

Abstract
Objective
The lack of therapies that inhibit valvular calcification and the conflicting outcomes of clinical studies regarding the impact of 3-hydroxy-3-methylglutaryl-coenzyme A (HMGCoA) reductase inhibitors on valve disease highlight the need for controlled investigations to characterize the interactions between HMG-CoA reductase inhibitors and valve tissue. Thus, we applied multiple in vitro disease stimuli to valvular interstitial cell (VIC) cultures and examined the impact of simvastatin treatment on VIC function.
Methods and Results
VICs were cultured on three different substrates that supported various levels of nodule formation. TGF-β1 was also applied as a disease stimulus to VICs on 2-D surfaces or encapsulated in 3-D collagen gels and combined with different temporal applications of simvastatin. Simvastatin inhibited calcific nodule formation in a dose-dependent manner on all materials, although the level of statin efficacy was highly substrate-dependent. Simvastatin treatment significantly altered nodule morphology, resulting in dramatic nodule dissipation over time, also in a substrate-dependent manner. These effects were mimicked in 3-D cultures, wherein simvastatin reversed TGF-β1-induced contraction. Decreases in nodule formation were not achieved via the HMG-CoA reductase pathway, but were correlated with decreases in ROCK activity.
Conclusions
These studies represent a significant contribution to understanding how simvastatin may impact heart valve calcification.
Keywords: heart valves, valvular interstitial cells, simvastatin, calcification, extracellular matrix
Calcification is the major cause of aortic heart valve failure and is the most common heart valve disorder in developed countries 1. Currently, there are no medical agents that are FDA-approved to halt the progression of aortic valve disease 2. Valvular interstitial cells (VICs) are fibroblast-like cells that comprise the bulk of the valve that are believed to be the predominant cell type involved in valve calcification 1, 3. Calcified heart valves are rich in activated VICs, also known as myofibroblasts, as well as osteogenic growth factors and cytokines, including bone morphogenetic proteins (BMPs) and transforming growth factor-beta1 (TGF-β1) 3-5. Diseased valves also display grossly altered extracellular matrix (ECM) composition and arrangement 6.
3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors have been associated with reduced mortality in patients with atherosclerotic disease 7-9. Despite the intended purpose of lowering LDL serum cholesterol levels, HMG-CoA reductase inhibitors appear to have pleiotropic, cholesterol-independent effects such as anti-oxidation, improvement of endothelial function, and modulation of pro-inflammatory cytokine production 10-12 . In some studies, this type of drug treatment was found to slow the progression of valvular stenosis in patients with atherosclerosis 13-16. However, the numerous - and often conflicting - clinical studies that have examined the relationship between HMG-CoA reductase inhibitor therapy and valvular disease ultimately demonstrate that the scientific and medical communities know relatively little about the nature of the interaction of HMG-CoA reductase inhibitors with heart valves 17, 18.
The majority of HMG-CoA reductase inhibitor/valve investigations have been retrospective clinical studies, which greatly limit the amount of obtainable information about the structural and molecular properties and characteristics of the diseased or drug-treated valves. Limitations inherent to in vivo studies introduce significant challenges in studying the progression of valvular disease through its intermediate stages. Moreover, complications such as patients with multiple types of cardiovascular disease, variable medication compliance, and a tissue that is difficult to evaluate without explantation, make it exceedingly difficult to characterize the relationship between valves and HMG-CoA reductase inhibitors. These issues highlight the need for a set of controlled in vitro experiments that determine whether and how VICs respond to treatments with HMG-CoA reductase inhibitors of varying duration and timing.
In the current study, we characterize the effects of simvastatin treatment on VIC function in 2-D and 3-D cultures of varying compositions. The results from these experiments will allow us to develop a better understanding of: (1) how simvastatin regulates VIC dysfunction, (2) the role of the extracellular environment in regulating VIC response to simvastatin, and (3) the limitations/capabilities of simvastatin in preventing or treating valve disease.
Methods
All reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Raw data were analyzed via ANOVA with a Tukey HSD post-test, and p-values <0.05 were considered statistically significant. All data are presented as mean ± standard deviation.
