

Abstract
The 1980 identification of nitric oxide (NO) as an endothelial cell-derived relaxing factor resulted in an unprecedented biomedical research of NO and established NO as one of the most important cardiovascular, nervous and immune system regulatory molecule. A reduction in endothelial cell NO levels leading to “endothelial dysfunction” has been identified as a key pathogenic event preceding the development of hypertension, metabolic syndrome, and cardiovascular disease. The reduction in endothelial NO in cardiovascular disease has been attributed to the action of oxidants that either directly react with NO or uncouple its substrate enzyme. In this report, we demonstrate that uric acid (UA), the most abundant antioxidant in plasma, reacts directly with NO in a rapid irreversible reaction resulting in the formation of 6-aminouracil and depletion of NO. We further show that this reaction occurs preferentially with NO even in the presence of oxidants peroxynitrite and hydrogen peroxide and that the reaction is at least partially blocked by glutathione. This study shows a potential mechanism by which UA may deplete NO and cause endothelial dysfunction, particularly under conditions of oxidative stress in which UA is elevated and intracellular glutathione is depleted.
Keywords: Uric acid, nitric oxide, cardiovascular disease, endothelial dysfunction, 6-aminouracil, glutathione
Introduction
Uric acid (UA) is generated in mammalian systems as an end-product of purine metabolism. In most mammals, UA is further degraded to allantoin by the enzyme uricase; however, this enzyme was mutated in hominoids five to fifteen million years ago.[1] This loss of uricase activity not only results in higher plasma UA levels, but also limits our ability to regulate UA, such that widely varied levels may be observed in man. UA possesses free-radical-scavenging properties[2,3] and is the most abundant antioxidant in human plasma.[4–6] It may also act as a prooxidant under conditions of oxidative stress.[7–10] Markedly increased levels of UA (>6.2 mg/dl or >370 μM) are known to cause gout and nephrolithiasis, but more importantly have been associated with increased risk for the development of cardiovascular disease, particularly hypertension, obesity/metabolic syndrome, and kidney disease.[11–15]
Recent experimental and clinical studies have linked elevated UA with endothelial dysfunction and a reduction in nitric oxide (NO) levels.[16] Experimental studies have reported that UA can reduce NO levels in endothelial cells in culture,[16] block acetylcholine-induced vasodilation of aortic rings,[16] and reduce circulating nitrites in experimental animals.[16] In humans a circadian rhythm has been identified in which UA and nitric oxide levels are inversely correlated.[17] Chronic hyperuricemia is also associated with endothelial dysfunction,[16] and reducing UA levels with xanthine oxidase inhibitors have been found to markedly improve endothelial function.[16] However, xanthine oxidase inhibitors also reduce oxidant generation, so the improvement in endothelial function could reflect a direct reduction of xanthine oxidase-associated oxidants as opposed to a reduction in uric acid per se.
While the importance of endothelial dysfunction in cardiovascular disease is well accepted, most evidence suggests that the primary mechanism by which this occurs is via oxidative stress in which superoxide either directly inactivates NO (by forming peroxynitrite), uncouples the endothelial NO synthase, or passively increases an inhibitory substrate (asymmetric dimethylarginine) by inhibiting the enzyme, dimethylarginine dimethylaminohydrolase.[18] In this regard, UA has been shown to help preserve endothelial NO levels via its role as an antioxidant, either by blocking the uncoupling of endothelial NO synthase by reacting with peroxynitrite,[18] or by preventing the oxidant-induced inactivation of extracellular superoxide dismutase.[19] A paradox then develops, for how can UA, which is the most abundant (about six times as ascorbate) antioxidant in plasma,[6] induce endothelial dysfunction in vivo?
In this study, we explored the possibility that UA might react directly with NO itself. Unlike most free radicals, NO has a low electron affinity; consequently, the chemistry of NO with anions is not dominated by electron transfer reactions.[20]
Materials and Methods
Preparation of Reagents
All chemicals and reagents, unless otherwise specified, were obtained from Sigma. 9-Methyluric acid and LiOH·1H2O were purchased from Fluka. Distilled, deionized water (metal ion free) and EDTA (500 mM) were purchased from Gibco. 1,3-15N2-Uric acid (15N2-UA) was purchased from Cambridge Isotopes. Peroxynitrite was purchased from Cayman Chemical. All the experiments were conducted shielded from light.
