

Abstract
The CD209 gene family that encodes C-type lectins in primates includes CD209 (DC-SIGN), CD209L (L-SIGN) and CD209L2. Understanding the evolution of these genes can help understand the duplication events generating this family, the process leading to the repeated neck region and identify protein domains under selective pressure. We compiled sequences from 14 primates representing 40 million years of evolution and from three non-primate mammal species. Phylogenetic analyses used Bayesian inference, and nucleotide substitutional patterns were assessed by codon-based maximum likelihood. Analyses suggest that CD209 genes emerged from a first duplication event in the common ancestor of anthropoids, yielding CD209L2 and an ancestral CD209 gene, which, in turn, duplicated in the common Old World primate ancestor, giving rise to CD209L and CD209. KA/KS values averaged over the entire tree were 0.43 (CD209), 0.52 (CD209L) and 0.35 (CD209L2), consistent with overall signatures of purifying selection. We also assessed the Toll-like receptor (TLR) gene family, which shares with CD209 genes a common profile of evolutionary constraint. The general feature of purifying selection of CD209 genes, despite an apparent redundancy (gene absence and gene loss), may reflect the need to faithfully recognize a multiplicity of pathogen motifs, commensals and a number of self-antigens.
Keywords: C-type lectins, HIV, Ebola, Mycobacteria, innate immunity, DC-SIGN
Introduction
The CD209 family of genes codes for DC-SIGN (dendritic cell-specific ICAM-grabbing non-integrin) and related proteins L-SIGN (for liver/lymph node ‘L’-SIGN, encoded by CD209L) and CD209L2. These homologous genes cluster on chromosome 19p13.3.
DC-SIGN is expressed on phagocytic cells such as dendritic cells and macrophages, whereas L-SIGN expression is restricted to lymph node sinus endothelia and hepatic sinusoidal endothelium. The third homologue, CD209L2, is absent in humans but present in other primates. In the Rhesus macaque, CD209L2 is expressed in liver, spleen, lymph node, heart and skin.1 One or more DC-SIGN-like proteins have been reported in mice, rats and dogs. These proteins are type-II C-type lectin receptors with roles as cell-adhesion receptors and in innate immunity as pathogen receptors.2–4 DC-SIGN and L-SIGN recognize a vast range of bacteria, mycobacteria, viruses and protozoa (reviewed in Koppel et al.2). Their function depends on a carbohydrate-recognition domain separated from a transmembrane region by a neck region made up of several 23-amino acid repeats. DC-SIGN has affinity for mannose oligosaccharides and fucose-containing moieties whereas L-SIGN binds only to mannose oligosaccharides (reviewed in Koppel et al.2). The neck region plays a role in the orientation and flexibility of the carbohydrate-recognition domain. The type and number of the neck-region repeats are important in dimer formation and stabilization of protein tetramers.5 Thus, neck-length variation could influence the pathogen-binding properties of these lectins. In previous analyses in primates,1 CD209 and CD209L have variable numbers of repeats. This contrasts with the single partial repeat that characterizes primate CD209L2, and that is a general feature of the neck region of other mammal CD209-like genes.
The importance of DC-SIGN family members in pathogen recognition as well as the importance of dendritic cells in pathogenesis of human immunodeficiency virus, Ebola and Mycobacterium tuberculosis infection, together with their particular genomic organization make these genes an excellent model system to investigate the mode and intensity of selective pressures that may act on pathogen defense genes.6 As a category, immunity- and defense-related genes have experienced by far the most positive selection in humans and other organisms.7
The aim of the present study was to extend the analysis of the distribution of the three recognized CD209 family genes in primates to thoroughly assess the evolutionary history of this gene family, to identify possible domains and amino acids under selective pressure, to date the gene-duplication events resulting in this family and to understand the evolutionary process giving rise to the neck repeat region. To trace the evolutionary history of these genes, gene-coding sequences are determined for a representative number of primates (hominoids, Old World and New World monkeys), followed by the analysis of amino-acid substitutional patterns in the framework of the accepted primate phylogeny.8 The analyses result in global estimates of the patterns of evolution (purifying, neutral, positive selective pressure) as well as shed light on episodes of adaptive evolution at specific sites.6
To help build a more contextual interpretation of the CD209 family data, we investigate in parallel the evolution of a second family of major innate immunity microbial sensors, namely the Toll-like receptor (TLR) gene family. TLRs are pattern-recognition receptors that identify specific pathogen-associated molecular patterns (PAMPs, conserved molecular patterns of pathogen structures). TLRs are located at the cell surface (TLR1, -2, -4, -5, -6 and -10) or intracellularly (TLR3, -7 and -8). Signalling through TLRs results in a type-1 interferon response and/or the production of pro-inflammatory cytokines.
