

Abstract
Hexavalent chromium [Cr(VI)] is a mutagen and carcinogen, and occupational exposure can lead to lung cancers and other adverse health effects. Genetic changes resulting from DNA damage has been proposed as an important mechanism that mediates chromate's carcinogenicity. Here we show that chromate exposure of human lung A549 cells increased global levels of di- and tri-methylated histone H3 lysine 9 (H3K9) and lysine 4 (H3K4) but decreased the levels of tri-methylated histone H3 lysine 27 (H3K27) and di-methylated histone H3 arginine 2 (H3R2). Most interestingly, H3K9 dimethylation was enriched in the human MLH1 gene promoter following chromate exposure and this was correlated with decreased MLH1 mRNA expression. Chromate exposure increased the protein as well as mRNA levels of G9a a histone methyltransferase that specifically methylates H3K9. This Cr(VI)-induced increase in G9a may account for the global elevation of H3K9 dimethylation. Furthermore, supplementation with ascorbate, the primary reductant of Cr(VI) and also an essential cofactor for the histone demethylase activity, partially reversed the H3K9 dimethylation induced by chromate. Thus our studies suggest that Cr(VI) may target histone methyltransferases and demethylases, which in turn affect both global and gene promoter specific histone methylation, leading to the silencing of specific tumor suppressor genes such as MLH1.
Keywords: chromium, epigenetic, histone, methylation
Introduction
Cr(VI) is widely utilized in many industries(Levy and Venitt, 1986; Langard, 1990), leading to occupational exposure and contamination of numerous drinking water supplies. Human exposure to Cr(VI) can induce a variety of cytotoxic and genotoxic effects eventually leading to cancer (Dayan and Paine, 2001; Costa and Klein, 2006). For example, epidemiological and risk assessment studies revealed a high incidence of lung cancer following occupational inhalation exposure to Cr(VI) (Gibb et al., 2000). However, the molecular mechanisms of carcinogenicity of Cr(VI) is still an area of intense investigation.
As a potent oxidant, the ability of Cr(VI) to induce oxidative stress and the formation of stable Cr-DNA adducts are thought to mediate a majority of its cytotoxic and genotoxic effects (Zhitkovich, 2005). After entering cells via a sulfate anion transporter, Cr(VI) is subjected to a series of metabolic reductions to form reactive Cr(V) and Cr(IV) intermediates as well as the final stable metabolite Cr(III) (Stearns and Wetterhahn, 1994; Zhitkovich, 2005). These reactive intermediates and final products, in combination with reactive oxygen species (ROS) generated from the reduction process, are able to induce the formation of stable Cr-DNA ternary adducts, protein-DNA cross-links, DNA-DNA crosslinks, and DNA single or double-strand breaks (Shi et al., 2004). The Cr(VI)-induced DNA damage can in turn impact upon DNA replication, transcription and translation, resulting in altered gene expression (Sugden and Stearns, 2000). Thus for many years genetic defects mediated by DNA damage have been considered as the key mechanism underlying the genotoxicity and carcinogenicity of Cr(VI).
Recently several studies highlighted the potential epigenetic effects of Cr(VI). The first evidence came from a study in a cell line expressing the bacterial gpt reporter gene (G12) (Klein et al., 2002). Exposure to potassium dichromate was able to induce DNA methylation in the G12 cells and silenced the expression of the gpt transgene (Klein et al., 2002). Recently, treating the Brassica napus L. plants with potassium dichromate induced genome-wide cytosine-hypermethylation in the CCGG-sequence (Labra et al., 2004). Consistent with this finding DNA methylation was increased in the promoter region of the tumor suppressor gene p16 (Kondo et al., 2006) and DNA mismatch repair (hMLH1) (Takahashi et al., 2005) gene in chromate-induced human lung cancers, as compared to the lung cancer in humans without chromate exposure. Since DNA methylation recruits various methylated DNA binding proteins which can inhibit the binding of specific transcription factors to the promoter, it was proposed that Cr(VI) might silence p16 and hMLH1 tumor suppressor genes by inducing DNA methylation in their respective promoter regions. In a more recent study chromate was able to cross-link histone deacetylase 1-DNA methyltransferase 1 complex to chromatin and produce histone silencing marks that prevented aryl hydrocarbon receptor (AHR)-mediated gene transactivation (Schnekenburger et al., 2007). Taken together, these studies suggested that other than genotoxic effects, chromate can also alter epigenetic marks which may contribute to its carcinogenic activity.
As an important epigenetic marker, DNA methylation can be induced in cooperation with other epigenetic modifications(Esteller, 2007). Recent studies revealed that DNA methyltransferases (DNMTs) were associated with the enzymes that modify histone acetylation and methylation (Dobosy and Selker, 2001; Ting et al., 2006), and their ability to induce DNA methylation was associated with the status of specific histone tail modification (Jackson et al., 2002; Tamaru et al., 2003). At least eight histone modifications have been identified. Acetylation, methylation, phosphorylation, and ubiquintination were among the most commonly studied (Strahl and Allis, 2000; Peterson and Laniel, 2004). Unlike DNA methylation in the promoter region that exclusively represses gene transcription, with the exception of histone acetylation, other histone modifications exhibit site-specific but not modification-specific activating or silencing marks. For example, in the promoter region methylation of histone H3 lysine 9 (H3K9) was associated with gene silencing, but methylation of histone H3 lysine 4 (H3K4) was associated with gene activation (Peterson and Laniel, 2004). H3K9 methylation has also been found to be a critical mark for the establishment of DNA methylation and long-term gene silencing (Tamaru et al., 2003; Jackson et al., 2004). Genetic ablation of G9a, a methyltransferase that specifically methylates H3K9, induced genome-wide and locus-specific DNA hypomethylation suggesting a crucial role of H3K9 methylation in the establishment of DNA methylation (Xin et al., 2003). On the other hand methylation of H3K4 protected gene promoters from de novo methylation of DNA (Ooi et al., 2007). Therefore, various histone modifications have distinct impacts on DNA methylation.
While studies on chromate-induced lung cancer suggested that Cr(VI) might silence tumor suppressor genes by inducing DNA methylation in their promoters (Takahashi et al., 2005; Kondo et al., 2006), very little is known about whether Cr(VI) can affect histone marks. In the present study we analyzed both global and gene promoter alterations in histone marks using human bronchial epithelials and lung carcinoma cells exposed to chromate. Our results demonstrated that chromate treatment increased H3K9 dimethylation in the human MLH1 gene promoter, coincident with decreased MLH1 mRNA expression. Thus the capacity of chromate to modulate histone methylation and subsequently silence specific tumor suppressor genes may underlie its carcinogenicity.
Material and Methods
Reagents
Potassium chromate (K2CrO4) was obtained from J. T. Baker Chemical Co. (Phillipsburg, NJ). Antibodies against mono-, di-, tri-methylated H3K9, tri-methylated H3K27, di-methylated H3K4, and G9a were obtained from Upstate Biotechnology, Inc. (Lake Placid, NY). Antibodies against di-methylated H3R2, mono- and tri-methylated H3K4 were purchased from Abcam (Cambridge, UK).