Simvatatin dose-response in varied culture environments
Valvular interstitial cells (VICs) were isolated from porcine aortic valves (Hormel, Inc. Austin, MN) by collagenase digestion and cultured as previously described 19. VICs (P2-P4) were seeded at a density of 50,000 cells/cm2 and cultured in low-serum (LS) medium (1% FBS) on unmodified tissue culture polystyrene (TCPS) or TCPS coated with adsorbed fibrin (FB, 1.5 μg/cm2) or laminin (2 μg/cm2) (prepared as in 20). These cells were then treated with 0.1-1 μmol/L simvastatin (clinical range is approximately 0.1-0.3 μmol/L 21), which was supplied in its active form, (EMD Biosciences, Inc., Gibbstown, NJ) in LS medium for 5 days. Addition of TGF-β1 (5 ng/mL) was performed as a positive control, and TGF-β1 (5 ng/mL) was also combined with simvastatin (1 μmol/L). Cultures were replenished with simvastatin every 48 hours. The number of calcific nodules formed after 5 days in culture was evaluated via microscopic observation (Olympus IX51) and mineralization staining with Alizarin Red S. A separate set of Day 5 samples was lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris-HCl) and cell number was quantified using the QuantIt PicoGreen assay kit (Invitrogen).
A similar simvastatin dose-response study was performed on VICs cultured in type I collagen gels (Inamed Biomaterials, Fremont, CA). Gels were prepared as described previously 22 with a cell density of 1×106 cells/mL and collagen concentration of 2.4 mg/mL. Gels were left in a ‘stressed’ configuration (i.e., adherent to the well walls) for five days, during which time they received 0.1-1 μmol/L simvastatin. On Day 5, gels were released from the sides of the wells, and gel contraction was measured every hour for 10 hours, and as needed thereafter.
Application of different simvastatin treatment regimens
VICs were cultured on TCPS, FB, or LN, and fed either regular LS medium or LS medium + 5 ng/mL TGF-β1 for 5 days, at which time nodules were counted. The culture conditions were then switched such that cells continued to receive either plain LS medium, or were administered 1 μmol/L simvastatin for another 5 days, at which point nodule counts were performed again (Day 10).
Nodule analysis
VICs were cultured on TCPS surfaces for 5 days in LS medium + 5 ng/mL TGF-β1, and a portion of the samples was harvested on Day 5. The remaining portion of the samples received 4-5 additional days of treatment, either in LS medium or in LS medium + 1 μmol/L simvastatin. On the last day of treatment (Day 5 or Day 10), cells were fixed in 10% formalin, rinsed with PBS, and sputter-coated with 40 nm of gold. Samples were then imaged on a white-light interferometer (New View 6300, Zygo, Middlefield, CT). Images were subjected to a low-pass filter and data fill through the Zygo MetroPro software package. These images were subsequently analyzed for nodule height and area, also with MetroPro.
Contraction assays
VICs were seeded in collagen gels, treated with 1 μmol/L simvastatin, 5 ng/ml TGF-β1, concurrent simvastatin + TGF-β1, or plain medium (all conditions 1% FBS), and then measured for contraction as described above. At time points of 1, 4, and 24 hours post-release, treatment conditions were ‘reversed’: simvastatin was switched to receive TGF-β1, statin + TGF-β1 was switched to TGF-β1, TGF-β1 was switched to simvastatin, and media-only remained untreated. Gel diameters were then measured for an additional 5 days in culture.
Mechanism of simvastatin action
Mevalonate (100 μmol/L) in LS medium was added directly to the wells, either alone or in conjunction with simvastatin (1 μmol/L), to VICs grown on all three surfaces: TCPS, LN, and FB. Cultures were replenished with mevalonate and/or simvastatin every 48 hours for a total of 5 days, at which point the number of calcific nodules in the cultures was counted.
Collagen gel samples were flash-frozen and mechanically digested in RIPA buffer. An ELISA-based assay kit (MBL Intl. Corp., Woburn, MA) was used to quantify Rho kinase (ROCK) in each sample per manufacturer’s instructions, with sample absorbances read at 540 nm (Synergy HT Microplate reader, Biotek Instruments, Inc., Winooski, VT). The ROCK values in each sample were normalized to DNA concentration (QuantIt PicoGreen assay kit).