Results
Homology and species distribution of CD209 family members
Phylogenetic analysis groups the various primate genes with high bootstrap values (Figure 1). Within a species, the percentage of identity at the nucleotide level (excluding the length-variable neck region) is 79.0 (range 78.3–79.6) for comparisons of CD209 and CD209L sequences, 78.8 (range 74.4–80.2) for comparisons of CD209 and CD209L2, and 72.1 (range 71.2–72.5) for comparisons of CD209L and CD209L2.
Figure 1.

Phylogenetic tree of CD209 family of genes in mammals. Bayesian estimation of the evolutionary relationships among coding sequences (excluding the neck repeat region) of CD209 family. Bootstrap values are indicated.
CD209L2 resembles the ancestral form, because the observed shorter neck region appears to be reminiscent of the shorter neck regions present in CD209-like sequences in other mammals. Analysis of the dog genome identifies a single CD209 homologue. Similarly, we identified by BLAST analysis of cow sequences the presence of a single CD209 homologue. The dog and cow CD209-like sequences are closely related to mouse CD209g and Signr8 (Figure 1). The rat genome codes for the same set of paralogues as mice (not shown). We and others1 have failed to amplify sequences of CD209 genes in prosimians. Our study confirms the previous observation that orangutan has a truncated form of CD209L and that CD209L2 is a pseudogene in the gorilla.1
The phylogenetic tree (Figure 1) supports a scenario where the three extant CD209 genes emerged from two duplication events; a first duplication event occurred in the common ancestor of anthropoids, yielding CD209L2 and an ancestral CD209 gene, which, in turn, duplicated in the common Old World primate ancestor, giving rise to CD209L and CD209.
Analysis of sequence evolution and selective pressures
Analysis of the substitutional pattern using several codon-based maximum likelihood procedures allowed the estimation of the number of non-synonymous (KA) over synonymous (KS) substitutions per site. Overall, all the three genes have values consistent with purifying selection (Figures 2a-c). The KA/KS values averaged over the entire tree were 0.43 (CD209), 0.52 (CD209L) and 0.35 (CD209L2). Due to the high variability in the number of neck-region repeats across the species and the difficulty of obtaining a reliable alignment, this region was not included in this analysis (a separate analysis of the neck region is presented below). Detailed analyses of the trees (Figures 2a-c) did, however, identify a number of branches with KA/KS values higher than 1.0, in particular in the gibbon lineage. There were no significant differences in the pattern of selective constrain among CD209 gene family among primates having all three or only two functional homologues (Figure 2d).
Figure 2.

Analysis of sequence evolution and selective pressure acting on CD209, CD209L and CD209L2. (a–c) KA/KS values and the estimated number of non-synonymous and synonymous substitutions (in parentheses) for each branch are indicated. Approximate divergence time in Mya is shown. Species in which a gene is absent or disabled by truncation or pseudogenization are identified by discontinued grey branches. (d) Differences in the degree of purifying selection among primates carrying two or three functional members of the CD209 family. KA and KS values obtained with PAML analysis are represented. Triangles represent values for primates carrying all three CD209 family members (CD209, CD209L and CD209L2). Black circles represent values for primates carrying two CD209 family members or having a copy disabled by truncation or pseudogenization. The upper left area represents positive selection; lower right area represents purifying selection. PAML, phylogenetic analysis by maximum likelihood.
