

Abstract
Purpose
Early clinical trials are underway exploring the direct oncolytic potential of reovirus. This study addresses whether tumor infection by reovirus is also able to generate bystander, adaptive anti-tumor immunity.
Experimental design
Reovirus was delivered iv. to C57Bl/6 mice bearing lymph node metastases from the murine melanoma, B16tk, with assessment of nodal metastatic clearance, priming of anti-tumor immunity against the tumour associated antigen (TAA) TRP-2, and cytokine responses. In an in vitro human system, the effect of reovirus infection on the ability of Mel888 melanoma cells to activate and load dendritic cells (DC) for cytotoxic T cell (CTL) priming was investigated.
Results
In the murine model, a single iv dose of reovirus reduced metastatic lymph node burden and induced anti-tumor immunity (splenocyte response to TRP-2, and IL-12 production in disaggregated lymph nodes). In vitro human assays revealed that uninfected Mel888 cells failed to induce DC maturation, or support priming of an anti-Mel888 CTL response. In contrast, reovirus infected Mel888 cells (reo-Mel) matured DC in a reovirus dose-dependent manner. When cultured with autologous peripheral blood lymphocytes, DC loaded with reo-Mel induced lymphocyte expansion, IFN-γ production, specific anti-Mel888 cell cytotoxicity, and cross primed CD8+ T cells specific against the human TAA MART-1.
Conclusion
Reovirus infection of tumor cells reduces metastatic disease burden and primes anti-tumour immunity. Future clinical trials should be designed to explore both the direct cytotoxic and immunotherapeutic effects of reovirus.
Keywords: Reovirus, melanoma, oncolytic virus, anti-tumor immunity
Introduction
Oncolytic viruses are self replicating, tumor selective viruses, which directly lyse cancer cells (1). Although most interest in both naturally occurring and genetically modified oncolytic viruses has focussed upon their direct oncolytic properties, there is accumulating evidence suggesting that tumor infection can also induce anti-tumor immunity (2-8).
The ability of the immune system to modify the immunogenicity and behaviour of clinically evident tumors, and the host of mechanisms by which tumors can induce a state of immune tolerance, is becoming increasingly recognised (9). Whilst a range of TAA have been identified (10), the presence of tumor-associated ‘danger’ signals is critical to the generation of an anti-tumor immune response (11). Tumors commonly lack such signals, and it has been proposed that successful tumor immunotherapy will be dependent on their provision (12). Oncolytic virotherapy is expected to promote an inflammatory ‘dangerous’ environment within the tumor, involving the release of pro-inflammatory cytokines, toll-like receptor (TLR) ligands, and an infiltration of innate immune cells (12-14). In addition, virally-induced tumor cell lysis can release a wide range of TAA into the tumor microenvironment for uptake by professional antigen presenting cells, such as DC, for adaptive T cell priming. The immune consequences of oncolytic viral therapy are, however, finely balanced, with many viruses possessing immune evasion strategies involving the inhibition of DC maturation and/or function (15, 16). The ability of DC to take up and cross-present TAA in an appropriate costimulatory context to T cells, is central to the generation of an effective adaptive anti-tumor cellular immune response (17).
Reovirus is a naturally occurring oncolytic virus, and is a ubiquitous member of the Reoviridae family of non-enveloped double stranded RNA (dsRNA) viruses. Reovirus can be isolated from the respiratory and enteric tracts of humans, but infection is usually asymptomatic (18, 19). Non-transformed cells are relatively resistant to infection, whilst transformed cells with activated ras signalling pathways are permissive to reovirus, via enhancement of viral uncoating, increased particle infectivity and apoptotic release of viral progeny (20). Ras signalling pathways are aberrant in most human tumours, involving either activating Ras mutations or altered upstream or downstream pathways (21), such that reovirus has a broad range of activity against human tumors including breast, colon, ovary, brain and haematological malignancies in pre-clinical models (22, 23). Oncolytic reovirus has already entered early phase clinical trials, administered intratumorally (24) and intravenously (25). The ability of reovirus infection of tumors to generate anti-tumor immunity has, however, not been fully addressed. In older murine studies combining reovirus with the chemotherapeutic agent BCNU, cured mice were protected from tumor rechallenge, implying an immune-mediated effect, although no mechanisms were defined (26).
This study first demonstrates the ability of intravenous reovirus monotherapy to induce anti-tumor immunity in addition to a reduction in metastatic disease burden, in an immunocompetent murine melanoma model of lymph node metastases. To determine the applicability of these findings to human cancer, we have also explored the immune consequences of reovirus infection of the human melanoma cell line Mel888. Reovirus-infected (but not uninfected) Mel888 cells activate DC phenotypically and functionally, in a contact dependent manner. Only DC loaded with reovirus-infected Mel888 cells prime an in vitro naïve CTL response against Mel888, including cross priming of CTL specific for the melanoma TAA, MART-1. These murine and human data support the role of reovirus as an immunogenic as well as directly cytotoxic therapy for human neoplasia, activating DC and priming effective anti-tumor immunity.
Materials and Methods
Reovirus
Reovirus Type 3 Dearing strain was provided by Oncolytics Biotech Inc. (Calgary, Canada), and stored in the dark at neat concentrations in phosphate buffered saline (PBS) at 4°C (maximum 3 months) or at -80°C (long term storage). Virus titre was determined by a standard plaque assay using L929 cells.
Murine in vivo assays
Murine Cells
Mouse B16-tk melanoma cells (H2-Kb) were derived from B16 cells by transducing them with a cDNA encoding the herpes simplex virus thymidine kinase (tk) gene (27). Cells were grown in DMEM (Life Technologies) supplemented with 10% (v/v) FCS (Life Technologies), L-glutamine (Life Technologies) and 1.25μg/ml puromycin selection. Cell lines were routinely tested for Mycoplasma and found to be free of infection.
In vivo studies
All procedures were approved by the Mayo Foundation Institutional Animal Care and Use Committee. C57BL/6 mice were purchased from Jackson Laboratories at 6-8 weeks of age. To establish subcutaneous (sc) tumors 5×105 B16-tk cells were injected in 100μl of PBS into the flanks of mice (subgroups of 3 mice in each experiment). 10 days later, 5×108pfu reovirus or PBS was administered intravenously (iv). Tumor draining lymph nodes and spleen were explanted after a further 10 days.
PCR screening for B16-tk tumor cells
Genomic DNA from lymph nodes was prepared with the DNeasy kit (Qiagen). 10ng of DNA was amplified by PCR with primers specific for HSVtk, which is stably integrated into the genome of B16tk tumor cells. As a control, PCR was performed with primers specific for a genomic fragment of the murine tyrosinase promoter (TyrP). In all experiments, a mock PCR (without added DNA) was performed to exclude contamination.