Reaction of UA, 15N2-UA, Lithium Urate, or 9-Methyluric Acid with NO [Reaction 1]
UA, 15N2-UA, lithium urate, or 9-methyluric acid [0.3 mM to 10 mM)] was added to potassium phosphate buffer (0.10 mM and 0.30 mM phosphate, pH 7.4 and 9.0) containing 0.1 mM EDTA (3.0 mL final volume). The reaction mixture was vortexed for 10 seconds and then purged with argon (TriGas) for 1 minute. After purging with argon, the reaction mixtures were placed on ice for 15 minutes. In a typical case, the reactions were conducted in triplicate by bubbling NO (Air Liquide, 99.998% pure NO) for 10, 30, or 90 seconds (2 mL/minute flow rate) through an ice-cold solution followed by incubation at room temperature for 3 minutes. The reaction mixtures were placed back on ice and aliquots of each reaction mixture were transferred to LC-MS vials on ice, and were stored at −80°C until LC-MS analysis could be performed. We also investigated reaction 1 at −20, 0, 25, and 60°C. The reactions of UA and 15N2-UA with NO were conducted as a pair of identical reactions to aid in the mass spectrometric identification of the products.
Reaction of UA with NO in the Presence of Peroxynitrite or Hydrogen Peroxide [Reactions 2 and 3]
UA (0.3 mM to 10 mM) was added to potassium phosphate buffer containing EDTA. One molar equivalent of peroxynitrite or hydrogen peroxide was then added to the ice-cold reaction mixture. The reaction mixture was vortexed for 10 seconds and then placed on ice and purged with argon for 1 minute. The reactions of UA with NO were performed by bubbling the ice-cold mixtures with NO for 10, 30, or 90 seconds (2 mL/minute). The reaction mixtures were allowed to incubate at room temperature for 3 minutes, at which time they were placed back on ice. Aliquots of each reaction mixture were transferred to vials for LC-MS analysis.
Reaction of UA with NO in the Presence of Antioxidants [Reactions 4–8]
UA was added to potassium phosphate buffer containing EDTA under the same conditions as outlined above. One molar equivalent of antioxidant [ascorbate (ASC), tetrahydrobiopterin (BH4), glutathione (GSH), cysteine (CYS), or tyrosine (TYR)] was then added to the reaction mixtures and vortexed for 10 seconds. Reactions were purged with argon, placed on ice, and then reacted with NO as outlined above. For the determination of protection of UA by various antioxidants 30 seconds bubbling time was used and the remaining UA was quantitated by LC-MS in the SRM mode.
Reaction of UA with NO in Human Plasma and Aortic Endothelial Cell (HAEC) Lysates [Reactions 9 and 10]
Reactions of NO with 15N2-UA human plasma or cell lysates were performed in a similar fashion to the reactions outlined above with the following exceptions: 15N2-UA (0.3–10 mM) was added to a mixture of human plasma or HAEC lysate in PBS (1:3 volumetric ratio). EDTA (final concentration 0.10 mM) was added to this mixture. In a typical reaction, these mixtures were then purged with argon, and bubbled with NO as above for 90 seconds (investigated at 30-, 60-, 90- 180-, and 360-second bubbling times). The reaction mixture was deproteinized with 3 ml of 10% trichloroacetic acid before LC-MS analysis. All other reaction conditions were the same as those outlined above.
Analyses of Reaction Mixtures
The reaction mixtures were analyzed by LC-MS in the fullscan (both positive and negative), MS/MS (tandem mass spectrometry) and SRM (single reaction monitoring) modes using APCI (atmospheric pressure chemical ionization) method. In addition, some samples also were further analyzed using the ESI (electrospray ionization) method. UA concentrations were determined by SRM mode using APCI method in the negative mode (reactions monitored for UA were m/z 166.9→95.9 at 25 V and 166.9→123.9 at 25 V and for 15N2-UA were m/z 168.9→96.9 at 25 V and 168.9→124.9 at 25 V). 6-AU, allantoin and triuret were quantified in the APCI positive mode using their respective SRM ions. The LC-MS/MS analyses of the reaction mixtures were carried out with an Agilent 1100 LC system and a ThermoFinnigan TSQ 7000 triple-quadrupole mass spectrometer equipped with APCI interface operated in positive-ion mode. LC analyses were performed in a gradient elution mode using Phenomenex Luna 5μ C18 100A (150 mm × 4.6 mm) column. The mobile phase used included ammonium acetate/acetic acid and methanol gradient or formic acid/methanol gradient. The mobile phase flow was 0.6 mL min−1, and the injection volume was 20 μL.