In more detailed analyses, we utilized models that allow for different KA/KS rates at different sites of the sequences, because adaptive evolution often occurs at a limited number of sites. This comparison revealed that the null model (which includes sites under purifying selection and neutrally evolving) could not be rejected for CD209 and C209L2. However, the alternative model that adds a third site class that allows for sites under positive selection provided a significantly better fit to the data with respect to CD209L with a P-value of 6.5E–05 (Table 1). The KA/KS for the additional site class is larger than one for L-SIGN (KA/KS∼5), suggesting adaptive protein evolution driven by positive selection at a small subset of sites (Table 1). Using a Bayesian approach,9 we analysed the site class under positive selection in L-SIGN in more detail. Only one residue, alanine in position 88, located in the first neck repeat is pinpointed to be under positive selection (posterior probability, P = 0.99). In addition, threonine 319 and alanine 393, in the C-lectin domain, are identified with lower confidence (P = 0.90 and P = 0.93 respectively). These two L-SIGN residues map at the protein surface away from the region that contains residues involved in carbohydrate interactions (Figure 3). The binding of small carbohydrate compounds by L-SIGN takes place principally at a calcium coordination site in the carbohydrate-recognition domain. The amino-acid residues involved directly in coordinating the calcium ion,10 Glu359, Asn361, Glu366 and Asp378, are fully conserved across all primates.
Table 1.
Codeml analyses using site-specific models
| Site-specific modelsa | ω0b | ω1c | ω2d | Log L | Sites with ω > 1e |
|---|---|---|---|---|---|
| CD209 | |||||
| M1a | 0.00 (56.44%) | 1.00 (43.55%) | −1999.16 | ||
| M2a | 0.07 (68.38%) | 1.00 (0.00%) | 1.36 (31.61%) | −1998.71 | N/Af |
| CD209L2 | |||||
| M1a | 0.23 (80.99%) | 1.00 (19.00%) | −1825.16 | ||
| M2a | 0.23 (80.99%) | 1.00 (13.43%) | 1.00 (5.57%) | −1825.16 | N/Af |
| CD209L | |||||
| M1a | 0.00 (68.62%) | 1.00 (31.37%) | −1786.23 | ||
| M2a | 0.00 (75.46%) | 1.00 (14.69%) | 5.28 (9.83%) | −1778.26 | 1 site |
The likelihood models used are described in the text.
Class of sites under purifying selection.
Class of sites evolving neutrally.
Class of sites that may show KA/KS > 1 positive selection.
Sites pinpointed to be under positive selection by Bayes Empirical Bayes analysis.
Test not applicable (M1 and M2a not significantly different).
Figure 3.
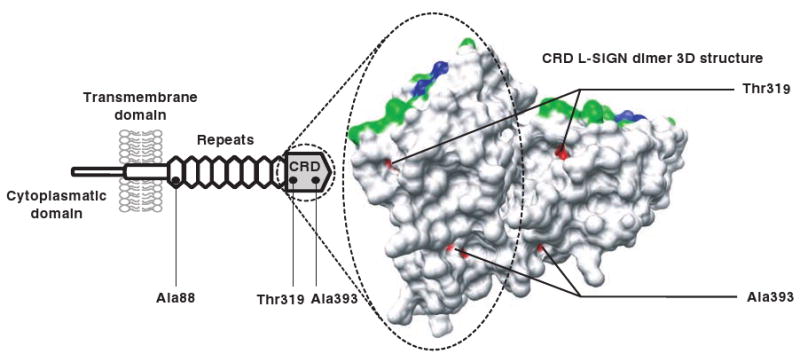
Structure of the carbohydrate-recognition domain of L-SIGN. Three residues were predicted to be under positive selection (in red): alanine 88 (in the neck repeat domain) and threonine 319 and alanine 393. The last two L-SIGN residues map at the protein surface away from recognized domains involved in carbohydrate interaction. Residues involved directly in coordinating the calcium ion are shown in blue, residues important for binding with carbohydrates are shown in green.