Puromycin-resistant colony outgrowth assay to detect metastatic B16-tk tumor cells
B16-tk tumor cells stably express the puromycin-resistance gene, allowing for growth in puromycin. To select for viable B16-tk cells present at resection, 1×106 cells from dissociated lymph nodes were plated in six-well plates at 1.25μg/ml puromycin. Every 2-3 days cultures were washed and fresh puromycin-containing medium added. Within 5-10 days, individual puromycin-resistant colonies were counted in wells.
Enzyme-linked immunosorbent assay analysis (ELISA) for interferon-γ (IFN-γ) secretion
1×106 day 10 splenocytes were plated into 24-well plates in triplicate and incubated at 37°C with 5μg/ml of appropriate peptide. Cell-free supernatants were collected after 48h and tested by specific ELISA for IFN-γ, according to the manufacturer's instructions (OptEIA IFN-γ kit; BD Biosciences). The synthetic, H-2Kb-restricted peptides TRP-2180-188 SVYDFFVWL and control ovalbumin SIINFEKL were synthesized at the Mayo Foundation Core Facility.
Statistics
The two-sample unequal variance Student's t-test was used for in vitro assays. Statistical significance was determined at the level of P<0.05.
Human in vitro assays
Cell culture
Human melanoma cell lines, Mel888, Mel624, Mewo, SK-Mel28, HT144, MM96, and non-melanoma tumor cell lines SW480, HCT116 (colorectal), MCF7 (breast), SKOV-3 (ovarian), EJ (bladder), and, SiHa (cervix), were grown in DMEM (Gibco BRL, Paisley, UK) supplemented with 10% (v/v) FCS (Harlan Sera-Labs, Crawley Down, UK) and 1% (v/v) L-glutamine (Gibco). Cells were routinely tested for Mycoplasma and found to be free of infection.
Human DC generation
Peripheral blood mononuclear cells (PBMCs) were obtained from buffy coats of healthy blood donors by Ficoll-Hypaque density centrifugation, and monocytes isolated by plastic adherence as previously described (28). Immature DC were generated by culture in DC media (RPMI 1640 (Gibco) supplemented with 10% (v/v) FCS and 1% L-glutamine and 800U/ml GMCSF and 500U/ml IL-4 (R&D Systems, Abingdon, UK)) for 5 days.
Reovirus infection of Mel888 cells and DC co-culture
Mel888 cells were seeded on day 1, and infected on day 2 at 0.1, 1 and 10pfu reovirus per cell. After 18h infection, Mel888 cells were harvested and cultured with DC at a 3:1 ratio in DC media. LPS 250ng/ml (Sigma) was added where appropriate as a positive control for DC activation. Co-cultures were harvested at 24h. Cell free supernatants were stored at -80°C. To test contact dependence, DC and tumor cells were separated by filters (0.4μm) in transwell plates (Corning, Schiphol Rijk, The Netherlands).
Flow cytometry
Flow cytometry was performed using a FACSCalibur (Becton-Dickinson). Anti-human HLA-DR-FITC, CD80-PE, CD83-PE, CD86-PE, CD40-PE (BD Pharmingen) were used for DC phenotype. DC were identified in the mixed DC/tumor cell population by gating on HLA-DR-FITC-positive cells (Mel888 cells are class II negative).
Cytokine detection
Levels of IFN-γ, IL-4, IL-6, IL-10, IL-12p70 and TNF-α in tissue culture supernatants were measured by ELISA using matched paired antibodies (all from BD Biosciences, except TNF-α from Biosource, Nivelles, Belgium) according to manufacturers' instructions.
DC viability
DC were labelled with 1μM CellTracker Green (Invitrogen), prior to co-culture with Mel888 cells, as above. DC and tumor cells were harvested and stained with propidium iodide (Sigma), prior to flow cytometry with analysis gated on labelled DC.
Phagocytosis assay
Living DC and Mel888 cells were labelled with 1μM CellTracker Green and 5μM CellTracker Red (Invitrogen) respectively, prior to co-culture at a 1:3 ratio. Double positive cells were enumerated by flow cytometry. Subsequent incubation for 1h with 75nM Lysotracker Blue (Invitrogen) was used to co-label late phagosomal and lysosomal structures. Living cells were visualised using a Zeiss Axiovert 200 inverted fluorescence microscope as previously described (29).
Generation of tumor-specific cytotoxic lymphocytes (CTL)
Immature DC were loaded with uninfected Mel888 cells or Mel888 infected for 18 hours with 0.1pfu/ cell reovirus at a 1:3 ratio. Tumor loaded DC were irradiated (30Gy), and mixed with autologous PBMCs at a 1:10-1:30 ratio. CTL media (RPMI supplemented with 7.5% (v/v) human AB serum (Sigma), 1% (v/v) L-glutamine, 1% v/v sodium pyruvate (Gibco), 1% (v/v) non-essential amino acids (Gibco), 1% (v/v) HEPES (Gibco), 20μM 2β-mercaptoethanol (Sigma)) was used in CTL cultures, supplemented with IL-7 (R&D Systems) 5ng/ml from day 1, and IL-2 (R&D Systems) 30U/ml on day 4 only. Cultures were restimulated using the same protocol at weekly intervals. Cells were harvested at day 14 or 21.
51Chromium (51Cr) cytotoxicity assay
Cytotoxicity was measured using a standard 4h 51Cr release assay (30). Unlabelled K562 cells added to tumor targets to reduce non-specific killing. 4h supernatants were counted in scintillation plates (Packard Biosciences, Groningen, The Netherlands). % lysis was calculated using the formula: % lysis= 100×(cpm experiment – cpm spontaneous release)/(cpm maximum release-cpm spontaneous release).
CD107 lymphocyte degranulation assay
Lymphocyte degranulation was measured as previously described (31). CTL and tumor targets were incubated at a 1:1 ratio with anti-CD107a-FITC and anti-CD107b-FITC antibodies (BD Biosciences), with brefeldin A (Sigma) added at 10μg/ml after 1 hour. After a further 4 hours, CTL were stained with anti-human CD8-PerCP and acquired by flow cytometry. To determine MHC class I restriction, a pan-MHC class I blocking antibody (Dako, Cambridgeshire, UK) or an isotype antibody (Dako) were added at 50μg/ml throughout CTL/ tumor target incubation.
Assessment of MART-1-specific lymphocytes
CTL were treated with Dead Cell Discrimination Kit (Miltenyi Biotec), labelled with MART-1-PE pentamer (ELAGIGITLV) or human negative control-PE pentamer (Proimmune), counterstained with CD8-FITC, and fixed in 1% PFA as per manufacturers' protocols. Analysis was performed by flow cytometry, gating on live lymphocytes by excluding cells labelled with Dead Cell Discrimator.