Results and Discussion
We investigated the direct reaction of NO (bubbled as NO gas[21,22]) with UA and labeled 1,3-15N2-uric acid (15N2-UA) under anaerobic conditions in several different media, including human plasma and endothelial cell lysates that mimic the physiological environment. The anaerobic conditions were selected to ensure that we are studying the direct reactions of NO with UA and not those of further oxidized N2O3 or NO2 (oxygen readily oxidizes NO[23]). The pH (7.4 and 9.0) or the concentration of buffer (0.10 and 0.30 mM), did not alter the product of the reaction. Typically, potassium phosphate was used as the buffer with the concentration of UA ranging from 0.3 mM (physiological level[6]) to 10 mM. In the range investigated, the concentration of UA had no effect on the products formed. The reaction scheme for the study is illustrated in Figure 1. The reactions have been performed in aqueous buffer (reaction 1); in aqueous buffer in the presence of an oxidant (peroxynitrite and H2O2) (reactions 2 and 3); in aqueous buffer in the presence of selected biologically important antioxidants (ASC, GSH, CYS, TYR, and BH4) (reactions 4 to 8); and in human plasma (reaction 9) or human aortic endothelial cell (HAEC) lysates (reaction 10). Also, the reaction between lithium urate (better solubility at physiological pH) and NO, as well as the reaction between 9-methyluric acid and NO has been studied in the media described in reactions 1 to 3. The ratios of UA to peroxynitrite or to hydrogen peroxide (reactions 2 and 3), and UA:antioxidant (reactions 4 to 8) used were 1:1 molar ratios. In a typical case, the reactions were conducted in triplicate by bubbling NO for 10, 30, or 90 seconds (2 mL/minute flow rate) through an ice-cold solution (cold increases NO solubility in water and reduces the amount that escapes during bubbling), followed by incubation at room temperature for 3 min. We also investigated reaction 1 at −20, 0, 25, and 60°C and the same product was formed at various temperatures. The optimum time for NO bubbling through the reaction media was determined to be 30 seconds (90 seconds for plasma and cell lysates) at which time UA reaction goes to completion, as reflected by the disappearance of the UA peak in the LC/MS analysis.
FIGURE 1.
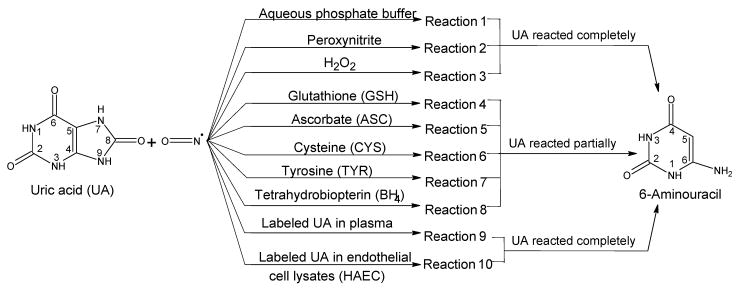
Summary of the reaction of NO with uric acid (UA) and labeled UA. The reactions 1 to 8 were conducted as a pair with labeled and unlabeled uric acid to aid in the mass spectrometry analysis and identification of products. The effect of multiple pHs (7.4 and 9.0) and temperatures (−20, 0, 25, and 60°C) were also investigated and did not alter the formation of 6-aminouracil as the product. Reactions 1 to 3 were also conducted with 9-methyluric acid. In addition, reactions 1 to 8 were also conducted with the more soluble lithium urate (physiological pH only). The quantifications were carried out with LC-MS/MS. In all the experiments reported, the NO bubbling was optimized (from preliminary studies) for the completion of the reaction of UA. Therefore, 100% or very close to 100% of UA is consumed in a given reaction. The optimum time for NO bubbling through the reaction media was determined to be 30 seconds (90 seconds for plasma and cell lysates) at which time UA reaction goes to completion, as reflected by the disappearance of the UA peak in the LC/MS analysis. The exceptions to the optimization of bubbling time were where the antioxidants were employed, in which case 30-second bubbling time was used irrespective of the extent of the reaction.