Analysis of the neck region
The neck region is characterized by a variable number of a conserved 23-amino acid repeats. In primates included in this work, CD209 and CD209L code five to nine repeats, a number that varies according to the species. The number of repeats may vary also within a species (apes and Old World monkeys).1 In contrast, the primate CD209L2 neck region contains a single (partial) repeat element, a general feature of the neck region of other mammal CD209-like genes—an exception being the mouse Cd209b (four repeats) and Cd209c (two repeats).
To better understand the evolutionary process resulting in the extended and variable length of the neck regions of primate CD209 and CD209L genes, we computed a phylogenetic tree considering all repeats in mammal CD209 family genes (Figure 4a). The C-terminal partial repeat of primate CD209 and CD209L, and the single partial repeat of CD209L2 were found to be most closely related to those of CD209-like repeats in other mammals. The first (proximal) repeat of primate CD209 and CD209L appears to result from a duplication of the last (distal) repeat. Although intermediate repeats have an unclear origin (that is the analysis does not differentiate whether they originated from the proximal or distal repeat), they are highly conserved within and across the various CD209 and CD209L genes, the only exception being the penultimate repeat of CD209L (Figure 4b).
Figure 4.

Analysis of the neck repeat region of DC-SIGN family in mammals. (a) Neighbor-joining tree of amino-acid sequences of the neck repeat region. Colour coding of circles and squares correspond to those described in (b). Black rhombus represents repeats of mouse SIGNR1–5, and dog and cow CD209-like. (b) Alignment of the neck region of DC-SIGN, L-SIGN and CD209L2 from bonobo (as representative of other primate repeat sequences). Repeats are numerated 1–8 (N- to C-terminal), colour coded and represented by circles for DC-SIGN and by squares for L-SIGN and CD209L2.
Comparison of evolutionary pattern of CD209 and TLR gene families
To better define the significance of the pattern of selection in the CD209 gene family, we performed an evolutionary genetic analysis of a second family of innate immunity receptors. For this, we selected five primate species, representatives of apes and Old and New World monkeys. We used sequence of TLR1 (95.7% of coding sequence), TLR2 (97.0%), TLR3 (97.1%), TLR4 (85.2%), TLR5 (65.2%), TLR6 (98.8%), TLR8 (36.0%) and the complete sequence of TLR7 and -10.
Overall, all nine TLR gene sequences had values consistent with purifying selection (Figure 5). The KA/KS values averaged over the entire tree were 0.50 (TLR1), 0.49 (TLR2), 0.23 (TLR3), 0.52 (TLR4), 0.59 (TLR5), 0.36 (TLR6), 0.34 (TLR7), 0.47 (TLR8) and 0.44 (TLR10).
Figure 5.

Phylogenetic trees of Toll-like receptor1, -2, -3, -4, -5, -6, -7, -8 and -10. KA/KS values and the estimated number of non-synonymous and synonymous substitutions (in parantheses) for each branch are indicated. Approximate divergence time in Mya is shown.
In the set of more detailed analyses using models that allow different KA/KS rates at different sites of the sequences (because adaptive evolution often occurs at a limited number of sites), we found that the null model (which includes sites under purifying selection and neutrally evolving) could not be rejected for TLR2, -3, -4, -5, -6, -7, -8 and -10. The alternative model (which adds a third site class that allows for sites under positive selection) provided a significantly better fit to the data with respect to TLR1, with a P-value of 2.46E–04. One per cent of TLR1 codons have a KA/KS ∼16.9, suggesting adaptive protein evolution driven by positive selection. Using a Bayesian approach,9 we analysed the site class under selective pressure in TLR1 in more detail. Two residues, tryptophan 61 located in the extracellular domain and serine 748 located in the Toll/interleukin-1 receptor domain, were predicted to be under positive selection (posterior probability, P>0.95).