Cell culture
Human lung carcinoma A549 cells were cultured in Ham's F-12K medium (Invitrogen, Carlsbad, CA). The medium was supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen). The cells were cultured at 37°C in an incubator with a humidified atmosphere containing 5% CO2. Human normal bronchial epithelial BEAS-2B cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS and 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen). For the ascorbate experiment, A549 cells were pretreated with 1 mM ascorbate for 2 hours. The cells were washed with PBS and then treated with different concentrations of chromate for 1 hour.
Whole cell lysates and Histone extraction
Whole cell lysates were extracted with ice-cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, PH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA) supplemented with protease inhibitor cocktail (Roche Applied Sciences). After lysis for 30 minutes on ice, lysates were clarified by centrifugation at 14,000 × g for 15 minutes at 4°C. Histones were extracted from cells as previously described (Chen et al., 2006). Briefly, cells were lysed with ice-cold RIPA buffer for 10 minutes, followed by centrifugation at 10,000 × g for 10 minutes. The pellets were washed once using a washing buffer [10 mM Tris-Cl, 37 mM EDTA (pH 7.4)], and histones were extracted with 0.4 N H2SO4 for 1.5 hours on ice. After centrifugation at 14,000 × g for 15 minutes, the histones were precipitated by ice-cold acetone overnight at −20°C and re-suspended in 4 M urea.
Western blot
The protein concentration was determined using the Bio-Rad DC protein assay (Bio-Rad, Hercules, CA). 50 μg total protein lysates or 5 μg histones were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad). Immunoblotting was performed using the indicated primary antibody followed by HRP-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The specific signals were detected by enhanced chemiluminescence using ECL reagent (Amersham Biosciences). Two or three independent experiments were performed for each data set. The intensity of signals was quantified using ImageJ software (version 1.37v; National Institutes of Health). The loading of histone in each lane were assessed by Bio-safe Coomassie stain (Bio-Rad).
RNA analysis
Total RNA was extracted from cells immediately after exposure by Trizol reagent (Invitrogen) following the manufacturer's protocol. For Northern blot analysis, 20 μg of total RNA was separated on formaldehyde-agarose gels, and transferred onto BrightStar-Plus positive charged nylon membranes (Ambion). PCR amplified G9a cDNA fragment was used as a probe. Probe labeling and subsequent hybridization were performed as described previously (Yan et al., 2003). For RT-PCR analysis, the reaction was performed using SuperScript III (Invitrogen). Semi-quantitative PCR was performed to amplify human MLH1 and β-actin using the following primer pairs: MLH1 :5′-CAGGTATTCAGTACACAATGCAGGC-3′(forward), 5′-CTACCAGACGATGGTTGATGAAGAG-3′ (reverse); β-actin: 5′-TCACCCACACTGTGCCCATCTACGA-3′ (forward), 5′-CAGCGGAACCGCTCATTGCCAATGG-3′ (reverse). PCR products were visualized by Ethidium bromide on 1% agarose gel.
Immunofluorescence staining
The cells were seeded in chamber slides (BD Falcon™, Bedford, MA), and exposed to the various chemicals. At selected time intervals, cells were fixed with 4% paraformaldehyde for 10 minutes, and permeabilized with 0.2% Triton X-100 for 5 minutes. Cells were then quenched with fresh 0.1% sodium borohydride for 5 minutes and blocked in a buffer containing 10% Goat serum, 1% BSA dissolved in PBS. The cells were incubated with antibodies against mono-, di- and tri-methylated H3K9 (1:250) overnight at 4°C, followed by incubation with Alexa Fluor 488 conjugated secondary antibody (Molecular Probes) for 1 hour at room temperature. The slides were mounted with ProLong Gold Anti-fade Reagent with DAPI (Molecular Probes). The signals were visualized and captured using a fluorescent microscopy (Model AX 70; Olympus, Melville, NY).
For dual-color immunofluorescence, fixed cells were stained with monoclonal antibody against H3K9me2 (1:1000, Abcam) and polyclonal antibody against H3K4me3 (1:5000, Abcam) sequentially, and followed with Alexa Fluor 594 conjugated anti-mouse secondary antibody and Alexa Fluor 488 conjugated anti-rabbit secondary antibody (Molecular Probe). The signals were visualized and scanned using Leica TSC SP5 confocal microscope.
In vitro histone demethylase reaction
The purification of recombinant Flag-JHDM2A from insect Sf9 cells is described elsewhere (29). Two micrograms of purified Flag-JHDM2A was incubated with 5 μg of core histones (Roche) in a buffer containing 50 mM Hepes-KOH pH 8.0, 100 μM FeCl3, 1 mM 2-oxoglutarate, 1 mM PMSF and varying concentrations of ascorbate. After 30 minutes incubation at 37°C the reaction was terminated by adding EDTA to a final concentration of 1 mM. The histones were then separated in 15% SDS-PAGE gels and transferred to PVDF membranes. After transferring the SDS-PAGE gels were stained with Biosafe Coomassie (Bio-Rad) to assess the amount of Flag-JHDM2A added into each reaction. The PVDF membranes were blotted with antibody against H3K9me2 (Upstate) to assess the enzymatic demethylation of H3K9me2 in core histones. The same membrane was then blotted with antibody against histone H3 (Sigma) to measure the loading of histones.
Chromatin immunoprecipitation (ChIP) assay
A549 cells were treated with chromate for 1 hour or 24 hours, and then crosslinked using formaldehyde. The ChIP assay was performed using EZ ChIP Kit (Upstate) according to the manufacturer's protocol. Antibodies against H3K9me2, H3K4me3 (Abcam) and normal IgG were used for immunoprecipitation. The primer pairs 5′-ACCGCTCGTAGTATTCGTGCTC-3′ and 5′-GTGGATGACGCCCAAAAGAAG-3′ were used for amplification of MLH1 promoter by PCR. Optimal PCR conditions for semi-quantitative measurement were 2 cycles of 94°C-30s, 62°C-30s, 72°-45s; and then followed by cycles with the same denature and extension conditions, but decreased annealing temperature: 60°C (3 cycles), 58°C (4 cycles), 52°C (22 cycles). PCR products were separated on 2% agarose gel and visualized by Ethidium bromide staining.
Statistical Analysis
For analysis of statistical significance, the Student's t-test was used for all statistical calculations, and P-values of <0.05 were considered to be statistically significant.
Results
Chromate modulates global H3K9 methylation
A number of studies have demonstrated that both H3K9 methylation and DNA methylation were present within the same promoter region of silenced genes suggesting that methylation of H3K9 was associated with DNA methylation and both marks acted to sequentially suppress gene expression. We assessed changes in the global levels of H3K9 methylation induced by Cr(VI).
Human lung carcinoma A549 cells were incubated with 5 or 10 μM of potassium chromate. Cr(VI) can be very rapidly reduced to Cr(III) after it enters the cell leaving less than 5% of the Cr(VI) in the cells within the first hour of exposure (Wei et al., 2004). After 1 hour of exposure to Cr(VI) A549 cells were harvested and cellular histones were isolated. The levels of H3K9 methylation was measured by Western blot using antibodies directed against mono-, di- and tri-methylated H3K9. As shown in Fig 1, chromate exposure significantly increased global levels of di-methylated H3K9 (Fig 1A, middle panel). The increase in tri-methylated H3K9 was less evident (Fig 1A, right panel) while mono-methylated H3K9 did not change (Fig 1A, left panel). Similar results were also observed by immunofluorescence staining (Fig 1B). These results indicated that exposure of A549 cells to Cr(VI) altered global H3K9 methylation.
