription factor X-box binding protein 1 (XBP1) controls the expression of genes required for the unfolded protein response, but on page 1492 in this issue, Lee et al. (2) establish an unexpected, independent role of XBP1 in regulating lipid synthesis in the liver. The finding provides new opportunities to develop treatments for conditions such as hyperlipidemia and fatty liver disease in humans.
XBP1 binds to promoter elements of genes that encode chaperone proteins that assist with protein folding in the endoplasmic reticulum. Global deletion of the Xbp1 gene in mice results in the deaths of embryos from anemia, secondary to reduced numbers of hematopoietic progenitor cells and hypoplastic fetal livers (3). XBP1 is also required for plasma cell differentiation and the development of cardiac muscle and secretory tissues.
To study the function of XBP1 in adult mice, Lee et al. deleted the Xbp1 gene exclusively in the adult mouse liver. This unexpectedly reduced liver fatty acid and cholesterol synthesis by ∼85 to 90%, thus lowering concentrations of plasma cholesterol and triglycerides. Much of the cholesterol and triglycerides transported in the blood are in apolipoprotein B (apoB) –containing particles secreted by the liver, so a block in apoB secretion would result in low plasma concentrations of these lipids. Given the established role of XBP1 in promoting protein folding and secretion from cells, the observed low blood cholesterol and triglyceride concentrations could be secondary to impaired secretion of apoB and its associated lipids from liver. Surprisingly, apoB secretion from liver was unaffected by the loss of XBP1; rather, the underlying mechanism of the hypolipidemia is the regulated expression of select genes involved in lipogenesis by XBP1.
Sterol regulatory element–binding protein-1c (SREBP-1c) and carbohydrate response element–binding protein (ChREBP) are transcription factors that stimulate fatty acid synthesis in response to insulin and high glucose concentrations in plasma, respectively. Action of SREBP-1c is enhanced by the transcription factor liver X receptor (LXR). SREBP-2 stimulates the expression of genes involved in cholesterol synthesis (4). SREBPs are generated as inactive precursors in the membrane of the endoplasmic reticulum (see the figure). To be active, the amino-terminal domain of an SREBP (nSREBP) must be released from the membrane to enter the nucleus and activate target genes. This process requires SREBP cleavage–activating protein (SCAP), which escorts SREBPs from the endoplasmic reticulum to the Golgi. There, two proteases (S1P and S2P) cleave SREBPs to release the amino terminus from the membrane (5).
Regulators of lipid synthesis and the unfolded protein response.
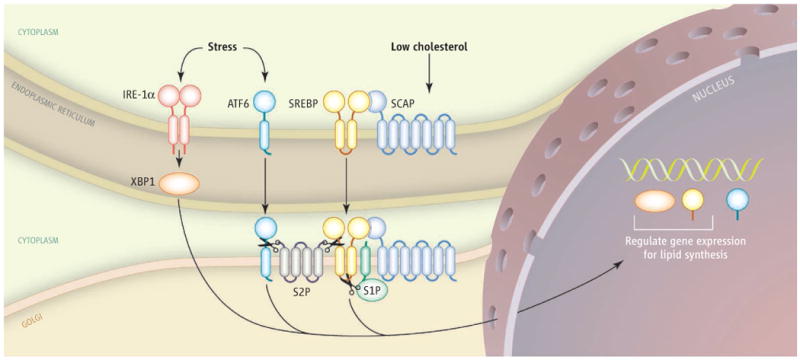
The transcription factors XBP1, ATF6, and SREBP require processing to active forms before entering the nucleus, where SERBPs and XBP1 control the expression of genes involved in lipogenesis and ATF6 activates XBP1. The activation of XBP1 and ATF6 in response to stress occurs independently of the regulation of SREBPs by cholesterol.
A transcription factor exhibits dual roles, regulating genes that respond to improperly folded proteins and genes that control lipid synthesis.
In mice that lack the Scap gene in liver, expression of nSREBPs and the expression of genes involved in fatty acid and cholesterol synthesis decrease (6), resulting in lower plasma triglyceride and cholesterol concentrations, similar to that observed in XBP1-deficient mice. Whereas SREBPs coordinately regulate all cholesterol and fatty acid biosynthetic genes, regulation of lipid synthesis by XBP1 appears to be more complex because the expression of nSREBP-1c or nSREBP-2 in XBP1-deficient livers was not reduced. This suggests that the decreased lipid synthesis rate is independent of SREBP-mediated transcriptional regulation.