Discussion
The evolutionary history of the CD209 gene family in primates is characterized by several episodes of gene duplication, recent gene deletion/truncation and elongation of the neck repeat region. Given the role of DC-SIGN and related proteins in pathogen uptake, it is plausible that these evolutionary changes have occurred in response to temporal selective pressures during primate evolution. However, the evolutionary analysis of various primate homologues and paralogues failed to identify widespread signs of positive selection, as assessed by global KA/KS values, by lineage and species-specific KA/KS values, or by site-specific analyses. Only more detailed analysis of some lineages suggested episodes of recent evolution (that is species-specific positive selection), in particular among gibbons. L-SIGN (encoded by CD209L) in humans may also be a relevant example of species-specific positive selection that has continued to operate during recent human evolution. Indeed, a study screening for the degree of sequence-based diversity in different human populations has shown that CD209L exhibits higher diversity in its coding region, as compared to its homologue CD209.11 These observations suggest that there has been an advantage for a higher diversity in CD209L with respect to CD209 within humans, probably driving CD209L to accumulate new mutations and eventually new functions, possibly compensating the recent loss of CD209L2 in humans.
The general pattern of purifying selection of the CD209 family of genes (KA/KS range 0.35–0.52) and TLR family of genes (KA/KS range 0.23–0.59) appears similar. These values are greater than the genome-wide average (KA/KS ∼0.2) for human–chimpanzee gene pairs.12 They are, however, much lower than the KA/KS estimates for intrinsic defense genes investigated in the context of human immunodeficiency virus pathogenesis, such as TRIM5α and APOBEC3G (KA/KS = 1.1 for both genes).13 Comparative analysis of the genes that encode APOBEC3G and TRIM5α has revealed the intensity of the selective pressures resulting from the long-standing conflict between retroviruses and their hosts.14,15 These two genes/proteins constitute a paradigm of pathogen-driven positive selection pressure, not only because of their global elevated KA/KS values, but also by the identification of precise residues and domains under strong positive selection. Analysis of APOBEC3G across primate species reveals many residues in the amino-terminal cytidine deaminase domain that are under positive selection, which coincide indeed with the proposed region of interaction with the human immunodeficiency virus-1 Vif protein. Analysis of the TRIM5α pinpoints a patch of amino acids that is under positive selective pressure at variable regions V1 and V2. The variable regions of TRIM5α have in turn evolved independently to recognize the various retroviral capsids.16
A second point of discussion concerns the length of the neck region that characterizes primate CD209 and CD209L among other CD209-like genes in mammals. In this study and in previous analyses in primates1, CD209 and CD209L have variable numbers of repeats. A single partial repeat characterizes primate CD209L2; a general feature of the neck region of other mammal CD209-like genes. The first and last repeat units of the neck region in CD209 and CD209L originate from a common ancestral motif. We studied only one individual per species, but Barreiro et al.11 studied the degree of polymorphism of the neck region in both CD209 and CD209L by genotyping 1064 individuals from 52 worldwide populations. Striking differences were observed between the two genes. Although minimal variation was observed for CD209, CD209L exhibited a strong variation in allelic frequencies of different neck lengths. In their population genetics study, the CD209 genes did not experience selective sweeps (that is directional selection, leading to rapid spread/fixation of an advantageous allele) but rather experienced purifying selection (CD209) or balancing selection (CD209L).11 Thus, these duplicated genes have evolved, and might still evolve, under completely different selective pressures.
The strong contrast observed in length variation of the neck region between the various genes may have consequences on function. A number of association studies in human populations have attempted to correlate length variation of the neck region and promoter variants with susceptibility to infectious diseases whose etiological agents are known to interact with one (or both) of these lectins. Results, in particular from the analysis of susceptibility to human immunodeficiency virus-1 or M. tuberculosis infection, remain under discussion.11,17–23
The usurpation of DC-SIGN and other family members by pathogens such as retroviruses, Ebola and Mycobacteria might have led to a pattern of distinctive patches or domains with the characteristic of positive selective pressure as a result of a genetic conflict.6,13 Against this expectation, the evolutionary analysis of the family suggests both a pattern of apparent redundancy of CD209 genes (gene absence and gene loss) and of positive selection in specific lineages, in the context of a general pattern of gene conservation. A similar pattern is identified for a second family of proteins of the innate immunity, the TLRs. This pattern is consistent with the concept put forward by Lynch and Conery24 proposing that most duplicated genes experience a brief period of relaxation of the selective constraint early in their history. Thereafter, most gene duplicates are silenced within a few million years, with a minority of duplicated genes subsequently experiencing strong purifying selection. The observed degree of purifying selection may be expected given the need to faithfully recognize various pathogen motifs, the inability of the pathogen to modify the molecular pattern,25 and in the case of the DC-SIGN family, the need to recognize the commensal flora, as well as ICAM-2, ICAM-3 adhesion molecules and other self-proteins.