Close examination of products derived from the reaction of UA with NO, by means of liquid chromatography-mass spectrometry (LC-MS), revealed that the dominant end-product has a molecular mass of 127 u and a chromatographic retention time of about 6 minute (Figure 2A). In the positive-ion mode, the signal of the protonated product (ion [127 + H]+) with m/z 128 was the predominant ion (Figure 2B). The reaction product left in reaction mixture was stable for at least a month at ambient temperature. When reactions (short time bubbling of NO) that contained unreacted UA were allowed to stand at ambient temperature, no further reaction occurred, suggesting that NO was completely consumed in the reaction, and that no additional NO became available from the medium or from further reaction of reaction products (for example, from reactions in which NO acted as a catalyst).
FIGURE 2.
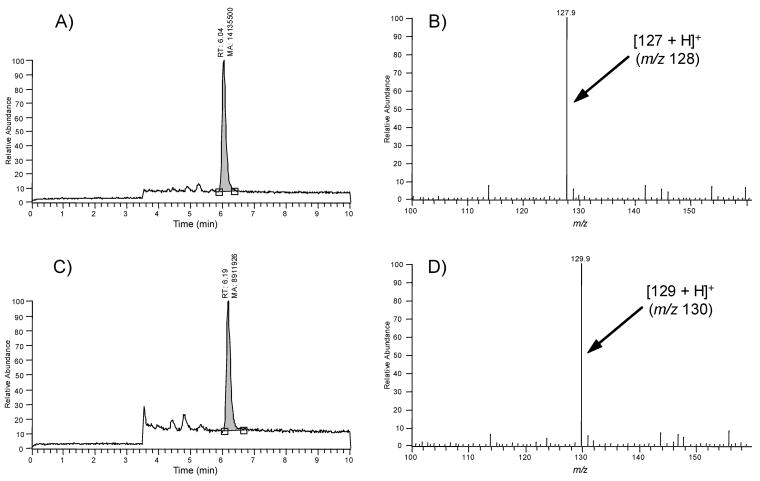
LC-MS analyses of the products of the reaction of UA (panels A and B) and labeled UA (15N2-UA; panels C and D). Panels A and C show the LC-MS trace (total ion current) and panels B and D show the corresponding mass spectra. The peak at 6.04 minutes in panels A and B are identical to commercially available 6-aminouracil. The formation of M+1 of 130 from 15N2-UA, clearly suggests that the detected end-product incorporated both labeled 15N atoms from the six-membered ring of the purine skeleton of UA.
Labeled 15N2-UA reacted with NO and generated a product with a molecular mass 129 u (ion [129 + H]+ with m/z 130) (Figure 2C and 2D). This result clearly suggests that the detected end-product incorporated both 15N atoms from the six-membered ring of the purine skeleton of UA. The examinations of the experimental data and that of molecular models allowed us to determine the molecular formula as C4H5O2N3 for the compound with the mass 127 u (or C4H5O2N1 15N2 for the compound with the mass 129 u) and the structure of the compound as an aminouracil. Aminouracil has two positional isomers: 5-aminouracil (5-AU) and 6-aminouracil (6-AU). To unambiguously distinguish one isomer from the other, commercially available samples of both isomers were subjected to tandem mass spectrometry, under collision-induced dissociation (CID) conditions at collision energies ranging from 15 to 40 eV. Fragmentation patterns of the aminouracils were found to be isomer-specific. Only one fragment ion with m/z 111 was common in the CID mass spectra of both aminouracils. Other detected fragment ions were different for the two isomers, and thus can be considered as diagnostic ions for isomer recognition. The main fragmentation patterns of protonated 5-AU are as follows: [127 + H]+ (m/z 128) → [128−NH3]+ (m/z 111) → [111−CO]+ (m/z 83) → [83−CO]+ (m/z 55). The main fragmentation patterns of protonated 6-AU are presented in Figure 3.
FIGURE 3.