Figure 1. Modulation of H3K9 methylation by Cr(VI).
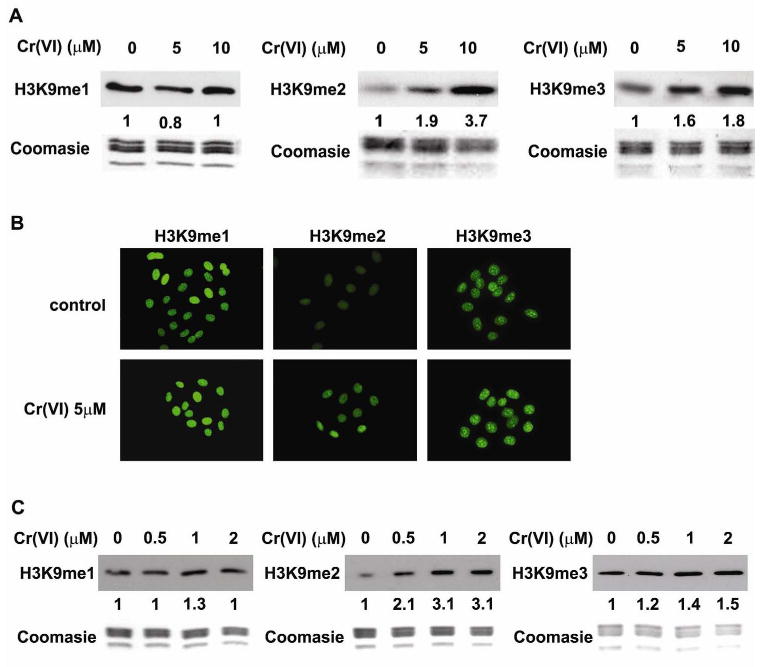
A549 cells were exposed to 5 or 10 μM of Cr(VI) for 1 hour and then the mono- (H3K9me1), di- (H3K9me2), and tri-(H3K9me3) methylation of H3K9 were studied by Western Blot (A) or immunofluorescent staining (B). (C) Beas-2B cells were exposed to 0.5, 1, and 2 μM of Cr(VI) for 1 hour, and the levels of H3K9 methylation were measured by Western Blot. The relative intensity of the bands was measured and is shown below the western blots in the figure. Coomassie blue staining was used to assess the equal loading of histones in Western blot analysis.
To investigate whether Cr(VI) modulates global H3K9 methylation in other cell types normal human bronchial epithelial (BEAS-2B) cells were also treated with various concentrations of Cr(VI). Compared to A549 cells BEAS-2B cells were more sensitive to Cr(VI) exposure and exhibited extensive cell death when treated with 10μM of Cr(VI). However at low doses of Cr(VI) the global level of H3K9 methylation di- and tri-methylation in BEAS-2B cells (Fig 1C) increased in a pattern similar to those observed in A549 cells.
Chromate alters the global level of methylation of H3K4, H3K27 and H3R2
Other than H3K9 at least 4 other lysine and 4 arginine residues in histone H3 tails can be methylated and these modifications play important roles in gene expression and chromatin remodeling.
The enrichment of methylated H3K4 in the nucleosome tail is normally associated with actively transcribed genes. While H3K4me3 was mostly located in the promoter region, H3K4me1 was enriched in the enhancer region and H3K4me2 can be enriched in both regions of actively transcribed genes (Heintzman et al., 2007). A549 cells were treated with 5 or 10 μM of potassium chromate for 1 hour and isolated histones were extracted and subjected to Western blot analysis with antibodies directed against mono-, di-, and tri-methylated H3K4. Interestingly while the global levels of mono-methylated H3K4 remained unchanged (Fig 2A), both di- and tri-methylated H3K4 were increased by 10 μM Cr(VI) (Fig 2B & 2C).
Figure 2. Modulation of H3K4, H3K27 and H3R2 methylation by chromate.
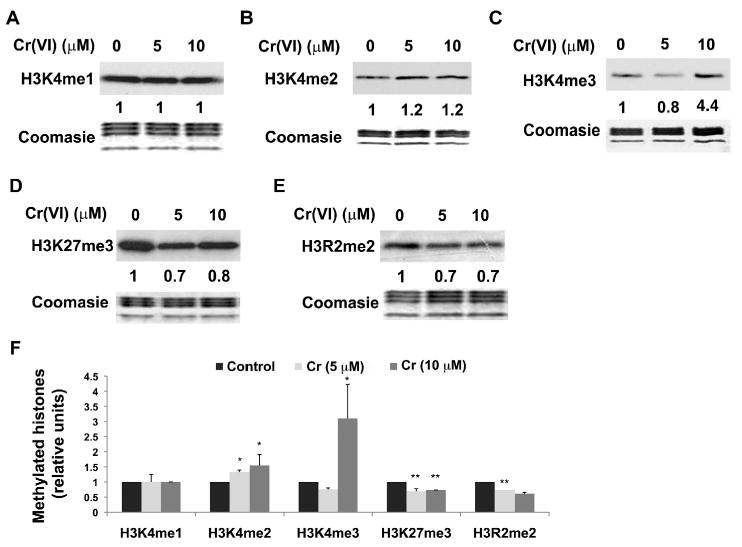
A549 cells were exposed to 5 or 10 μM Cr(VI) for 1 hour. The global levels of mono-(H3K4me1, A), di- (H3K4me2, B), tri- (H3K4me3, C) methylated H3K4, tri-methylated H3K27 (H3K27me3, D) and di-methylated H3R2 (H3R2me2, E) were measured using specific antibodies. The relative intensity of the bands was measured and the numbers were shown below the corresponding bands. The results from replicate experiments were plotted as the mean ± SE (error bars, n=3). Coomassie blue staining was used to assess the equal loading of the histones. Statistical significance of difference between control and chromium-treated cells was analyzed using Student's t-test. * P < 0.05, ** P < 0.01.
We also measured the level of methylation of H3K27 and H3R2 following Cr(VI) exposure. Methylation at these two sites has been found to be associated with gene repression. Cr(VI) exposure decreased the levels of both tri-methylated H3K27 (Fig 2D) and di-methylated H3R2 (Fig 2E). Thus our study demonstrated that Cr(VI) exposure was able to change global histone methylation in several important residues including H3K9, H3K4, H3K27 and H3R2.
Distinct localization of H3K9me2 and H3K4me3
It was of interest that Cr(VI) can increase global H3K9me2 which is associated with gene repression and also increase H3K4me3, which is associated with gene activation. To investigate the location of these epigenetic marks in chromatin we performed dual-immunofluorescent staining for both H3K9me2 and H3K4me3 and then analyzed the staining by confocal microscopy (Fig 3). Interestingly, while both H3K9me2 (red) and H3K4me3 (green) increased after 24 hours of Cr(VI) exposure, the enrichment patterns of each mark were quite different: H3K9me2 was primarily enriched at the periphery of the nucleus whereas H3K4me3 was exclusively enriched in the center of the nucleus. Thus the results revealed distinct localization of chromate induced H3K9me2 and H3K4me3 marks in chromatin.
Figure 3. Distinct localization of H3K9me2 and H3K4me3 in Cr(VI)-exposed cells.