Consistent with the normal concentration of nSREBPs in XBP1-deficent livers, expression of only three genes involved in fatty acid and triglyceride synthesis decreased—acetyl–coenzyme A (CoA) carboxylase 2 (ACC2), stearoyl–CoA desaturase 1, and acyl–CoA:diacylglycerol acyltransferase 1. By contrast, the expression of genes encoding enzymes (ACC1 and fatty acid synthase) that synthesize long-chain fatty acids was not reduced. Similarly, the expression of genes encoding 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase and HMG-CoA synthase, key cholesterol biosynthetic enzymes, was unchanged in the absence of XBP1. Nevertheless, the rates of hepatic fatty acid and sterol synthesis were markedly reduced. These observations raise the possibility that in addition to the transcriptional changes observed in the XBP1-deficient livers, posttranscriptional regulatory mechanisms may contribute to the phenotype. One such modification that alters both cholesterol and fatty acid synthesis is phosphorylation and inactivation of HMG–CoA reductase (7) and ACCs (8) by adenosine 5′-monophosphate (AMP)–activated protein kinase (AMPK). AMPK is activated by stress that raises the cellular ratio of AMP to ATP (adenosine 5′-triphosphate), and it modulates many processes involved in cellular energetics. Whether such stress is present in XBP1-deficient livers is not yet known.
Although the fields of lipid metabolism and the unfolded protein response have developed largely in parallel, indirect links between the disciplines have emerged. For example, activating transcription factor 6 (ATF6) is activated through transport from the endoplasmic reticulum to the Golgi, where it is cleaved by S2P, the same protease that cleaves SREBPs (9). ATF6 activates Xbp1 (among other targets), which in turn increases phospholipid synthesis, a process that may support the creation of new membrane in endoplasmic reticulum, allowing it to handle the stress of accumulated, improperly folded proteins (10). Another link between the two fields was revealed through studies of mice deficient in leptin, a hormone that controls appetite and fat metabolism. These mice are obese, insulin-resistant, and have high concentrations of triglycerides in their livers. These fatty livers also show molecular evidence of endoplasmic reticulum stress, which includes activation of RNA-activated protein kinase–like eukaryotic initiation factor 2α kinase (PERK) and inositol-requiring enzyme-1α (IRE-1α), resident membrane proteins of the organelle (11). PERK phosphorylates eukaryotic translational initiation factor 2α, which attenuates protein translation. Activated IRE-1α excises a portion of the messenger RNA encoding XBP1, which then is translated into the XBP1 protein that translocates to the nucleus to activate target genes (12, 13). IRE-1α also triggers a signaling pathway (the c-Jun amino-terminal kinase cascade) that blocks insulin signaling (14), thus establishing a causal link between endoplasmic reticulum stress, cellular insulin resistance, and excess triglyceride accumulation in liver.
Even though XBP1 is well known as a transcriptional regulator of the unfolded protein response, its role in regulating lipid synthesis in liver appears to be independent of stress in the endoplasmic reticulum. XBP1 is present in the nucleus of normal mouse liver and its expression is induced after fructose feeding, suggesting that an alternative mechanism for regulating XBP1 exists in hepatocytes. Further studies are required to reveal this mechanism and to determine whether the regulation of lipid synthesis by XBP1 and SREBPs occur independently or in concert.
References
- 1.Zhao L, Ackerman SL. Curr Opin Cell Biol. 2006;18:444. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Science. 2008;320:1492. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reimold AM, et al. Genes Dev. 2000;14:152. [PMC free article] [PubMed] [Google Scholar]
- 4.Horton JD, Goldstein JL, Brown MS. J Clin Invest. 2002;109:1125. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldstein JL, Rawson RB, Brown MS. Arch Biochem Biophys. 2002;397:139. doi: 10.1006/abbi.2001.2615. [DOI] [PubMed] [Google Scholar]
- 6.Matsuda M, et al. Genes Dev. 2001;15:1206. doi: 10.1101/gad.891301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato R, Goldstein JL, Brown MS. Proc Nat Acad Sci USA. 1993;90:9261. doi: 10.1073/pnas.90.20.9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carling D, Zammita VA, Hardie DG. FEBS Lett. 1987;223:217. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- 9.Ye J, et al. Mol Cell. 2000;6:1355. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 10.Sriburi R, et al. J Biol Chem. 2007;282:7024. doi: 10.1074/jbc.M609490200. [DOI] [PubMed] [Google Scholar]
- 11.Özcan U, et al. Science. 2004;306:457. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 12.Calfon M, et al. Nature. 2002;415:92. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 13.Lee K, et al. Genes Dev. 2002;16:452. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urano F, et al. Science. 2000;287:664. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]