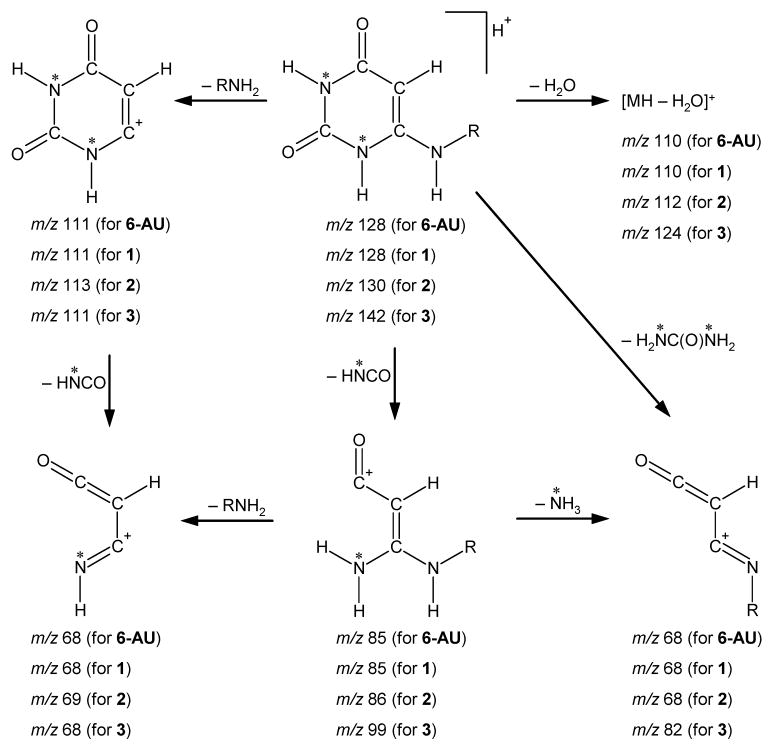
Main fragmentation patterns of protonated 6-aminouracils under CID conditions. (Notes: 6-AU represents standard 6-aminouracil; 1 represents compound produced from UA; 2 produced from labeled 1,3-15N2-UA; 3 produced from 9-methyluric acid; asterisk represents 15N atom (for labeled compound 2); R = H for 6-AU, 1 and 2; and R = CH3 for compound 3.).
The MS/MS data generated by the analyses of the end-products of the reactions of UA with NO (reactions 1 to 10) were found to be identical with those recorded for standard 6-AU (Figure 3). Furthermore, the fragmentation generated from the reaction product of 1,3-15N2-UA with NO is perfectly described by the same scheme (Figure 3). Consequently, the presented data confirm that UA can react directly with NO producing 6-AU. The reactions (reactions 1 to 3) of NO with 9-methyluric acid produced N-methylaminouracil with a molecular mass 141 u, demonstrating that the N9–Me fragment has been preserved in the end-product. This confirms that the nitrogen-9 (Figure 1) atom in the purine skeleton of UA becomes the amino-group nitrogen in 6-AU.
The reactions of UA with NO in the presence of peroxynitrite or hydrogen peroxide also generated 6-AU in a rapid, selective reaction in which UA preferentially reacted with the NO rather than the oxidants already in solution. Under the described reaction conditions, Li urate behaved similarly to UA.
The reactions of NO with 15N2-UA in human plasma and endothelial cell (HAEC) lysates produced only labeled 6-AU. Figure 4 illustrates the identification of labeled 6-AU from plasma reactions of 15N2-UA with NO. The formation of 6-AU in human plasma (representing extracellular compartment), as well as in human cell lysates (representing intracellular compartment) underscores the biological significance of this reaction and the measurement of 6-AU in human biological samples provides a means of determining the extent of the direct reaction of NO with UA in these biological systems.
FIGURE 4.
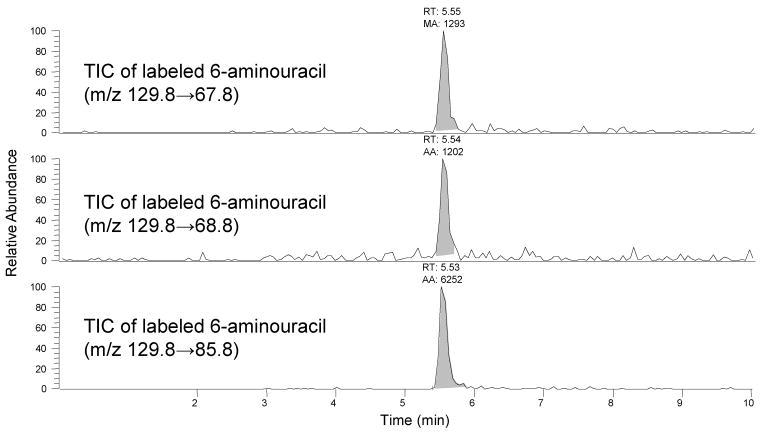
LC-MS/MS identification of 15N labeled 6-aminouracil (6-AU) (retention Time = 5.55 minutes) from plasma reactions of 15N2-UA with NO. The three panels show the total ion chromatograms (TIC) of three specific SRMs of the labeled 6-AU. The presence of all three labeled SRMs in the plasma reaction product is confirmatory of the formation of labeled 6-AU from 15N2-UA and NO.