Results
Dose-dependence and ECM-dependence of simvastatin treatment
Increasing dosages of simvastatin administered to VIC cultures resulted in fewer calcified nodules, as shown in Figure 1. This dose-dependent trend was found on both ECM-coated surfaces (FB, LN), as well as the TCPS control (Figure 1a). We have previously shown that FB and LN coatings create nodule-conducive environments 20, leading to higher numbers of nodules when compared to the TCPS control. Figure 1 demonstrates that the efficacy of simvastatin treatment was dependent upon the type of culture substrate. Significant decreases in nodule number on LN and FB were achieved with lower simvastatin concentrations compared to TCPS. The highest simvastatin dosage resulted in an 80% reduction of nodules on LN, but only a 45% reduction on FB and TCPS. The trend of simvastatin efficacy, in terms of potency in nodule reduction, was: LN>FB>TCPS. Interestingly, the TGF-β1/simvastatin combination was the most effective at inhibiting nodule formation on all three culture substrates. Medium supplemented with TGF-β1 was used as a positive control, and for all substrates, yielded much greater nodule numbers than any simvastatin condition in Figure 1a (>400% increase in nodule number; not shown).
Figure 1.
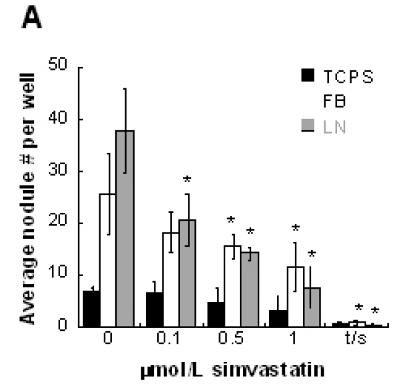
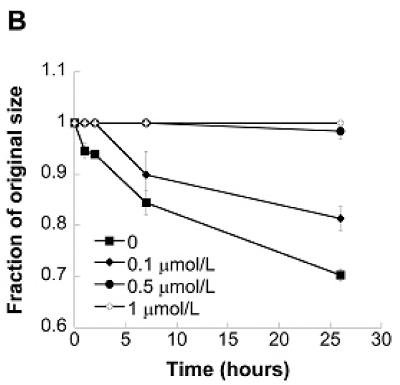
(A) Simvastatin reduced the formation of nodules in VIC cultures in a dose-dependent and ECM-dependent manner (fibrin - FB, laminin - LN, and tissue culture polystyrene - TCPS). * p < 0.01 vs. untreated control; ‘t/s’ refers to concurrent administration of 5 ng/mL TGF- β1 + 1 μmol/L simvastatin. (B) Simvastatin also reduced the contractile response of VICs cultured in collagen gels in a dose-dependent manner.
The dose-dependent trend seen in 2-D experiments was replicated in a 3-D environment, as shown in Figure 1b. VIC-seeded collagen gels exhibited steadily decreasing contraction upon receiving increasing dosages of simvastatin. The highest simvastatin dosage (1 μmol/L) completely inhibited gel contraction, while gels that received 0.5, 0.1, and 0 μmol/L simvastatin contracted to 95%, 81.3%, and 70.3% of their original sizes, respectively.
Combined effects of simvastatin treatment and the ECM on VIC nodule formation
Further experiments were performed to explore administration of various simvastatin treatment regimens in combination with nodule-inducing stimuli. Specifically, VICs were cultured on TCPS (Figure 2a), FB (Figure 2b), or LN (Figure 2c) with or without TGF-β1 supplementation for 5 days, followed by either simvastatin or no simvastatin treatment for the subsequent 5 days. When the nodule number on Day 10 was significantly less than the nodule number on Day 5, this occurrence was labeled ‘regressive’. This regressive phenomenon was observed on all three surfaces (TCPS, FB, and LN), but only when the cells had received TGF-β1 for the first 5 days; no regressive effects were observed in cultures that were untreated during the initial culture period. An assay for apoptosis in nodule-regressive cultures (Supplementary Material, Figure 1b) verified that the observed dissipation of nodules was not due to an increase in apoptotic events. The magnitude of the regressive effects also differed across the different substrates. Notably, simvastatin was most effective when applied to TGF-β1-treated VICs on LN, where nodule number was reduced by approximately 10-fold between days 5 and 10. In comparison, the regressive action was much more modest on TCPS and FB, with 1.3-fold and 2-fold reductions in nodule number, respectively. In one instance (FB without TGF-β1 pre-treatment, Figure 2b), statin treatment did not lower nodule numbers compared to Day 5 values, but did result in less nodule formation than found on the untreated control at Day 10; this occurrence was labeled an ‘inhibitory’ action of simvastatin.