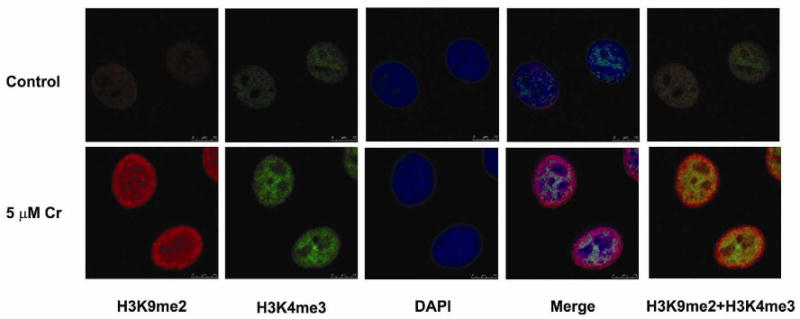
H3K9me2 (red) was primarily enriched at the periphery of the nucleus, and tri-methylated H3K4 (green) was exclusively enriched in the center of nucleus. A549 cells were exposed to 5 μM chromate for 24 hours, and cells were stained with antibodies against H3K9me2 and H3K4me3. The nucleus was counterstained by DAPI (blue).
Chromate induces H3K9 methylation in the MLH1 promoter and represses its expression
It was previously reported that MLH1 gene expression was down-regulated by increased DNA methylation in the promoter region in chromate exposed workers that developed lung cancers (Takahashi et al., 2005). Since H3K9me2 in the promoter region has been associated with DNA methylation and gene silencing, we used a chromatin immunoprecipitation (ChIP) assay to study whether chromate increased the level of H3K9me2 in the MLH1 promoter. As shown in Fig 4A, treatment with chromate for 1 hour increased the amount of H3K9me2 but not H3K4me3 at the MLH1 promoter. The increased H3K9me2 in the MLH1 promoter region was more evident when cells were treated with chromate for a longer time interval (24 hours) (Fig 4A). In contrast the gene activating mark H3K4me3 was slightly decreased in the MLH1 promoter after 24 hours of chromate. The enrichment in the gene silencing mark (H3K9me2) at the MLH1 gene promoter led us to investigate the level of MLH1 expression. A549 cells were exposed to chromate for 1 or 2 days and then MLH1 gene expression was analyzed by semi-quantitative RT-PCR. Consistent with the increased silencing mark (H3K9me2) and the decreased activation mark (H3K4me3) in the promoter region MLH1 mRNA levels were decreased in chromate exposed cells (Fig 4B) in a time and dose dependent manner. Similar results were seen in BEAS-2B cells (Fig 4C). Thus chromate exposure induced H3K9me2 silencing mark in MLH1 gene promoter and repressed its expression.
Figure 4. Chromate induced enrichment of H3K9 dimethylation in MLH1 promoter and repressed MLH1 mRNA expression.
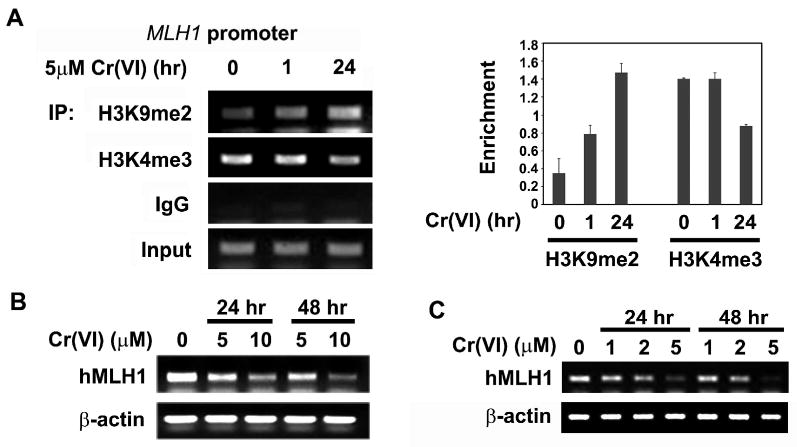
(A) Occupancy of H3K9 dimethylation at human MLH1 promoter. A549 cells were exposed to 5 μM Cr(VI) for 1hour or 24 hours and then chromatin immunoprecipitation analysis of MLH1 promoter was performed using antibodies against H3K9me2 and H3K4me3. Normal mouse IgG was used as negative control. The ChIP results were quantitated and plotted as the mean ratio of precipitated DNA to input DNA ± SE (error bars, n=3). (B) Cr(VI) exposure suppressed MLH1 mRNA levels in A549 cells. A549 cells were exposed to 5 μM or 10 μM Cr(VI) for 24 or 48 hours, MLH1 mRNA level was measured by semi-quantitative RT-PCR. β-actin was used to assess the equal amount of RNA samples. (C) Cr(VI) exposure suppressed MLH1 mRNA levels in BBEAS-2B cells. BEAS-2B cells were exposed to 1, 2, and 5 μM Cr(VI) for 24 or 48 hours, MLH1 mRNA level was measured by semi-quantitative RT-PCR. β-actin was used to assess the equal amount of RNA samples.
Effects of Chromate on G9a level in A549 cells
We next investigated whether Cr(VI) exposure could increase the global levels of dimethylated H3K9 by affecting the respective known methytransferases or demethylases that modify this mark. Among several well-defined histone methyltransferases G9a mediates most of the cellular H3K9 di-methylation (Tachibana et al., 2002) while JHDM2A respesented a major histone demethylase responsible for demethylation of H3K9me2 (Yamane et al., 2006). Since Cr(VI) treatment significantly increased global H3K9me2 levels, we examined whether any of these histone modifying enzymes were altered by chromate treatment. Following exposure of A549 cells to 5 or 10 μM of chromate the protein levels of G9a increased while it decreased with nickel exposure as previously reported (Fig 5A) (Chen et al., 2006). Consistent with increased G9a protein the levels of H3K9me2 were also increased at 24 hours after chromate exposure (Fig 5B). This result indicated that chromate treatment was associated with an increased level of histone methyltransferase which may be responsible for the elevation in global H3K9me2 levels. The levels of JHDM2A protein remained unchanged following chromate exposure (data not shown).
Figure 5. Cr(VI) modulated the levels of histone methyltransferase G9a.
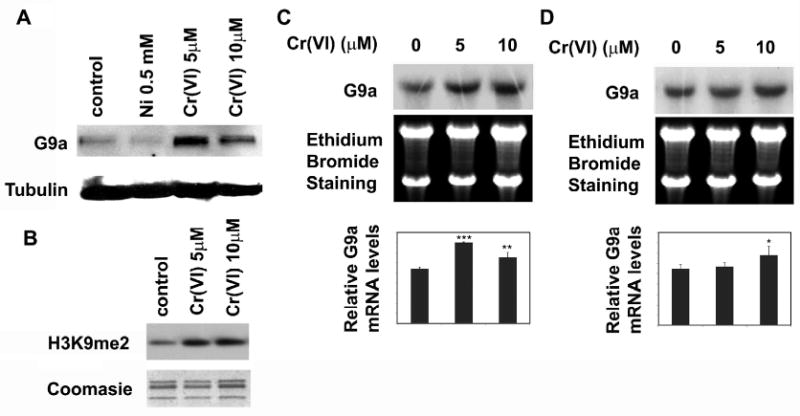
(A) Cr(VI) exposure increased G9a protein levels. A549 cells were treated with 5 or 10 μM of Cr(VI) for 24 hours. Total protein lysates were extracted and analyzed with antibody against G9a. Antibody against tubulin was used to assess the loading of proteins. (B) A549 cells were exposed to 5 or 10 μM Cr(VI) for 24 hours. The global levels of H3K9me2 were measured using specific antibodies. Coomassie blue staining was used to assess the equal loading of the histones. (C and D) Chromate exposure increased G9a mRNA levels after 24 hr exposure. A549 cells were treated with 5 or 10 μM Cr(VI) for 1 (D) or 24 (C) hours. G9a mRNA levels were analyzed by Northern blotting. The ethidium bromide staining of 28S and 18S RNA was performed to assess the loading of RNA samples. The relative intensity of the bands was measured and plotted as the mean ratio of G9a mRNA to 18S RNA ± SE (error bars). * P < 0.05, ** P < 0.01, *** P < 0.001.