Results
Intravenous reovirus reduces lymph nodes metastases in vivo
A murine model of lymph node metastasis from an established tumour was used as recently described (5). In this model, the readouts are the clearance of metastases from lymph nodes draining the primary tumor, cytokine production and associated generation of an immune response against a melanoma TAA (TRP-2). In the current work, a melanoma cell line encoding HSV-tk was used (27), with tumor detection measured by rtPCR for the HSVtk transgene and puromycin-resistant tumor colony outgrowth. B16-OVA, as previously studied (5) was not suitable, as the OVA-transfected line (unlike parental B16 and B16-tk) is relatively resistant to oncolysis by reovirus (data not shown). The mechanism of the resistance of B16-OVA to reovirus is unclear, although the mechanisms underlying sensitivity to reovirus are known to be complex (20). 10 days after seeding with sc B16-tk tumours, mice were treated iv with 5×108 pfu reovirus or PBS; 10 days later tumour draining lymph nodes (LN) and spleen were isolated for analysis.
Semi-quantitative PCR for the HSVtk transgene indicated that iv delivery of reovirus alone induced a reduction (though not clearance) of the numbers of B16-tk cells that could be detected in the draining lymph nodes (Figure 1A). These results were supported by a significant reduction in the number of puromycin resistant B16-tk colonies grown from dissociated tumour draining lymph node cultures exposed to puromycin following treatment with iv reovirus (for pooled results of 2 independent experiments p=0.015) (Figure 1B). These data suggest that direct iv reovirus is able to significantly reduce the tumor burden in lymph nodes draining a primary B16-tk tumor in immune competent mice.
Figure 1. Intravenously administered reovirus can reduce lymph node metastatic melanoma burden, prime anti-tumor immunity and induce pro-inflammatory cytokines.
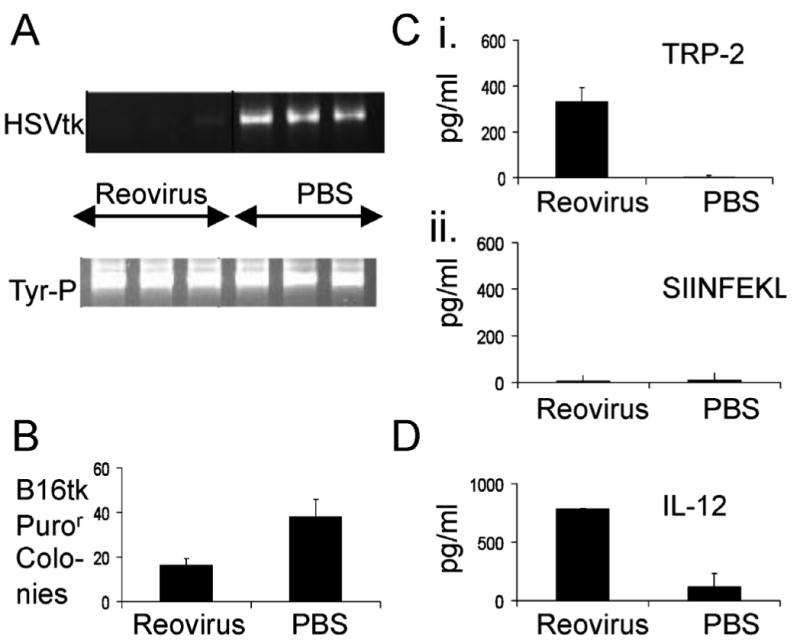
C57Bl/6 mice were seeded with sc B16-tk tumors (5×105 cells). 10 days later, mice were treated intravenously with 5×108 pfu reovirus or with PBS. 10 days after that, tumor draining LN and spleen were explanted, and LN were dissociated and plated overnight in culture. A. Total genomic DNA from 106 LN cells was screened with primers specific for the HSVtk gene. Equal loading of DNA was confirmed using primers specific for a genomic fragment in the tyrosinase gene promoter. B. 106 cells from the dissociated LN cultures were seeded in medium containing 1.25μg/ml puromycin to select for viable B16-tk cells which were present in the LN at the time of resection. Within 5-10 days individual puror colonies appear, which were counted. C. Spenocytes recovered at day 10 were pulsed, in triplicates of 750000, with the synthetic TRP-2 180-188 SVYDFFVWL peptide (i) or with the irrelevant H-2Kb-restricted Ova SIINFEKL peptide (ii). 48h later supernatants were assayed by ELISA for IFNγ. D. Explanted tumor draining LN were dissociated and plated overnight in culture. Supernatants were assayed for IL-12. Data shown representative of one of two independent experiments.
Reovirus oncolysis in lymphoid organs primes antitumor immunity and induces IL-12 in lymph nodes following in vivo delivery
The splenocytes recovered at day 10 were pulsed with synthetic tyrosinase-related protein-2180-188 (TRP-2) peptide and an irrelevant SIINFEKL epitope of the ovalbumin antigen, and supernatants assayed for IFN-γ after 48h (Figure 1C). A single iv dose of reovirus was able to prime significant anti-TRP-2 immune responses (Figure 1C); no specific T cell responses were seen towards the irrelevant SIINFEKL epitope (Figure 1C). These data suggest that iv delivery of reovirus is effective at priming anti tumor immunity, through the breaking of tolerance to self tumor antigens. To investigate this further, the production of Th1 cytokines from explanted tumor draining LN was examined. Reovirus induced significant levels of IL-12 (Figure 1D), although neither IL-6 nor TNF-α were detected (data not shown).
Taken together, these data indicate that in vivo a single dose of iv reovirus can reduce tumor metastatic burden, and induce priming of an anti tumor immune response in a fully immune competent murine system. To progress these studies towards clinical application, we next tested whether the observations made in the murine model also applied to human in vitro systems.
Effect of reovirus-infected Mel888 cells upon human DC phenotype, function and viability
DC are the key antigen presenting cells regulating adaptive immunity, and the interaction between tumour cells and DC is critical in determining the ability of DC to generate an effective immune response (32). To test the immunological consequences of reovirus infection of tumour cells in a human in vitro system, and to translate the murine data towards human application, the effect of reovirus infection of the human melanoma cell line Mel888 upon DC phenotype, cytokine secretion and viability was first examined. Recent data have confirmed that reovirus is cytotoxic to human melanoma cells, that infected cells secrete inflammatory cytokines (33), and that free reovirus directly matures DC (34). However, the effect of reovirus infection on the interaction between tumor cells and DC has not previously been addressed. First, immature DC were co-cultured for 24 hours with control Mel888 cells or Mel888 cells which had been infected with 0.1, 1 and 10pfu/cell reovirus 18 hours previously; LPS was used as a positive control for DC activation. DC maturation was examined by surface expression of CD86, CD80, CD83, CD40, and MHC class II (Figure 2A), and secretion of the inflammatory cytokines IL12-p70, TNF-α, and IL-6 (Figure 2B).
Figure 2. Analysis of human DC phenotype, cytokine secretion and viability after co-culture with reovirus-infected Mel888 cells.