Figure 2.
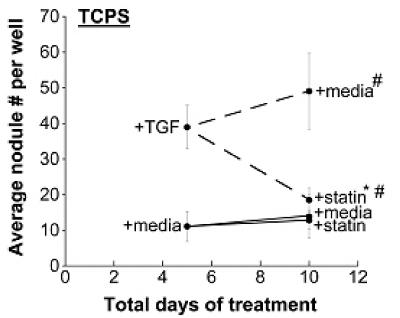
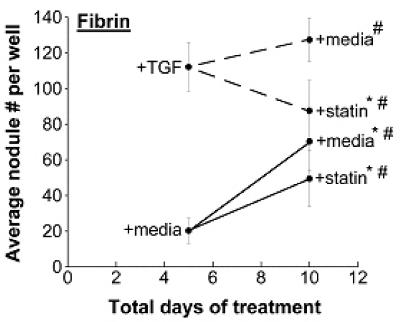
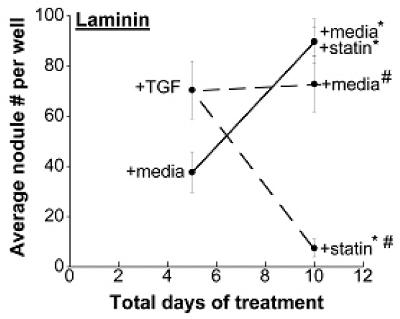
Different time courses of statin and TGF-β1 treatment were applied to VICs cultured on TCPS, fibrin, and laminin, followed by measurement of nodules at Days 5 and 10. * p < 0.002 compared to the corresponding Day 5 condition, # p < 0.002 vs. Day 10 paired condition.
Analysis of nodule characteristics and topography
The regressive results in Figure 2 raised questions regarding the fate of nodules following statin treatment. Thus, topographical imaging techniques were employed to examine nodule height and area over time, and time-course photomicrographs were captured to track nodule morphology upon simvastatin treatment. Figure 3 shows the gradual dissipation of a representative nodule from cultures treated with 1 μmol/L simvastatin for 5 days. In this experiment, nodules were formed during an initial 5-day TGF-β1 pre-treatment, mimicking the regressive conditions documented in Figure 2.
Figure 3.
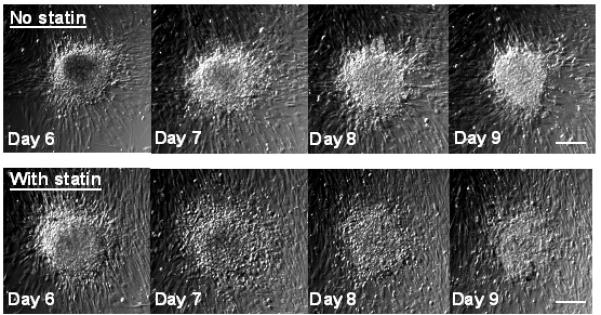
Brightfield photomicrographs tracking individual nodules over time. Nodules were induced via treatment of VICs with TGF-β1 for 5 days on TCPS, and then switched to either medium alone or medium+simvastatin for Days 5-9. Scale bars = 100 μm.
A topographical analysis of nodules was performed on Day 5 and Day 9 samples from the aforementioned experiment. As seen in Figure 4a, the topography of nodules treated with 1 μmol/L simvastatin changed dramatically when compared to both the original Day 5 nodules and Day 9 nodules that did not receive simvastatin. This morphological observation is confirmed quantitatively in Figure 4b, in which the simvastatin-treated nodules exhibited a more dramatic drop in maximum height when compared to their untreated counterparts. This drop in height was accompanied by an increase in area (Figure 4c). These data validate the images in Figures 3 and 4a in which simvastatin-treated nodules appear to flatten and dissipate over the course of statin treatment.