Materials and methods
Primates
CD209 gene family coding sequences from primates were generated by amplification and sequencing of genomic DNA: bonobo (Pan paniscus CD209 EU041926, CD209L EU041931, CD209L2 EU041934), chimpanzee (Pan troglodytes CD209L2 EU041935), bornean orangutan (Pongo pygmaeus CD209L EU041932, CD209L2 EU041936), lar gibbon (Hylobates lar CD209L2 EU041937), nomascus (Hylobates leucogenys CD209 EU041927, CD209L EU041933, CD209L2 EU041938), siamang (Hylobates syndactylus CD209L2 EU041939), red baboon (Papio hamadryas CD209L2 EU041940), African green monkey (Cercopithecus (chlorocebus) aethiops CD209 EU041928, CD209L2 EU041941), owl monkey (Aotus trivirgatus CD209 EU41930). Common marmoset (Callithrix jacchus jacchus CD209 EU041929, CD209L2 EU041942) sequences were obtained by amplification and sequencing of cDNA from liver. For cotton-top tamarin (Saguinus Oedipus) and golden headed lion (Leontopithecus rosalia chrysomelas), partial sequences were obtained for CD209 and CD209L2 from cDNA of peripheral blood. Other primate and mammal sequences were downloaded from the NCBI database, human (Homo sapiens CD209 AF290886, CD209L BC038851), chimpanzee (CD209 AY078913, CD209L AH011538), gorilla (Gorilla gorilla CD209 AY078906, CD209L AH011537), bornean orangutan (CD209 AY078905), lar gibbon (CD209 AH011540, CD209L AH011531), siamang (CD209 AY078878, CD209L AH011532), black crested gibbon (Hylobates concolor CD209 AY078885, CD209L AH011533), rhesus monkey (Macaca mulatta CD209 NH_001032870, CD209L2 AY074781), red baboon (CD209 AY078864), mouse (Mus musculus Cd209a AF373408, Cd209b AF373409, Cd209c AF373410, Cd209d AF373411, Cd209e AF373412, Cd209g XM_284376, Signr8 XM_284386) and dog (Canis lupus familiaris CD209-like XM_542118). A cow (Bos taurus) CD209-like was identified by BLAST search on is entire genome (UCSC Genome Bioinformatics, www.genome.ucsc.edu).
Toll-like receptor family of gene sequences were generated by amplification and sequencing of genomic DNA of lar gibbon (Hylobates lar TLR1 EU488847, TLR2 EU488848, TLR3 EU488849, TLR4 EU488850, TLR5 EU488851, TLR6 EU488852, TLR7 EU488853, TLR8 EU488854, TLR10 EU488855), cotton-top tamarin (Saguinus Oedipus TLR1 EU488856, TLR2 EU488857, TLR4 EU488859, TLR5 EU488860, TLR6 EU488861, TLR7 EU488862, TLR8 EU488863, TLR10 EU488864) and cDNA from common marmoset (Callithrix jacchus jacchus TLR3 EU488858). Human sequences were downloaded from the NCBI database (TLR1NM_003263, TLR2NM_003264, TLR3NM_003265, TLR4NM_138554, TLR5Nm_003268, TLR6NM_006068, TLR7NM_016562, TLR8NM_138636, TLR10NM_030956). Chimpanzee (P. troglodytes) and rhesus monkey (M. mulatta) sequences were obtained by BLAST search on their entire genome (UCSC Genome Bioinformatics, www.genome.ucsc.edu).