DC were incubated with LPS (250ng/ml), uninfected Mel888 cells or Mel888 cells infected with 0.1, 1 and 10pfu reovirus/ Mel888 cell, at a 1:3 ratio for 24 hours. A. Surface expression of DC phenotypic markers, CD86, CD80, CD83, CD40 and MHC class II, was examined by flow cytometry. Median fluorescence intensity is shown in each plot. Contact dependence of phenotypic changes was examined using a 0.4μm transwell. Data shown is one representative of at least four independent experiments. B. Levels of IL-12p70, IL-6 and TNFα in supernatant were determined by ELISA. Data shown is one representative of six independent experiments. C. The proportion of non-viable DC was examined by flow cytometry, gating on DC labelled with CellTracker Green, and stained with propidium iodide. Data shown is representative of two independent experiments.
Uninfected Mel888 cells had little effect upon the immature DC phenotype. In contrast, reovirus-infected Mel888 cells induced DC maturation in a virus dose-dependent fashion (Figure 2A). Although phenotypic changes were minimal with Mel888 cells infected with reovirus 0.1pfu/cell, the DC phenotype following co-culture with Mel888 cells infected at 10pfu/cell was similar to that induced by LPS. To explore the mechanisms behind the maturation of DC phenotype, transwell experiments were performed to test whether this effect required cell-cell contact, or was mediated by a soluble factor. As seen in Figure 2A, the upregulation of a representative activation marker, CD86, induced by reovirus-infected Mel888 cells was almost completely abrogated, indicating that DC maturation was dependent on contact between melanoma cells and DC.
As shown in Figure 2B immature DC, as expected, produced very low levels of IL-12p70, TNF-α and IL-6. Co-culture of DC with uninfected Mel888 cells did not affect production of these cytokines, whilst reovirus infection of Mel888 cells elicited a dose dependent increase in all three.
Several oncolytic viruses including vaccinia virus (35) and herpes simplex virus-1 (36) have been reported to adversely effect DC viability. Therefore, the effect of reovirus-infected Mel888 cell co-culture upon DC viability was examined by propidium iodide staining of DC. As shown in Figure 2C, there was some loss of DC viability, although toxicity was minimal at a reovirus dose of 0.1pfu/Mel888 cell.
Reovirus does not alter phagocytic uptake of Mel888 cells into DC late endosomes/lysosomes
The infection of tumour cells by several viruses has been reported to enhance the phagocytosis of these cells by DC (7, 8). Therefore, phagocytic assays were performed using differential cell labelling of DC and tumor cells with fluorescent dyes, to allow analysis of uptake of Mel888 cells +/- reovirus infection by DC. As shown in Figure 3A, any association of tumour cells with DC was low after 40 minutes co-culture, but after 4 hours the majority of DC were associated with material from tumor cells; reovirus infection of Mel888 cells did not alter association of tumour with DC. Since flow cytometry is unable to determine whether DC were phagocytosing material from Mel888 cells, as opposed to closely associating with such material or adhering to intact tumour cells, fluorescence microscopy was also performed as shown in Figure 3B. Images demonstrated uptake of Mel888 material (red), into DC (green), confirming phagocytosis. These images were similar regardless of whether Mel888 cells were infected with reovirus. In order to address the nature of the subcellular compartments to which tumor material localised after uptake by DC, microscopy was carried out after the incubation of cocultures with Lysotracker Blue, a dye which labels acidic late endosomal and lysosomal structures. Within DC, internalised tumour cell material clearly co-localised with Lysotracker Blue labelled compartments, consistent with phagocytic uptake into appropriate compartments for priming (37).
Figure 3. DC phagocytose uninfected and reovirus-infected Mel888 cells equally efficiently.
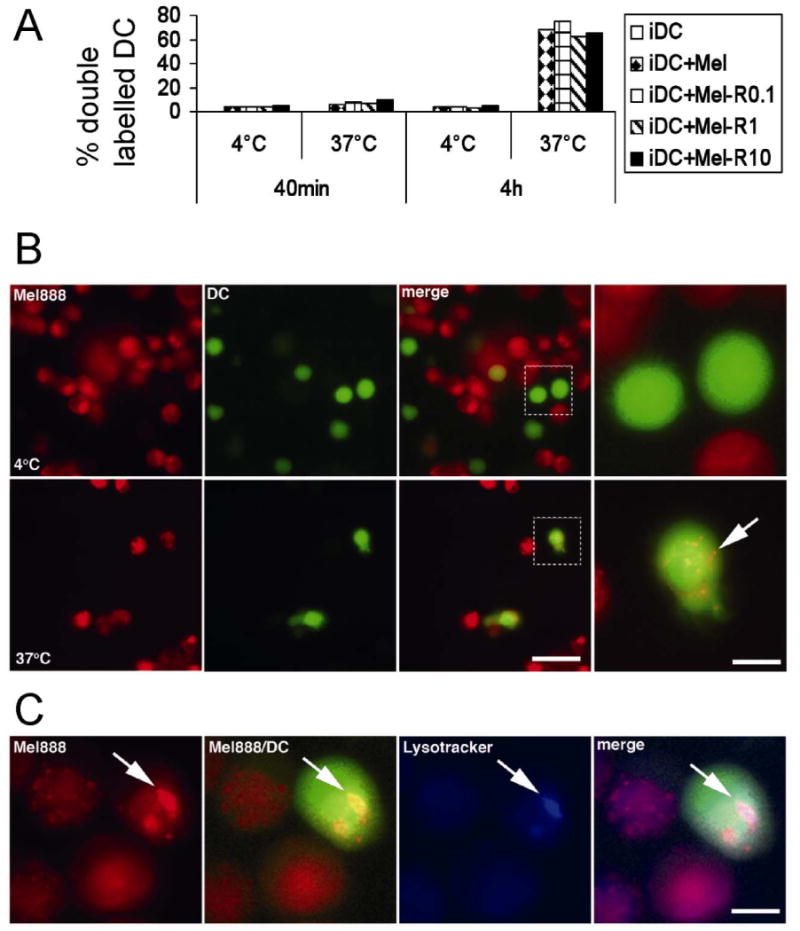
DC labelled with CellTracker Green were co-cultured at 4°C and 37°C at a 1:3 ratio with Mel888 cells or Mel888 cells infected with 0.1, 1 or 10pfu reovirus and labelled with CellTracker Red. A. Uptake of Mel888 by DC was examined by flow cytometry after co-culture for 40 minutes or 4 hours. The percentage of DC double-labelling red and green was assessed. Results shown are representative of six independent experiments. B. The uptake of Mel888 tumor material by DC after 4h at 37°C was confirmed by fluorescence microscopy of living cultures. At 4°C no Mel888 derived material (red) was observed within DC (green), whereas at 37°C Mel888 material was observed to be internalised within DC (arrow). Bar=40μm. In zoomed image bar=10μm. C. Mel888-derived material internalised within DC localised to late endosomal/ lysosomal structures, as shown by colocalisation of red dyed material with Lysotracker Blue labelled intracellular compartments, within green labelled DC (arrow). Bar=10μm. Images shown are representative of data obtained from 2 independent donors. Similar images were obtained following 4h coculture of DC with uninfected or reovirus-infected Mel888 cells.