Preliminary in vivo studies were also conducted by investigating rats infused with 15N2-UA, as well as by the analyses of urine samples from hypertension and preeclamptia patients for UA and its metabolites. These studies indicated the in vivo formation of 6-AU. Although we have not detected thus far, it is possible that intermediates from the reaction of UA with NO in vivo could be diverted to other products or that 6-AU is further metabolized. This does not alter the important fact that NO can be rapidly depleted by the direct reaction with UA. Further studies to characterize these potential pathways are on going. The control studies of NO reactions with UA metabolites allantoin and triuret demonstrated that neither allantoin nor triuret reacted with NO to produce 6-AU.
We also investigated the reactions of UA (and Li urate) with NO (30-second bubbling of NO) in the presence of the following antioxidants: ASC, BH4, GSH, CYS, and TYR (1:1:1 molar ratio of UA to antioxidant to NO). The concentrations of UA remaining were determined by LC-MS as a measure of the extent of protection of UA by a given antioxidant. Among the antioxidants tested, one equivalent of GSH affords the best protection of UA from the reaction with NO. Thus, under conditions of low oxidative stress and high GSH availability it is possible for GSH to partially block the UA-NO reaction. Conversely, under conditions of oxidative stress, the direct reaction of NO with UA could be a significant source of NO depletion and endothelial dysfunction.
While this work was in progress (we had discovered the reaction of NO with UA in July 2005; see Figure 5), Suzuki[24] reported the formation of nitrosated UA from the reaction of UA with NO. Suzuki's studies were conducted under aerobic conditions and under these conditions the main nitrosating species is thought to be N2O3 and not NO.[21] We conducted our studies (except the in vivo studies) under anaerobic conditions to ensure that we are studying the direct reactions of NO with UA. LC-MS analyses of our reaction products with APCI and ESI sources in the positive and negative modes did not reveal the presence of nitrosated UA. It is possible that the compound reported by Suzuki[24] is a labile intermediate (the author reported a half life of 2.2 minutes in phosphate buffer) of the reaction of N2O3 (or other oxidized NO species such as NO2) and not that of the reaction of NO with UA. In contrast, we have identified stable 6-aminouracil from the reaction of NO with UA.
FIGURE 5.
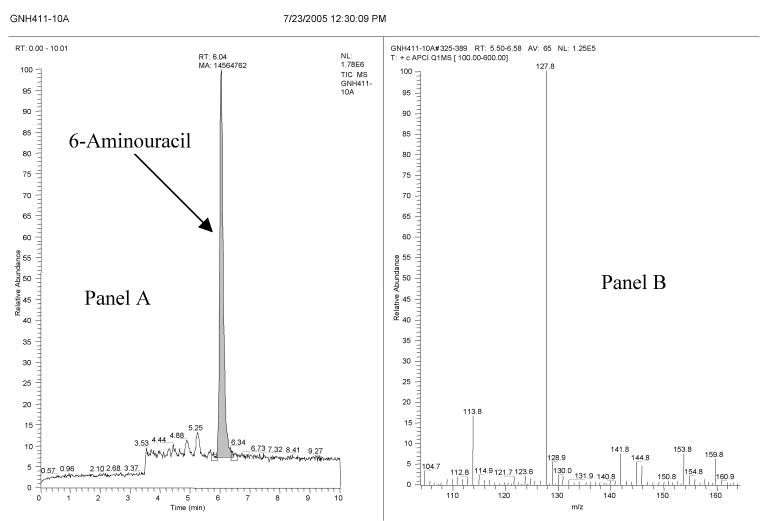
LC-MS analysis of the products from the reaction of uric acid (panels A and B) with NO. The figure contains the original date stamp of the analysis to support the discovery claim. Panel A shows the LC-MS trace (total ion current) and panel B shows the corresponding mass spectra (M+1 ion = 128). The peak at 6.04 minutes in panel A is identical to the commercially available 6-aminouracil. In addition, two small unidentified peaks (at 4.88 and 5.25 minutes) were present in the LC-MS trace.