Total RNA was extracted from chromate treated cells and then G9a mRNA was measured using Northern blotting to address whether the increased G9a protein level was due to the transcriptional regulation of the G9a gene by chromate. As shown in Fig 5C, G9a mRNA levels increased after 24 hours of Cr(VI) treatment. Interestingly, although H3K9me2 increased within 1 hr of chromate exposure (Fig 1), the G9a RNA level at that time interval did not exhibit any significant change (Fig 5D). These results suggested that in addition to the increased G9a level other factors may also be involved in the Cr-induced H3K9me2.
Effect of ascorbate on chromate-induced H3K9me2 in A549 cells
Intermediates in the reduction of Cr(VI) and ROS produced during its reduction have been considered to play a role in the cytotoxic and genotoxic effects of Cr(VI). Ascorbate and glutathione were the principal cellular reductants of Cr(VI). Originally ascorbate was considered as an anti-oxidant that protected cells from the cytotoxicity and genotoxicity of Cr(VI). However, recent studies suggested that ascorbate acted as a pro-oxidant in Cr(VI)-treated cells and dramatically enhanced Cr(VI) induced DNA double-strand breakage and mutagenesis (Martin et al., 2006; Reynolds et al., 2007; Reynolds and Zhitkovich, 2007). Previous studies by Mattagajasingh et al. demonstrated that Cr(VI) exposure decreased intracellular ascorbate levels in a dose-dependent manner (Mattagajasingh and Misra, 1995). We reasoned that a reduction of intracellular ascorbate by Cr(VI) would adversely affect the activity of the Jmjc-domain-containing histone demethylases which use Fe(II), 2-oxoglutarate and oxygen as co-factors to catalyze an oxidative demethylation reaction on histone lysines. JHDM2A is believed to require reduced ascorbic acid to catalyze the reduction of oxidized Fe formed during the reaction (Schneider and Shilatifard, 2006; Tian and Fang, 2007). An in vitro demethylation assay was employed to examine the ascorbate requirement for JHDM2A activity. Purified FLAG-tagged JHDM2A was incubated with decreasing concentrations of ascorbate and its activity was then measured using an in vitro histone demethylation assay with dimethylated H3K9 as the substrate. As shown in Fig 6a, JHDM2A demethylated H3K9me2 in an ascorbate-dependent manner and its activity was inhibited when the molar ratio of ascorbate to Fe (100 μM) fell below 1.
Figure 6. The role of ascorbate in Cr(VI) induced H3K9 dimethylation.
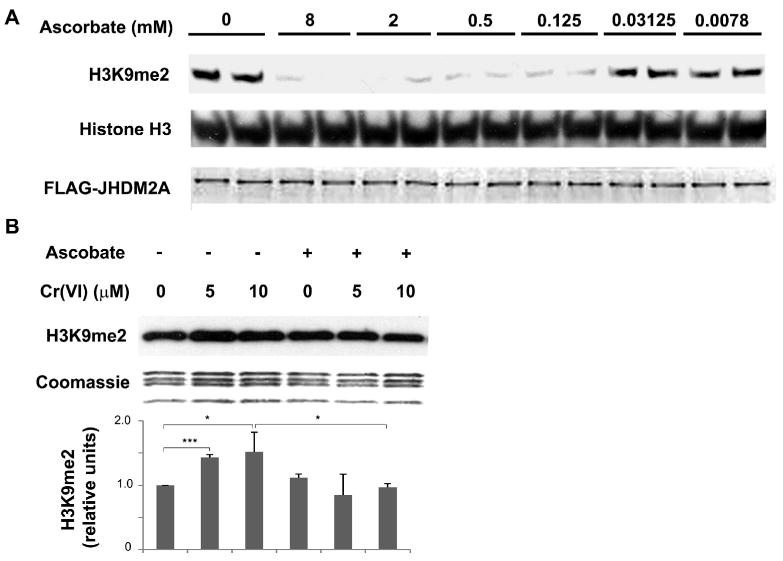
(A) JHDM2A demethylated H3K9me2 in an ascorbate-dependent manner. Purified recombinant Flag-JHDM2A was incubated with core histones in a buffer containing 100 μM Fe3+, 1 mM 2-oxoglutarate, and increased concentrations of ascorbate as indicated. After reaction, the loss of H3K9me2 in histones was assessed using Western blot. The same membrane was blotted with anti-histone H3 antibody to assess the loading of histones. The bottom figure shows the amount of recombinant Flag-JHDM2A added into each reaction. (B) A549 cells were pretreated with 1 mM of ascorbate for 2 hr. After washing with PBS, the cells were exposed to various concentration of chromate for 1 hr. The levels of di-methylated H3K9 were analyzed using Western blot. Coomassie blue staining was used to assess the loading of the histones. The relative intensity of the bands was measured and plotted as means ± SE (error bars). * P < 0.05, ** P < 0.01, *** P < 0.001.
We next tested whether Cr(VI) induced H3K9 di-methylation could be reversed by increasing intracellular ascorbate level. The intracellular level of ascorbate was modulated by pretreating cells with 1mM of ascorbate for 2 hours prior to Cr(VI) treatment. While ascorbate alone did not change H3K9me2 levels, ascorbate pretreatment completely blocked the Cr(VI)-induced H3K9 dimethylation (Fig 6b). This result further supports the notion that the effect of Cr(VI) on H3K9 methylation involved a loss of reduced ascorbic acid in the cell which was required for JHDM2A activity.
Discussion
It is known that environmental factors have a significant impact upon the epigenetic program of gene expression. Previous studies in our laboratory demonstrated that carcinogenic nickel compounds induced a broad range of epigenetic changes which likely mediate its effects on gene silencing and cell transformation (Zhang et al., 2003; Costa et al., 2005; Chen et al., 2006; Ke et al., 2006). In the current study we report that Cr(VI), another important mutagen and carcinogen also affected various histone methylation profiles. Cr(VI) exposure of A549 cells increased the global levels of H3K9me2, H3K9me3 and H3K4me3, but decreased those of H3K27me3 and H3R2me2. The enrichment of methylated H3K4 is normally associated with actively transcribed genes while methylated H3K9, H3K27me3 and H3R2me2 are repressive marks. The changes on both active and repressive histone marks suggested that the gene transcription process can be differently modulated in response to Cr(VI) exposure and this is supported by several other studies demonstrating diverse changes in gene expression profiles following Cr(VI) exposure (Ye and Shi, 2001; Izzotti et al., 2002; Gavin et al., 2007).