Priming of tumour-specific cytotoxic T cells by DC loaded with reovirus-infected Mel888 cells
Next, the ability of reovirus-infected versus uninfected Mel888 cells to prime a human naïve T cell response was determined. DC were loaded for 24 hours with uninfected Mel888 cells (Mel-DC) or Mel888 cells infected with 0.1pfu/cell reovirus (reo-Mel-DC). Autologous PBMC were then co-cultured with tumour-loaded DC, and restimulated with further DC loaded in the same way weekly. Despite minor effects upon DC phenotype and function (Figure 2A and B), this low reovirus concentration of 0.1pfu/Mel888 cell was selected for these longer term priming cultures due to the lack of toxicity to DC (Figure 2C), and to avoid overwhelming DC with mounting viral antigen during replication. In addition, recent insights have suggested that phenotypic maturation per se is not the distinguishing feature of immunogenic versus tolerogenic DC (38).
In order to monitor PBMC proliferation during naïve priming, trypan blue exclusion was used to determine the number of viable cells each week. The results of two donors representative of results in >10 experiments are shown in Figure 4A. Consistent with previous data (30), PBMC stimulated with Mel-DC did not undergo any expansion. In contrast, stimulation with reo-Mel-DC consistently yielded more effector cells after two weeks of culture.
Figure 4. Naïve priming of human tumour-specific cytotoxic lymphocytes by DC loaded with reovirus-infected Mel888 cells.

PBMC were incubated with autologous DC loaded overnight with Mel888 cells or Mel888 cells infected with 0.1pfu reovirus per Mel888 cell (at 1:3 ratio), restimulated 7 days later, and assayed at 14 days. A). Lymphocyte proliferation was determined by trypan blue exclusion. B). Cytotoxicity of lymphocytes primed in the presence or absence of reovirus was determined by Cr51 release assay using Mel888 cells and a range of melanoma and non-melanoma cell lines as targets. Two experiments representative of at least six independent donors are shown in A and B. C. The specificity of the activity of lymphocytes primed by DC loaded with reovirus-infected Mel888 cells towards Mel888 cells was further tested using the CD107 lymphocyte degranulation assay, towards a larger panel of melanoma and non-melanoma cell lines. D. A pan-MHC class I blocking antibody and isotype control was used to test the MHC class I restriction of degranulation, measured by the CD107 assay, towards Mel888 cells. The percentage of CD8 T cells degranulating is shown. Data representative of at least four independent experiments.
The activity of CTL generated by Mel-DC and reo-Mel-DC after 2 weeks of culture towards Mel888 cells and other melanoma and non-melanoma targets, was determined first using a standard 51Cr release assay. Figure 4B shows the pattern of target killing in two donors, representing results typical of 6 independent experiments. CTL generated by stimulation by Mel-DC showed little cytotoxic activity. In contrast, CTL generated using reo-Mel-DC consistently exhibited high levels of specific cytotoxicity towards Mel888 cells, with up to 80% lysis observed. As shown, no significant cytotoxicity was observed towards a limited range of other cell lines in this 51Cr release assay. In order to further assess the degree of specificity of these CTL, their degranulation was assessed using a CD107 expression assay (31), in response to a wider panel of melanoma and non-melanoma cell lines. As shown in Figure 4C, reo-Mel-DC generated CTL exhibited no significant cross reactivity to 12 other cell lines. Furthermore, CTL activity was MHC class I restricted, as shown by significant reduction in the levels of degranulation against Mel888 cells in the presence of a pan-MHC class I blocking antibody (Figure 4D).
Cytokine production in CTL priming cultures
The profile of cytokines produced within the CTL priming cultures was investigated to determine whether reo-Mel-DC polarised the immune response towards a Th1 or Th2 direction. Figure 5 demonstrates that priming with reo-Mel-DC was associated with the production of high levels of the Th1 cytokine IFN-γ (up to 10,000pg/ml). In contrast, IFN-γ was barely detectable when Mel-DC were used in priming cultures. Low levels of TNF-α and IL-6 were inconsistently produced, and no significant production of the Th2 cytokines IL-4 and IL-10 was found in either priming condition (data not shown). The high levels of IFN-γ produced are indicative of a Th1 skew induced by reo-Mel-DC.
Figure 5. Cytokine production in CTL cultures.
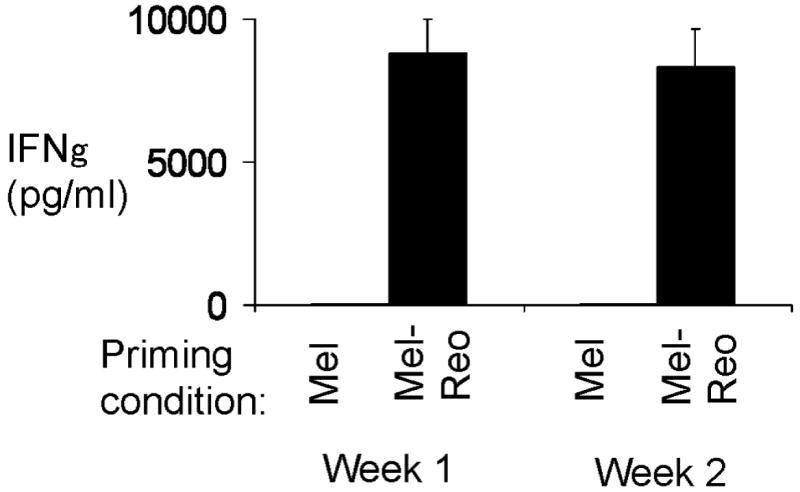
A). Levels of IFN-γ in CTL cultures were determined by ELISA each week. Data from one donor representative of nine shown.