The clinical significance of the finding that uric acid can react with nitric oxide to form 6-aminouracil remains to be established. However, it could provide a mechanism for how uric acid may inhibit endothelial function under conditions of oxidative stress in which intracellular glutathione is depleted. It may also provide a logical explanation for why UA can function as an antioxidant to block the effects of exogenous peroxynitrite to uncouple endothelial NO synthase[18] yet in the presence of high concentrations of UA favor the reduction of intracellular NO levels.[16] It also provides an explanation for why UA infusions into healthy individuals (likely with good intracellular glutathione stores) do not cause endothelial dysfunction acutely.[25] Finally, it could explain the recent report[26] that allopurinol improves endothelial function by blocking oxidative stress rather than by lowering serum UA concentrations. In this case, by blocking oxidative stress, the allopurinol could be restoring intracellular glutathione stores back to a healthy level. This increased concentration of intracellular glutathione would reduce the scavenging of NO by UA independent of serum UA concentrations. It is important to recognize that UA may also have other mechanisms by which it participates in the induction of endothelial dysfunction. However, given the preferential scavenging of NO by UA, even in the presence of peroxynitrite, this potentially could be a significant pathway. The stable end-product, 6-AU, may serve as a marker for detecting the direct reaction of NO with UA in vivo and its contribution to endothelial dysfunction under conditions of oxidative stress. Clearly, more studies are needed to address the importance of this reaction under pathophysiological conditions.
Acknowledgments
This work was supported by Gatorade and by NIH grants DK-52121, HL-68607 and MO1-RR00082.
References
- 1.Wu X, Muzny DM, Lee CC, Caskey CT. Two idependent mutational events in the loss of urate oxides during hominoid evolution. J Mol Evol. 1992;34:78–84. doi: 10.1007/BF00163854. [DOI] [PubMed] [Google Scholar]
- 2.Kuzkaya N, Weissmann N, Harrison DF, Dikalov S. Interactions of peroxynitrite with uric acid in the presence of ascorbate and thiols: implications for uncoupling endothelial nitric oxide synthase. Biochem Pharmacol. 2005;70:343–354. doi: 10.1016/j.bcp.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Robinson KM, Morre JT, Beckman JS. Triuret: A novel product of peroxynitrite-mediated oxidation of urate. Arch Bioch Biophys. 2004;423:213–217. doi: 10.1016/j.abb.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci USA. 1981;78:6853–6862. doi: 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kand'ar P, Zakova P, Muzakova V. Monitoring of antioxidant antioxidant properties properties of uric uric acid acid in humans humans for a consideration consideration measuring measuring of levels levels of allantoin allantoin in plasma plasma by liquid liquid chromatographychromatography. Clin Chim Acta. 2006;365:249–256. doi: 10.1016/j.cca.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Hediger MA. Kidney function: Gateway to a long life? Nature. 2002;417:393–395. doi: 10.1038/417393a. [DOI] [PubMed] [Google Scholar]
- 7.Bagnati M, Perugini C, Cau C, Bordone R, Albano E, Bellomo G. When and why a water-soluble antioxidant becomes pro-oxidant during copper-induced low-density lipoprotein oxidation: A study using uric acid. Biochem J. 1999;340:143–152. [PMC free article] [PubMed] [Google Scholar]
- 8.Sanguinetti SM, Batthyany C, Trostchansky A, Botti H, Lopez GI, Wikinski RLW, Rubbo H, Schreier LE. Nitric oxide inhibits prooxidant actions of uric acid during copper-mediated LDL oxidation. Arch Biochem Biophys. 2004;423:302–308. doi: 10.1016/j.abb.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 9.Kittridge KJ, Willson RL. Uric acid substantially enhances the free radical-induced inactivation of alcohol dehydrogenase. FEBS Lett. 1984;170:162–164. doi: 10.1016/0014-5793(84)81391-5. [DOI] [PubMed] [Google Scholar]