Several lines of evidence suggested that Cr(VI) was able to alter epigenetic homeostasis and these changes may play a role in promoting carcinogenesis. First, Cr(VI) was able to induce genome-wide or gene-specific DNA methylation changes in cultured mammalian cells (Klein et al., 2002) and plants (Labra et al., 2004). DNA methylation induced by Cr(VI) very likely accounted for the silencing of gpt transgene by chromate in G12 cells (Klein et al., 2002). Second, studies on human lung cancers in workers exposed to chromate revealed an increased DNA methylation level in the promoter region of two tumor suppressor genes p16 and MLH1 which likely accounted for the decreased expression of their mRNA (Takahashi et al., 2005; Kondo et al., 2006). Third, the studies reported herein indicated that Cr(VI) was able to significantly increase the level of H3K9me2 which was known to be crucial for DNA methylation (Tamaru et al., 2003; Jackson et al., 2004) (Kondo et al., 2003). Finally, Cr(VI) exposure increased H3K9me2 occupancy at the promoter region of human MLH1 gene which correlated with decreased MLH1 gene expression in A549 cells.
The human MLH1 gene is often epigenetically silenced by aberrant DNA hypermethylation within its promoter region in many different cancers (Imai and Yamamoto, 2008). Recently, other factors including histone modification (McGarvey et al., 2006; Xiong et al., 2006; Meng et al., 2007) and nucleosome occupancy (Lin et al., 2007) at the promoter region have been implicated in the regulation of MLH1 gene expression. Enrichment of the repressive histone marks, H3K9me1, H3K9me2, H3K9me3, H3K27me2, and H3K27me3 were found at the hypermethylated MLH1 promoter in MLH1 silenced cells compared with the unmethylated active promoter in MLH1 expressing cells (McGarvey et al., 2006). Treatment of MLH1 silenced cells with 5-aza-2′-deoxycytidine (5-Aza-dC), a DNA methyltransferase inhibitor, not only reactivated MLH1 expression in those cells but also enriched the active histone marks H3K9Ac and H3K4me2 in its promoter (McGarvey et al., 2006; Xiong et al., 2006). Interestingly, the occupancy of H3K9me2 in the promoter was strikingly reduced after 5-Aza-dC treatment while the other repressive marks H3K9me3, H3K27me2 and H3K27me3 remained unchanged (McGarvey et al., 2006). These studies indicated that both DNA methylation and histone modification are actively involved in epigenetic silencing of MLH1 and H3K9me2 is an important mark dynamically associated with DNA methylation and gene expression. Here we analyzed the changes of H3K9me2 and H3K4me3 occupancy at the MLH1 promoter during chromate-induced silencing of the active MLH1 gene which differs from studies that re-activated silenced MLH1 gene in cancer cells. Our findings demonstrate that the enrichment of H3K9me2 at MLH1 promoter is highly correlated with MLH1 gene inactivation supporting the previous reports that H3K9me2 is important for both initiation and maintenance of gene silencing (McGarvey et al., 2006; Xiong et al., 2006). We do not know whether acute exposure of chromate is able to induce DNA hypermethylation in MLH1 promoter. Further analysis of the DNA methylation status following chromate exposure will provide more insights into its epigenetic regulation and help us understand correlations among different histone marks, DNA methylation and gene silencing.
The mechanism by which Cr(VI) modulates histone methylation is not presently fully understood. Our studies suggested that multiple processes may be targeted by Cr(VI). First, increasing cellular levels of ascorbate partially reversed Cr(VI)-induced H3K9 dimethylation suggesting the epigenetic effect of chromate was in part dependent on the ascorbate levels in cells. Once inside the cell Cr(VI) is reduced mainly by ascorbate to Cr(III) and this process depletes cellular ascorbic acid reserves that are needed for dioxygenase enzymes such as JHDM2A a H3K9 demethylase. Ascorbate catalyzes the production of DNA-ascorbate-Cr adducts. Thus depending on the cellular context, ascorbate can either act as an anti-oxidant to protect cells from the cytotoxicity and genotoxicity of Cr(VI) or as a pro-oxidant promoting Cr(VI) induced DNA double-strand breakage and mutagenesis (Martin et al., 2006; Reynolds et al., 2007; Reynolds and Zhitkovich, 2007). Our data indicated that excess ascorbate could inhibit the excess global H3K9me2 induced by Cr(VI) which in turn supported its protective role against Cr-induced epigenetic changes. Moreover using an in vitro demethylase activity assay, we demonstrated the crucial role of ascorbate concentration in JHDM2A histone demethylase activity. It is possible that Cr(VI) decreased ascorbate levels sufficiently to inhibit JHDM2A demethylase activity leading to elevated global H3K9me2. This could explain our observation that H3K9 methylation was increased within one hour. Both protein and mRNA levels of G9a, an enzyme responsible for H3K9 dimethylation, were increased by Cr(VI). G9a is known to associate with DNA methyltransferase and promote DNA methylation (Esteve et al., 2006). Thus increased G9a may have two functions: maintaining high level of H3K9me2 and recruiting DNA methyltransferase to specific promoters. Following Cr(VI) exposure both an early event such as reduced ascorbic acid levels as well as a later event involving G9a up-regulation cooperated to inactivate tumor suppressor genes, such as MLH1. This is supported by the existence of DNA hypermethylation in certain gene promoters in lung cancers that arise in chromate workers. In addition to negative regulation of target gene transcription, recent study reported the enrichment of H3K9me2, 3 and other silencing marks in the site of DSB, suggesting its role in DNA repair (O'Hagan et al., 2008). Cr(VI) exposure increased both DSB and H3K9 methylation. It is not clear whether the two changes correlate each other in Cr(VI) exposed cells. One possible function of increased H3K9 methylation may be involved in silencing the damaged genes to prevent the initiation of transcription before the completion of DNA repair.
DNA double strand break (DSB), a major secondary lesion generated by mismatch repair system, is an early event of Cr(VI) exposure and responsible for Cr(VI) induced cell apoptosis (Ha et al., 2004; Xie et al., 2005; Reynolds et al., 2007; Reynolds and Zhitkovich, 2007). MLH1 is required for DSB formation as MLH1-deficient cells have less DSB and are more resistant to cytotoxicity of Cr(VI) (Peterson-Roth et al., 2005). Our observation of increased H3K9me2 levels in the MLH1 promoter region and consequently down-regulation of MLH1 expression, suggested that Cr(VI) exposure might trigger a protective response in cells through the epigenetic mechanism. Though the epigenetic marks changed rapidly (within 1 h of Cr exposure), the actual changes on mRNA and protein levels started rather late (24-48 h) and may not be able to affect the early DSB formation, but rather have a late impact on cell survival and malignant transformation. In addition, the presence of ascorbic acid can partially reverse Cr(VI)-induced H3K9me2, which may rescue MLH1 levels in exposed cells and subsequently enhance DSB formation and cell apoptosis. This is supported by the finding that ascorbic acid induces the MLH1 mRNA level in non-tumorigenic human HaCat cells (Catani et al., 2002), as well as the recent report that cellular vitamin C can only increase Cr(VI) toxicity in MLH1 proficient cells but not MLH1 deficient cells (Reynolds and Zhitkovich, 2007). Previous works from our group have shown the ability of nickel (Chen et al., 2006) and arsenite (Zhou et al., 2008) to change methylation of histone. Although the three chemicals can induce similar effects on the overall level of H3K9me2, the mechanisms mediating their effects are likely different. First, chromate-induced changes of histone modification occurred very early, about 1 hour after exposure, whereas nickel and arsenite require 24 hours to change histone marks. Second, nickel exposure down-regulated G9a mRNA and protein levels; thus the increase of H3K9me2 by nickel was largely through the inhibition of H3K9 demethylation (Chen et al., 2006). In contrast arsenite up-regulated G9a mRNA and protein which may account for arsenite induced H3K9 dimethylation (Zhou et al., 2008). Our data indicated that chromate probably affect both histone methyltransferase and demethylase. The inhibition of chromate-induced H3K9 dimethylation by ascorbate suggested demethylase activity is likely the target of chromate. Third, chromate likely inactivated the demethylase through depleting reduced ascorbate in cells, since pretreatment of ascorbate reversed this inhibition. Nickel is able to directly bind and replace the Fe(II) from H3K9me2 demethylase JHDM2A thus inactivating H2K3 demethylation (Chen, unpublished data).