Molecular analysis
Exons were amplified by primers designed for the flanking intron regions (for genomic DNA), and for 3′- and 5′-UTR regions (for cDNA). HotStarTaq Master Mix (Qiagen AG, Hombrechtikon, Switzerland) was used for PCR amplification of fragments smaller than 1 kb and PrimeSTAR DNA polymerase (TAKARA Bio Inc. Shiga, Japan) for fragments larger than 1 kb. All primer sequences are presented in Supplementary Table S1. Sequences were aligned using MUSCLE.26 Coding regions were aligned according to their corresponding amino-acid sequences using the European Molecular Biology Open Software Suite package.27 To perform phylogenetic analysis, we use a Bayesian inference of phylogeny with Mr Bayes 3.28 Different phylogenetic trees were performed at nucleic acid and amino acid levels, with different lengths of sequence (entire sequences, sequences without the neck repeat region, sequences consisting solely of the carbohydrate-recognition domain and of the neck repeat region).
Evolutionary analyses
To trace the evolutionary history of the CD209 and TLR families, we analysed their substitutional patterns in the framework of the accepted primate phylogeny8 using several codon-based maximum likelihood procedures as implemented in the codeml tool of the phylogenetic analysis by maximum likelihood program package.29 To obtain an overview of the coding-sequence evolution, we estimated the number of non-synonymous (KA) over synonymous (KS) substitutions per site (averaged over the entire sequence) for each branch of the trees using the free-ratio model of codeml.29 In more detailed analyses, we utilized models that allow for different KA/KS rates at different sites of the sequences, because adaptive evolution often occurs at a limited number of sites.30 We first compared a null model (‘M1a’9,31), which assumes two site classes (sites under purifying selection and neutrally evolving sites), to an alternative model (‘M2a’9,31), which adds a third site class that allows for sites with KA/KS >1, using likelihood ratio tests.32
Supplementary Material
Acknowledgments
We thank Keith Mansfield and Kuei-Chin Lin from the New England Primate Center, and Charles Buillard and Eugène Chabloz from the Zoo of Servion for materials. This work was funded by the Swiss National Science Foundation and a grant for interdisciplinary research from the Faculty of Biology and Medicine of the University of Lausanne. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Supplementary Information accompanies the paper on Genes and Immunity website (http://www.nature.com/gene)
References
- 1.Bashirova AA, Wu L, Cheng J, Martin TD, Martin MP, Benveniste RE, et al. Novel member of the CD209 (DC-SIGN) gene family in primates. J Virol. 2003;77:217–227. doi: 10.1128/JVI.77.1.217-227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koppel EA, van Gisbergen KP, Geijtenbeek TB, van Kooyk Y. Distinct functions of DC-SIGN and its homologues L-SIGN (DC-SIGNR) and mSIGNR1 in pathogen recognition and immune regulation. Cell Microbiol. 2005;7:157–165. doi: 10.1111/j.1462-5822.2004.00480.x. [DOI] [PubMed] [Google Scholar]
- 3.Wu L, Kewalramani VN. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol. 2006;6:859–868. doi: 10.1038/nri1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Figdor CG, van KY, Adema GJ. C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol. 2002;2:77–84. doi: 10.1038/nri723. [DOI] [PubMed] [Google Scholar]
- 5.Feinberg H, Guo Y, Mitchell DA, Drickamer K, Weis WI. Extended neck regions stabilize tetramers of the receptors DC-SIGN and DC-SIGNR. J Biol Chem. 2005;280:1327–1335. doi: 10.1074/jbc.M409925200. [DOI] [PubMed] [Google Scholar]