Cross priming of CTL with specificity towards a defined melanoma TAA by reo-Mel-DC
Although reo-Mel-DC generate CTL with specific MHC class I-restricted cytotoxicity towards Mel888 cells, it was not clear whether the anti-Mel888 activity was directed towards TAA in this allogeneic system. DC loaded with whole tumour cells in this system have access to a host of TAA (many of which are likely to be as undefined) in addition to non-tumour antigens (including viral) and allo-antigens. We therefore wished to address whether the reo-Mel-DC-induced CTL polyclonal population included cells specific for a TAA. MART-1 is one defined HLA-A2 restricted melanoma TAA expressed by Mel888 cells. Mel888 cells are HLA-A2 negative and therefore cannot present MART-1 directly to T cells (30). Hence, any expansion of T cells with specificity towards MART-1 on co-culture with Mel888-loaded HLA-A2+ve DC (from the same donor as the T cells) must represent cross priming against MART-1 mediated by DC. In view of the host of different antigens present in these priming cultures, the frequency of CTL generated with activity towards a particular TAA is likely to be very low. For this reason, priming cultures were performed as previously but with a third identical stimulation step, and the frequency of MART-1-specific CD8+ T cells generated after 2 and 3 weeks was determined using a MART-1-specific pentamer. As shown in Figure 6, a small but significant expansion of MART-1 specific T cells was seen in reo-Mel-DC cultures, whereas no expansion of pentamer +ve CD8+ T cells was seen in Mel-DC primed cultures. This suggests that reovirus infection of human tumour cells is able to support the priming of an effective anti-tumour CTL response which includes CD8+ T cells directed against relevant TAA.
Figure 6. Cross priming of MART-1-specific CTLs by DC loaded with reovirus-infected Mel888 cells.
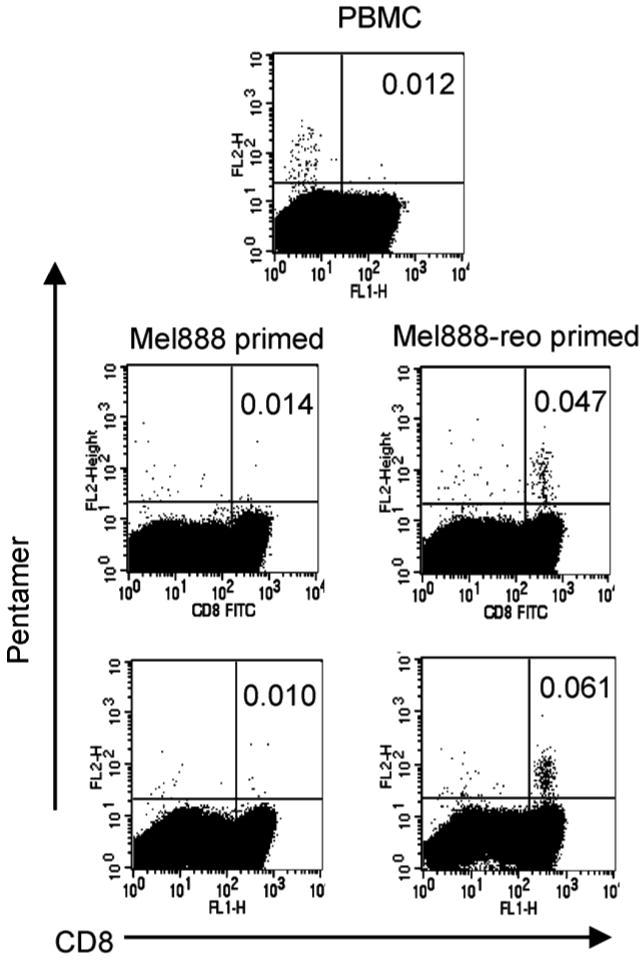
A2+ve PBMCs were incubated with autologous DC loaded overnight with uninfected Mel888 cells (an A2-ve cell line) or reovirus-infected Mel888 cells, as in Figure 5. A total of 3 stimulations were performed at 7 day intervals, with analysis on day 21. Initial PBMC and the lymphocyte populations generated after 2 and 3 weeks in culture were examined by flow cytometry after labelling with a MART-1-specific pentamer and a negative control (which labelled no detectable cells - not shown). Figures represent the percentage of CD8+ T cells labelled with the MART-1 pentamer. Representative of 5 independent experiments.
Discussion
The interplay between host anti-viral and anti-tumor immune responses is complex during oncolytic virotherapy. Anti-viral responses limit the intratumoral replication and spread of viruses, but also play an important role in reducing normal tissue toxicity by providing a barrier to normal tissue infection (39). In contrast, the development of anti-tumor immunity may enhance the efficacy of virotherapy (2-8). Virus-induced oncolysis is likely to release a wide range of tumor antigens from whole tumor cells, which may be taken up and cross presented by infiltrating DC, and virally infected cells can be more effective at delivering non-viral antigen for in vivo cross priming of APC than non-infected cells (40). DC are known to be important mediators of early viral recognition, via pattern recognition receptors (PRRs) such as toll-like receptors (TLRs), which respond to viral RNA and DNA (41).
Reovirus is a promising naturally occurring oncolytic virus, which has already entered phase I and II clinical trials. In the data reported here, we have for the first time explored the ability of reovirus infection of tumor cells to support generation of adaptive anti-tumor immunity. Melanoma was chosen as the disease target for this study, due to the susceptibility of melanoma to reovirus (33), and the potential immunogenicity of melanomas (42, 43). Initially, intravenous reovirus was administered using an established murine model of B16 melanoma lymph node metastasis (5). In this system, a single iv dose of reovirus reduced lymph node metastatic burden (Figure 1A and B), and generated a response against the self TAA TRP-2 (Figure 1C) (44). In addition, iv reovirus was associated with production of IL-12 (a cytokine with a key role in immune priming (45)), in tumor draining lymph nodes (Figure 2C).
Although murine models allow assessment of in vivo interactions between viruses and components of the immune system, they are limited in their application to human systems (46). For this reason, it was important to determine whether these findings also held true in a human in vitro model. Mel888 cells were chosen for testing, as they are inherently immunosuppressive (30), although potentially able to provide TAA for cross priming DC in an appropriate immunostimulatory context (47).
Experiments co-culturing tumor cells with DC confirmed that Mel888 cells alone are unable to induce DC phenotypic maturation or induce cytokine secretion. In contrast, reovirus infected Mel888 cells activated DC phenotypically and functionally (Figure 2A and B). The IL-12p70 and TNFα secreted may in particular promote priming in vivo, with IL-12p70 linking the innate and adaptive arms of the immune system, activating NK cells and directing the differentiation of Th1 helper T cells (45). These data demonstrate for the first time that reovirus-infected tumor cells activate DC. It is significant, in this context, that many viruses, including oncolytic viruses, conversely interfere with DC function (15, 16, 48, 49)
Transwell experiments showed that DC maturation induced by reo-Mel888 is dependent upon direct DC contact with reovirus-infected tumor cells, as opposed to a soluble factor (Figure 2A). Although previous reports with oncolytic viruses have demonstrated an enhancement of phagocytosis of tumor by DC following tumor infection (7, 8), we found no increased DC uptake of reovirus-infected tumor cells compared with non-infected cells (Figure 3A and B). Previous studies have shown that cross priming by cells infected with dsRNA viruses requires phagocytosis of infected material, and signalling via the dsRNA receptor, TLR-3 (40). Colabelling experiments (Figure 3C) demonstrated that tumor material taken up by DC co-localised with acidic late endosomal/ lysosomal compartments. Significantly, TLR-3 engagement has been shown to occur in an acidic environment, in such late endosomal/lysosomal compartments (50). Overall, this would be consistent with a role for TLR-3 receptor interactions in DC maturation in response to reovirus-infected Mel888 cells, and we are now further addressing these mechanisms in our laboratory.