- 10.Aruoma OI, Halliwell B. Inactivation of alpha-antiproteinase by hydroxyl radicals. The effect of uric acid. FEBS Lett. 1989;244:76–80. doi: 10.1016/0014-5793(89)81166-4. [DOI] [PubMed] [Google Scholar]
- 11.Johnson RJ, Feig DI, Kang DH, Herrera-Acosta J. Resurrection of uric acid as a causal risk factor in essential hypertension. Hypertension. 2005;45:18–20. doi: 10.1161/01.HYP.0000150785.39055.e8. [DOI] [PubMed] [Google Scholar]
- 12.Nakagawa T, Hu H, Zharikov S, Tuttle KR, Ahort RA, Glushakova O, Ouyang X, Feig DI, Block ER, Herrera-Acosta J, Patel JM, Johnson RJ. Uric acid a causal factor for fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290:F625–F631. doi: 10.1152/ajprenal.00140.2005. [DOI] [PubMed] [Google Scholar]
- 13.Masuo K, Kawaguchi H, Mikani H, Ogihara T, Tuck ML. Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension. 2003;42:474–480. doi: 10.1161/01.HYP.0000091371.53502.D3. [DOI] [PubMed] [Google Scholar]
- 14.Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Takishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertension Res. 2001;24:691–697. doi: 10.1291/hypres.24.691. [DOI] [PubMed] [Google Scholar]
- 15.Tomita M, Mizuno S, Yamanaka H, Hosoda Y, Sakuma K, Matuoka Y, Odaka M, Yamaguchi M, Yosida H, Morisawa H, Murayama T. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol. 2000;10:403–409. doi: 10.2188/jea.10.403. [DOI] [PubMed] [Google Scholar]
- 16.Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, Krotova K, Block ER, Prabhakar S, Johnson RJ. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005;67:1739–1742. doi: 10.1111/j.1523-1755.2005.00273.x. [DOI] [PubMed] [Google Scholar]
- 17.Kanabrocki EL, Sothern RB, Messmore HL, Roitman-Johnson B, McCormick JB, Dawson S, Brenner FW, Third JL, Nemchausky BA, Shirazi P, Scheving LE. Circadian relationship of serum uric acid and nitric oxide. J Am Med Assoc. 2000;283:2240–2241. doi: 10.1001/jama.283.17.2240. [DOI] [PubMed] [Google Scholar]
- 18.Forstermann U. Janus-faced role of endothelial NO synthase in vascular disease: Uncoupling of oxygen reduction from NO synthesis and its pharmacological reversal. Biol Chem. 2006;387:1521–1533. doi: 10.1515/BC.2006.190. [DOI] [PubMed] [Google Scholar]
- 19.Goldstone AB, Liochev SI, Fridovich I. Inactivation of copper, zinc superoxide dismutase by H2O2: Mechanism of protection. Free Radic Biol Med. 2006;41:1860–1863. doi: 10.1016/j.freeradbiomed.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 20.Chacko SA, Wenthold PG. The negative ion chemistry of nitric oxide in gas phase. Mass Spectrom Rev. 2006;25:112–116. doi: 10.1002/mas.20060. [DOI] [PubMed] [Google Scholar]
- 21.Kharitonov VG, Sundquist AR, Sharma VS. Kinetics of nitrosation of thiols by nitric oxide in the presence of oxygen. J Biol Chem. 1995;270:28158–28164. doi: 10.1074/jbc.270.47.28158. [DOI] [PubMed] [Google Scholar]
- 22.Patel JM, Zhang J, Block ER. Nitric oxide-induced inhibition of lung endothelial cell nitric oxide synthase via interaction with allosteric thiols: Role of thioredoxin in regulation of catalytic activity. Am J Respir Cell Mol Biol. 1996;15:410–419. doi: 10.1165/ajrcmb.15.3.8810647. [DOI] [PubMed] [Google Scholar]
- 23.Hoehn T, Gratopp A, Raehse K, Koehne P. Effects of hyperoxia and nitric oxide on endogenous nitric oxide production in polymorphonuclear leukocytes. Neonatology. 2005;94:132–137. doi: 10.1159/000119723. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki T. Nitrosation of uric acid induced by nitric oxide under aerobic conditions. Nitric Oxide. 2007;16:266–273. doi: 10.1016/j.niox.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 25.Waring WS, Adwani SH, Breukels O, Webb DJ, Maxwell SR. Hyperuricaemia does not impair cardiovascular function in healthy adults. Heart. 2004;90:155–159. doi: 10.1136/hrt.2003.016121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.George J, Carr E, Davies J, Belch JJ, Struthers A. High dose allopurinol improves endothelial function by profoundly reducing vaxcular oxidative stress and not by lowering uric acid. Circulation. 2006;114:2508–2516. doi: 10.1161/CIRCULATIONAHA.106.651117. [DOI] [PubMed] [Google Scholar]