In summary, this is the first report indicating that exposure of cultured lung cancer cells and non-tumorigenic bronchial epithelial cells to Cr(VI) was able to induce both global and gene specific changes in histone methylation. These changes affected marks that activate and repress gene expression, leading to DNA hypermethylation in tumor suppressor genes and DNA hypomethylation in other genes that may promote tumorigenesis as well as tumor progression. Thus, further analysis using primary lung epithelial cells will provide more insights into the epigenetic mechanism underlying tumorigenicity of Cr(VI). A more detailed analysis of genome-wide and gene-specific changes in these active and repressive marks will significantly add to our understanding of chromate cytotoxicity, genotoxicity, and carcinogenicity.
Acknowledgments
We would like to thank T. Kluz for the technical assistance and Dr. T.P. Ellen for his comments on the manuscript. This work was supported by grant numbers ES000260, ES014454, ES005512, from the National Institutes of Environmental Health Sciences, and grant number CA16087 from the National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Catani MV, Costanzo A, Savini I, Levrero M, de Laurenzi V, Wang JY, Melino G, Avigliano L. Ascorbate up-regulates MLH1 (Mut L homologue-1) and p73: implications for the cellular response to DNA damage. Biochem J. 2002;364:441–447. doi: 10.1042/BJ20011713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ke Q, Kluz T, Yan Y, Costa M. Nickel ions increase histone H3 lysine 9 dimethylation and induce transgene silencing. Mol Cell Biol. 2006;26:3728–3737. doi: 10.1128/MCB.26.10.3728-3737.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Davidson TL, Chen H, Ke Q, Zhang P, Yan Y, Huang C, Kluz T. Nickel carcinogenesis: epigenetics and hypoxia signaling. Mutat Res. 2005;592:79–88. doi: 10.1016/j.mrfmmm.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Costa M, Klein CB. Toxicity and carcinogenicity of chromium compounds in humans. Crit Rev Toxicol. 2006;36:155–163. doi: 10.1080/10408440500534032. [DOI] [PubMed] [Google Scholar]
- Dayan AD, Paine AJ. Mechanisms of chromium toxicity, carcinogenicity and allergenicity: review of the literature from 1985 to 2000. Hum Exp Toxicol. 2001;20:439–451. doi: 10.1191/096032701682693062. [DOI] [PubMed] [Google Scholar]
- Dobosy JR, Selker EU. Emerging connections between DNA methylation and histone acetylation. Cell Mol Life Sci. 2001;58:721–727. doi: 10.1007/PL00000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20:3089–3103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin IM, Gillis B, Arbieva Z, Prabhakar BS. Identification of human cell responses to hexavalent chromium. Environ Mol Mutagen. 2007;48:650–657. doi: 10.1002/em.20331. [DOI] [PubMed] [Google Scholar]
- Gibb HJ, Lees PS, Pinsky PF, Rooney BC. Lung cancer among workers in chromium chemical production. Am J Ind Med. 2000;38:115–126. doi: 10.1002/1097-0274(200008)38:2<115::aid-ajim1>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Ha L, Ceryak S, Patierno SR. Generation of S phase-dependent DNA double-strand breaks by Cr(VI) exposure: involvement of ATM in Cr(VI) induction of gamma-H2AX. Carcinogenesis. 2004;25:2265–2274. doi: 10.1093/carcin/bgh242. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Imai K, Yamamoto H. Carcinogenesis and microsatellite instability: the interrelationship between genetics and epigenetics. Carcinogenesis. 2008;29:673–680. doi: 10.1093/carcin/bgm228. [DOI] [PubMed] [Google Scholar]
- Izzotti A, Cartiglia C, Balansky R, D'Agostini F, Longobardi M, De Flora S. Selective induction of gene expression in rat lung by hexavalent chromium. Mol Carcinog. 2002;35:75–84. doi: 10.1002/mc.10077. [DOI] [PubMed] [Google Scholar]
- Jackson JP, Johnson L, Jasencakova Z, Zhang X, PerezBurgos L, Singh PB, Cheng X, Schubert I, Jenuwein T, Jacobsen SE. Dimethylation of histone H3 lysine 9 is a critical mark for DNA methylation and gene silencing in Arabidopsis thaliana. Chromosoma. 2004;112:308–315. doi: 10.1007/s00412-004-0275-7. [DOI] [PubMed] [Google Scholar]
- Jackson JP, Lindroth AM, Cao X, Jacobsen SE. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002;416:556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- Ke Q, Davidson T, Chen H, Kluz T, Costa M. Alterations of histone modifications and transgene silencing by nickel chloride. Carcinogenesis. 2006;27:1481–1488. doi: 10.1093/carcin/bgl004. [DOI] [PubMed] [Google Scholar]
- Klein CB, Su L, Bowser D, Leszczynska J. Chromate-induced epimutations in mammalian cells. Environ Health Perspect. 2002;110 5:739–743. doi: 10.1289/ehp.02110s5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Takahashi Y, Hirose Y, Nagao T, Tsuyuguchi M, Hashimoto M, Ochiai A, Monden Y, Tangoku A. The reduced expression and aberrant methylation of p16(INK4a) in chromate workers with lung cancer. Lung Cancer. 2006;53:295–302. doi: 10.1016/j.lungcan.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Shen L, Issa JP. Critical role of histone methylation in tumor suppressor gene silencing in colorectal cancer. Mol Cell Biol. 2003;23:206–215. doi: 10.1128/MCB.23.1.206-215.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labra M, Grassi F, Imazio S, Di Fabio T, Citterio S, Sgorbati S, Agradi E. Genetic and DNA-methylation changes induced by potassium dichromate in Brassica napus L. Chemosphere. 2004;54:1049–1058. doi: 10.1016/j.chemosphere.2003.10.024. [DOI] [PubMed] [Google Scholar]
- Langard S. One hundred years of chromium and cancer: a review of epidemiological evidence and selected case reports. Am J Ind Med. 1990;17:189–215. doi: 10.1002/ajim.4700170205. [DOI] [PubMed] [Google Scholar]
- Levy LS, Venitt S. Carcinogenicity and mutagenicity of chromium compounds: the association between bronchial metaplasia and neoplasia. Carcinogenesis. 1986;7:831–835. doi: 10.1093/carcin/7.5.831. [DOI] [PubMed] [Google Scholar]