By priming and restimulating autologous PBMC with DC loaded with reovirus-infected Mel888 cells, in the absence of other maturation factors, we have demonstrated lymphocyte proliferation (Figure 4A) and effective naïve CTL priming (Figure 4B) towards Mel888 cell targets, with minimal activity towards other melanoma and non-melanoma targets (Figure 4B and C). Importantly, in this allogeneic system, the response included cross priming of CTL specific for a melanoma TAA, MART-1 (Figure 6). Although the levels of MART-1-specific CTL generated were low, this is unsurprising in a system primed with whole tumor cells containing a host of antigens. Moreover, lymphocyte degranulation towards Mel888 cell targets was highly specific and MHC class I dependent (Figure 5C, D). Notably, in the absence of reovirus infection, Mel888 cells consistently failed to expand lymphocytes or prime an anti-tumor response (Figure 4A and B). High levels of the Th1 cytokine, IFN-γ, accumulated in reovirus-infected Mel888 cell primed CTL cultures (Figure 5), consistent with generation of a Th1 response. These data are consistent with the detection of the Th1 cytokine, IL-12, from lymphocytes disaggregated from tumor draining lymph nodes in the in vivo murine model following iv reovirus (Figure 1D). It is interesting to note that effective anti-tumor priming in the human in vitro system took place with a reovirus dose (0.1pfu/ Mel888 cell) which caused minimal DC maturation after 24 hour co-culture (Figure 2A and B). Furthermore, these DC did not show any further increase in maturation over 48h (data not shown). This reovirus dose was chosen in view of the detrimental effect of higher reovirus doses upon DC viability (Figure 2C), and also to maintain relevance to human therapy, in which several obstacles are likely to limit the dose which can be delivered to tumors (1).
In summary, we have shown that reovirus infection of tumor cells is immunogenic, activating DC, and providing an antigen source in an appropriate dangerous context to prime a naïve CTL response towards TAA, in both in vivo murine and in vitro human model systems. These findings provide a powerful rationale for the design of future clinical studies with reovirus, and other oncolytic viruses, to explore both their cytotoxic and immunotherapeutic activity. Combination therapy manipulating virus delivery, anti-viral and anti-tumor immune responses, provide an encouraging avenue for future pre-clinical and clinical development.
Acknowledgments
RJP, FE, EEM, AAM are supported by grants from Cancer Research UK
RV is supported by a US National Institutes of Health grant CA R01107032-02, Mayo Foundation, Richard M. Schulze Family Foundation
Footnotes
Statement of clinical relevance: Oncolytic viruses are able to selectively lyse cancer cells, and represent a promising novel approach to anti-cancer therapy. Despite widespread interest in their direct anti-cancer activity, only limited attention has been applied to the critical interaction between viral therapy and the immune system. Anti-viral immune responses can limit the efficacy of oncolytic virotherapy by viral clearance, whilst in contrast viral oncolysis may release tumor associated antigens in combination with ‘danger’ signals leading to the generation of anti-tumour immunity. Reovirus is a naturally occurring oncolytic virus, currently in phase I and II clinical trials. This study demonstrates that reovirus infection of tumor cells is immunogenic, activating dendritic cells, and providing a source of tumour associated antigens in an ‘dangerous’ context to prime adaptive anti-tumor immune responses. On this basis future clinical trials of oncolytic virotherapy should be designed to explore immunotherapeutic as well as direct cytotoxic efficacy.
References
- 1.Parato KA, Senger D, Forsyth PAJ, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005 doi: 10.1038/nrc1750. advanced online publication. [DOI] [PubMed] [Google Scholar]
- 2.Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther. 1999;10:385–93. doi: 10.1089/10430349950018832. [DOI] [PubMed] [Google Scholar]
- 3.Li H, Dutuor A, Fu X, Zhang X. Induction of strong antitumor immunity by an HSV-2-based oncolytic virus in a murine mammary tumor model. J Gene Med. 2007;9:161–9. doi: 10.1002/jgm.1005. [DOI] [PubMed] [Google Scholar]
- 4.Li H, Dutuor A, Tao L, Fu X, Zhang X. Virotherapy with a type 2 herpes simplex virus-derived oncolytic virus induces potent antitumor immunity against neuroblastoma. Clin Cancer Res. 2007;13:316–22. doi: 10.1158/1078-0432.CCR-06-1625. [DOI] [PubMed] [Google Scholar]
- 5.Qiao J, Kottke T, Willmon C, et al. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat Med. 2008;14:37–44. doi: 10.1038/nm1681. [DOI] [PubMed] [Google Scholar]
- 6.Diaz RM, Galivo F, Kottke T, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–8. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 7.Moehler MH, Zeidler M, Wilsberg V, et al. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum Gene Ther. 2005;16:996–1005. doi: 10.1089/hum.2005.16.996. [DOI] [PubMed] [Google Scholar]
- 8.Greiner S, Humrich JY, Thuman P, Sauter B, Schuler G, Jenne L. The highly attenuated vaccinia virus strain modified virus Ankara induces apoptosis in melanoma cells and allows bystander dendritic cells to generate a potent anti-tumoral immunity. Clin Exp Immunol. 2006;146:344–53. doi: 10.1111/j.1365-2249.2006.03177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prestwich RJ, Errington F, Hatfield P, et al. The Immune System - is it Relevant to Cancer Development, Progression and Treatment? Clin Oncol (R Coll Radiol) 2007 doi: 10.1016/j.clon.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 10.Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother. 2005;54:187–207. doi: 10.1007/s00262-004-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 12.Matzinger P. An innate sense of danger. Semin Immunol. 1998;10:399–415. doi: 10.1006/smim.1998.0143. [DOI] [PubMed] [Google Scholar]
- 13.Zeng J, Fournier P, Schirrmacher V. Induction of interferon-alpha and tumor necrosis factor-related apoptosis-inducing ligand in human blood mononuclear cells by hemagglutinin-neuraminidase but not F protein of Newcastle disease virus. Virology. 2002;297:19–30. doi: 10.1006/viro.2002.1413. [DOI] [PubMed] [Google Scholar]
- 14.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 15.Pollara G, Kwan A, Newton PJ, Handley ME, Chain BM, Katz DR. Dendritic cells in viral pathogenesis: protective or defective? International Journal of Experimental Pathology. 2005;86:187–204. doi: 10.1111/j.0959-9673.2005.00440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenne L, Schuler G, Steinkasserer A. Viral vectors for dendritic cell-based immunotherapy. Trends Immunol. 2001;22:102–7. doi: 10.1016/s1471-4906(00)01813-5. [DOI] [PubMed] [Google Scholar]