- 6.Yang Z. The power of phylogenetic comparison in revealing protein function. Proc Natl Acad Sci USA. 2005;102:3179–3180. doi: 10.1073/pnas.0500371102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nielsen R, Hellmann I, Hubisz M, Bustamante C, Clark AG. Recent and ongoing selection in the human genome. Nat Rev Genet. 2007;8:857–868. doi: 10.1038/nrg2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodman M. The genomic record of Humankind's evolutionary roots. Am J Hum Genet. 1999;64:31–39. doi: 10.1086/302218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Z, Wong WS, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 10.Snyder GA, Colonna M, Sun PD. The structure of DC-SIGNR with a portion of its repeat domain lends insights to modeling of the receptor tetramer. J Mol Biol. 2005;347:979–989. doi: 10.1016/j.jmb.2005.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barreiro LB, Patin E, Neyrolles O, Cann HM, Gicquel B, Quintana-Murci L. The heritage of pathogen pressures and ancient demography in the human innate-immunity CD209/CD209L region. Am J Hum Genet. 2005;77:869–886. doi: 10.1086/497613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner A. Rapid detection of positive selection in genes and genomes through variation clusters. Genetics. 2007;176:2451–2463. doi: 10.1534/genetics.107.074732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortiz M, Bleiber G, Martinez R, Kaessmann H, Telenti A. Patterns of evolution of host proteins involved in retroviral pathogenesis. Retrovirology. 2006;3:11. doi: 10.1186/1742-4690-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawyer SL, Emerman M, Malik HS. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004;2:E275. doi: 10.1371/journal.pbio.0020275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawyer SL, Wu LI, Emerman M, Malik HS. Positive selection of primate TRIM5{alpha} identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci USA. 2005;102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldschmidt V, Ciuffi A, Ortiz M, Brawand D, Munoz M, Kaessmann H, et al. Antiretroviral activity of ancestral TRIM5alpha. J Virol. 2008;82:2089–2096. doi: 10.1128/JVI.01828-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barreiro LB, Quintana-Murci L. DC-SIGNR neck-region polymorphisms and HIV-1 susceptibility: From population stratification to a possible advantage of the 7/5 heterozygous genotype. J Infect Dis. 2006;194:1184–1185. doi: 10.1086/507845. [DOI] [PubMed] [Google Scholar]
- 18.Wichukchinda N, Kitamura Y, Rojanawiwat A, Nakayama EE, Song H, Pathipvanich P, et al. The polymorphisms in DC-SIGNR affect susceptibility to HIV type 1 infection. AIDS Res Hum Retroviruses. 2007;23:686–692. doi: 10.1089/aid.2006.0212. [DOI] [PubMed] [Google Scholar]
- 19.Martin MP, Lederman MM, Hutcheson HB, Goedert JJ, Nelson GW, van KY, et al. Association of DC-SIGN promoter polymorphism with increased risk for parenteral, but not mucosal, acquisition of human immunodeficiency virus type 1 infection. J Virol. 2004;78:14053–14056. doi: 10.1128/JVI.78.24.14053-14056.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H, Hwangbo Y, Holte S, Lee J, Wang C, Kaupp N, et al. Analysis of genetic polymorphisms in CCR5, CCR2, stromal cell-derived factor-1, RANTES, and dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin in seronegative individuals repeatedly exposed to HIV-1. J Infect Dis. 2004;190:1055–1058. doi: 10.1086/423209. [DOI] [PubMed] [Google Scholar]
- 21.Gramberg T, Zhu T, Chaipan C, Marzi A, Liu H, Wegele A, et al. Impact of polymorphisms in the DC-SIGNR neck domain on the interaction with pathogens. Virology. 2006;347:354–363. doi: 10.1016/j.virol.2005.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olesen R, Wejse C, Velez DR, Bisseye C, Sodemann M, Aaby P, et al. DC-SIGN (CD209), pentraxin 3 and vitamin D receptor gene variants associate with pulmonary tuberculosis risk in West Africans. Genes Immun. 2007;8:456–467. doi: 10.1038/sj.gene.6364410. [DOI] [PubMed] [Google Scholar]
- 23.Vannberg FO, Chapman SJ, Khor CC, Tosh K, Floyd S, Jackson-Sillah D, et al. CD209 genetic polymorphism and tuberculosis disease. PLoS ONE. 2008;3:e1388. doi: 10.1371/journal.pone.0001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 25.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 26.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 28.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 29.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 30.Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Z, Nielsen R, Goldman N, Pedersen AM. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.