- Lin JC, Jeong S, Liang G, Takai D, Fatemi M, Tsai YC, Egger G, Gal-Yam EN, Jones PA. Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island. Cancer Cell. 2007;12:432–444. doi: 10.1016/j.ccr.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BD, Schoenhard JA, Hwang JM, Sugden KD. Ascorbate is a pro-oxidant in chromium-treated human lung cells. Mutat Res. 2006;610:74–84. doi: 10.1016/j.mrgentox.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Mattagajasingh SN, Misra HP. Alterations in the prooxidant and antioxidant status of human leukemic T-lymphocyte MOLT4 cells treated with potassium chromate. Mol Cell Biochem. 1995;142:61–70. doi: 10.1007/BF00928914. [DOI] [PubMed] [Google Scholar]
- McGarvey KM, Fahrner JA, Greene E, Martens J, Jenuwein T, Baylin SB. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 2006;66:3541–3549. doi: 10.1158/0008-5472.CAN-05-2481. [DOI] [PubMed] [Google Scholar]
- Meng CF, Zhu XJ, Peng G, Dai DQ. Re-expression of methylation-induced tumor suppressor gene silencing is associated with the state of histone modification in gastric cancer cell lines. World J Gastroenterol. 2007;13:6166–6171. doi: 10.3748/wjg.v13.i46.6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol. 2004;14:R546–551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Peterson-Roth E, Reynolds M, Quievryn G, Zhitkovich A. Mismatch repair proteins are activators of toxic responses to chromium-DNA damage. Mol Cell Biol. 2005;25:3596–3607. doi: 10.1128/MCB.25.9.3596-3607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds M, Stoddard L, Bespalov I, Zhitkovich A. Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair. Nucleic Acids Res. 2007;35:465–476. doi: 10.1093/nar/gkl1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds M, Zhitkovich A. Cellular vitamin C increases chromate toxicity via a death program requiring mismatch repair but not p53. Carcinogenesis. 2007;28:1613–1620. doi: 10.1093/carcin/bgm031. [DOI] [PubMed] [Google Scholar]
- Schneider J, Shilatifard A. Histone demethylation by hydroxylation: chemistry in action. ACS Chem Biol. 2006;1:75–81. doi: 10.1021/cb600030b. [DOI] [PubMed] [Google Scholar]
- Schnekenburger M, Talaska G, Puga A. Chromium Cross-Links Histone Deacetylase 1-DNA Methyltransferase 1 Complexes to Chromatin, Inhibiting Histone-Remodeling Marks Critical for Transcriptional Activation. Mol Cell Biol. 2007;27:7089–7101. doi: 10.1128/MCB.00838-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Hudson LG, Liu KJ. Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free Radic Biol Med. 2004;37:582–593. doi: 10.1016/j.freeradbiomed.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Stearns DM, Wetterhahn KE. Reaction of chromium(VI) with ascorbate produces chromium(V), chromium(IV), and carbon-based radicals. Chem Res Toxicol. 1994;7:219–230. doi: 10.1021/tx00038a016. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Sugden KD, Stearns DM. The role of chromium(V) in the mechanism of chromate-induced oxidative DNA damage and cancer. J Environ Pathol Toxicol Oncol. 2000;19:215–230. [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–1791. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Kondo K, Hirose T, Nakagawa H, Tsuyuguchi M, Hashimoto M, Sano T, Ochiai A, Monden Y. Microsatellite instability and protein expression of the DNA mismatch repair gene, hMLH1, of lung cancer in chromate-exposed workers. Mol Carcinog. 2005;42:150–158. doi: 10.1002/mc.20073. [DOI] [PubMed] [Google Scholar]
- Tamaru H, Zhang X, McMillen D, Singh PB, Nakayama J, Grewal SI, Allis CD, Cheng X, Selker EU. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat Genet. 2003;34:75–79. doi: 10.1038/ng1143. [DOI] [PubMed] [Google Scholar]
- Tian X, Fang J. Current perspectives on histone demethylases. Acta Biochim Biophys Sin (Shanghai) 2007;39:81–88. doi: 10.1111/j.1745-7270.2007.00272.x. [DOI] [PubMed] [Google Scholar]
- Ting AH, McGarvey KM, Baylin SB. The cancer epigenome--components and functional correlates. Genes Dev. 2006;20:3215–3231. doi: 10.1101/gad.1464906. [DOI] [PubMed] [Google Scholar]
- Wei YD, Tepperman K, Huang MY, Sartor MA, Puga A. Chromium inhibits transcription from polycyclic aromatic hydrocarbon-inducible promoters by blocking the release of histone deacetylase and preventing the binding of p300 to chromatin. J Biol Chem. 2004;279:4110–4119. doi: 10.1074/jbc.M310800200. [DOI] [PubMed] [Google Scholar]
- Xie H, Wise SS, Holmes AL, Xu B, Wakeman TP, Pelsue SC, Singh NP, Wise JP., Sr Carcinogenic lead chromate induces DNA double-strand breaks in human lung cells. Mutat Res. 2005;586:160–172. doi: 10.1016/j.mrgentox.2005.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Z, Tachibana M, Guggiari M, Heard E, Shinkai Y, Wagstaff J. Role of histone methyltransferase G9a in CpG methylation of the Prader-Willi syndrome imprinting center. J Biol Chem. 2003;278:14996–15000. doi: 10.1074/jbc.M211753200. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Dowdy SC, Eberhardt NL, Podratz KC, Jiang SW. hMLH1 promoter methylation and silencing in primary endometrial cancers are associated with specific alterations in MBDs occupancy and histone modifications. Gynecol Oncol. 2006;103:321–328. doi: 10.1016/j.ygyno.2006.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Yan Y, Kluz T, Zhang P, Chen HB, Costa M. Analysis of specific lysine histone H3 and H4 acetylation and methylation status in clones of cells with a gene silenced by nickel exposure. Toxicol Appl Pharmacol. 2003;190:272–277. doi: 10.1016/s0041-008x(03)00169-8. [DOI] [PubMed] [Google Scholar]
- Ye J, Shi X. Gene expression profile in response to chromium-induced cell stress in A549 cells. Mol Cell Biochem. 2001;222:189–197. [PubMed] [Google Scholar]
- Zhang Q, Salnikow K, Kluz T, Chen LC, Su WC, Costa M. Inhibition and reversal of nickel-induced transformation by the histone deacetylase inhibitor trichostatin A. Toxicol Appl Pharmacol. 2003;192:201–211. doi: 10.1016/s0041-008x(03)00280-1. [DOI] [PubMed] [Google Scholar]
- Zhitkovich A. Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI) Chem Res Toxicol. 2005;18:3–11. doi: 10.1021/tx049774+. [DOI] [PubMed] [Google Scholar]
- Zhou X, Sun H, Ellen TP, Chen H, Costa M. Arsenite alters global histone H3 methylation. Carcinogenesis. 2008;29:1831–1836. doi: 10.1093/carcin/bgn063. [DOI] [PMC free article] [PubMed] [Google Scholar]