- 17.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 18.Norman KL, Lee PWK. Not all viruses are bad guys: the case for reovirus in cancer therapy. Drug Discovery Today. 2005;10:847. doi: 10.1016/S1359-6446(05)03483-5. [DOI] [PubMed] [Google Scholar]
- 19.Rosen L, Evans HE, Spickard A. Reovirus infections in human volunteers. Am J Hyg. 1963;77:29–37. doi: 10.1093/oxfordjournals.aje.a120293. [DOI] [PubMed] [Google Scholar]
- 20.Marcato P, Shmulevitz M, Pan D, Stoltz D, Lee PW. Ras Transformation Mediates Reovirus Oncolysis by Enhancing Virus Uncoating, Particle Infectivity, and Apoptosis-dependent Release. Mol Ther. 2007;15:1522–30. doi: 10.1038/sj.mt.6300179. [DOI] [PubMed] [Google Scholar]
- 21.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 22.Hirasawa K, Nishikawa SG, Norman KL, Alain T, Kossakowska A, Lee PW. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002;62:1696–701. [PubMed] [Google Scholar]
- 23.Alain T, Hirasawa K, Pon KJ, et al. Reovirus therapy of lymphoid malignancies. Blood. 2002;100:4146–53. doi: 10.1182/blood-2002-02-0503. [DOI] [PubMed] [Google Scholar]
- 24.Morris DF, Paterson A. A phase I clinical trial evaluating intralesional reolysin (reovirus) in histologically confirmed malignancies. Proc Am Soc Clin Oncol. 2002;24a:92. [Google Scholar]
- 25.White CL, Twigger KR, Vidal L, et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther. 2008 doi: 10.1038/gt.2008.21. [DOI] [PubMed] [Google Scholar]
- 26.Steele TA, Cox DC. Reovirus type 3 chemoimmunotherapy of murine lymphoma is abrogated by cyclosporine. Cancer Biother. 1995;10:307–15. doi: 10.1089/cbr.1995.10.307. [DOI] [PubMed] [Google Scholar]
- 27.Vile RG, Hart IR. Use of tissue-specific expression of the herpes simplex virus thymidine kinase gene to inhibit growth of established murine melanomas following direct intratumoral injection of DNA. Cancer Res. 1993;53:3860–4. [PubMed] [Google Scholar]
- 28.Romani N, Reider D, Heuer M, et al. Generation of mature dendritic cells from human blood. An improved method with special regard to clinical applicability. J Immunol Methods. 1996;196:137–51. doi: 10.1016/0022-1759(96)00078-6. [DOI] [PubMed] [Google Scholar]
- 29.Langford KJ, Askham JM, Lee T, Adams M, Morrison EE. Examination of actin and microtubule dependent APC localisations in living mammalian cells. BMC Cell Biol. 2006;7:3. doi: 10.1186/1471-2121-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Errington F, Jones J, Merrick A, et al. Fusogenic membrane glycoprotein-mediated tumour cell fusion activates human dendritic cells for enhanced IL-12 production and T-cell priming. Gene Ther. 2006;13:138–49. doi: 10.1038/sj.gt.3302609. [DOI] [PubMed] [Google Scholar]
- 31.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 32.Melief CJ. Mini-review: Regulation of cytotoxic T lymphocyte responses by dendritic cells: peaceful coexistence of cross-priming and direct priming? Eur J Immunol. 2003;33:2645–54. doi: 10.1002/eji.200324341. [DOI] [PubMed] [Google Scholar]
- 33.Errington F, White CL, Twigger KR, et al. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther. 2008 doi: 10.1038/gt.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Errington F, Steele L, Prestwich R, et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol. 2008;180:6018–26. doi: 10.4049/jimmunol.180.9.6018. [DOI] [PubMed] [Google Scholar]
- 35.Engelmayer J, Larsson M, Subklewe M, et al. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J Immunol. 1999;163:6762–8. [PubMed] [Google Scholar]
- 36.Mikloska Z, Bosnjak L, Cunningham AL. Immature monocyte-derived dendritic cells are productively infected with herpes simplex virus type 1. J Virol. 2001;75:5958–64. doi: 10.1128/JVI.75.13.5958-5964.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dani A, Chaudhry A, Mukherjee P, et al. The pathway for MHCII-mediated presentation of endogenous proteins involves peptide transport to the endo-lysosomal compartment. J Cell Sci. 2004;117:4219–30. doi: 10.1242/jcs.01288. [DOI] [PubMed] [Google Scholar]
- 38.Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6:476–83. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 39.Qiao J, Wang H, Kottke T, et al. Cyclophosphamide Facilitates Antitumor Efficacy against Subcutaneous Tumors following Intravenous Delivery of Reovirus. Clin Cancer Res. 2008;14:259–69. doi: 10.1158/1078-0432.CCR-07-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schulz O, Diebold SS, Chen M, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 41.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 42.Nathanson Spontaneous regression of malignant melanoma: a review of the literature on incidence, clinical features, and possible mechanisms. Natl Cancer Inst Monogr. 1976;44:67–76. [PubMed] [Google Scholar]
- 43.Ferradini L, Mackensen A, Genevee C, et al. Analysis of T cell receptor variability in tumor-infiltrating lymphocytes from a human regressive melanoma. Evidence for in situ T cell clonal expansion. J Clin Invest. 1993;91:1183–90. doi: 10.1172/JCI116278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engelhorn ME, Guevara-Patino JA, Noffz G, et al. Autoimmunity and tumor immunity induced by immune responses to mutations in self. Nat Med. 2006;12:198–206. doi: 10.1038/nm1363. [DOI] [PubMed] [Google Scholar]
- 45.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 46.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 47.Bateman AR, Harrington KJ, Kottke T, et al. Viral fusogenic membrane glycoproteins kill solid tumor cells by nonapoptotic mechanisms that promote cross presentation of tumor antigens by dendritic cells. Cancer Res. 2002;62:6566–78. [PubMed] [Google Scholar]
- 48.Bai L, Koopmann J, Fiola C, Fournier P, Schirrmacher V. Dendritic cells pulsed with viral oncolysates potently stimulate autologous T cells from cancer patients. Int J Oncol. 2002;21:685–94. doi: 10.3892/ijo.21.4.685. [DOI] [PubMed] [Google Scholar]
- 49.Jenne L, Hauser C, Arrighi JF, Saurat JH, Hugin AW. Poxvirus as a vector to transduce human dendritic cells for immunotherapy: abortive infection but reduced APC function. Gene Ther. 2000;7:1575–83. doi: 10.1038/sj.gt.3301287. [DOI] [PubMed] [Google Scholar]
- 50.de Bouteiller O, Merck E, Hasan UA, et al. Recognition of double-stranded RNA by human toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem. 2005;280:38133–45. doi: 10.1074/jbc.M507163200. [DOI] [PubMed] [Google Scholar]