Figure 4.
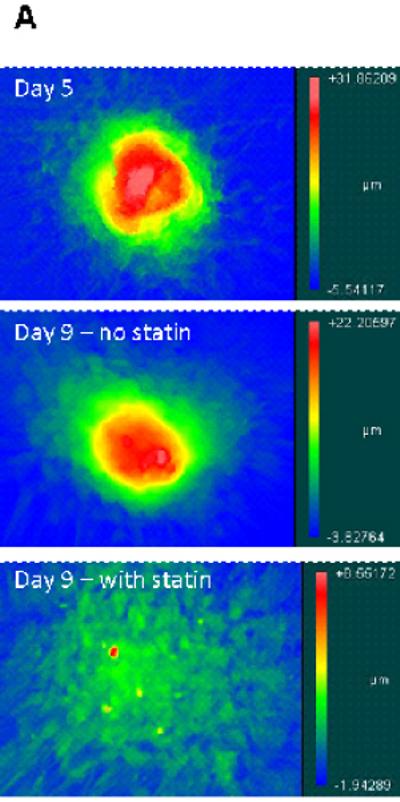
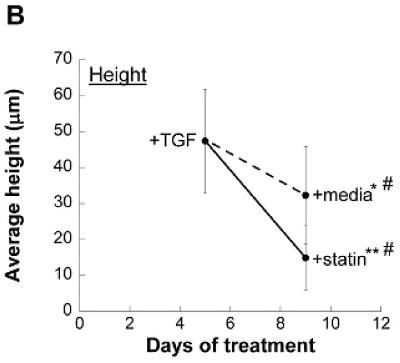
Nodules were induced via TGF-β1 treatment for 5 days and then switched to either plain medium or medium+simvastatin. (A) Representative topographical images of nodules at Days 5 and 9. (B) Average nodule height, and (C) Average nodule area at Days 5 and 9. * p < 0.03 vs. Day 5 condition, ** p < 0.0001 vs. Day 5 condition, # p < 0.0008 vs. Day 9 paired condition. n ≥ 16.
Impact of simvastatin on cell contractility
The impact of simvastatin treatment on VIC contractility was also measured in an effort to better understand the mechanism behind simvastatin’s inhibition of nodule formation. It is clearly shown in Figure 5a that simvastatin application, either alone or in conjunction with TGF-β1, drastically reduced contraction. To test the reversibility of this contraction, simvastatin was applied at various time points to the gels that received an initial 5 days of TGF-β1 treatment. This condition was intended to mimic the regressive scenarios described above for 2-D conditions. Figures 5b and 5c show that simvastatin application at either 1 or 4 hours led to a partial reversal of gel contraction, while simvastatin applied at 24 hours (Figure 5d) was not able to reverse contraction.
Figure 5.
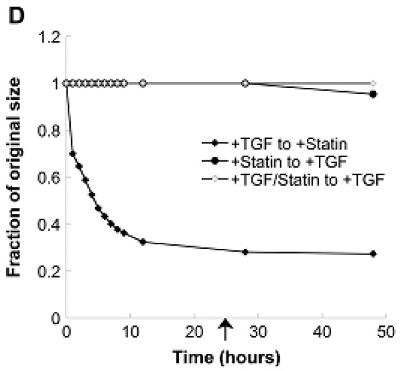
(A) Combinations of a pro-calcific agent (TGF-β1) and simvastatin were applied to VICs cultured in collagen gels for 5 days. In (B)-(D), initial TGF-β1 or simvastatin treatment was followed by a “reversed” condition applied at t=1 hr (B), t=4 hr (C), or t=24 hr (D) after the commencement of gel contraction, and then sustained for an additional 5 days. Arrow indicates time of application of reversed conditions.
To test the reversibility of simvastatin treatment (i.e., contraction resistance), TGF-β1 treatment was applied to the gels that had initially received simvastatin. However, application of TGF-β1 at either 1, 4, or 24 hours after gel release did not cause contraction of the gels initially treated with simvastatin.
Mechanism of simvastatin regulation of VIC function
To investigate the mechanism of simvastatin’s inhibition of VIC nodule formation, mevalonate, a downstream metabolite of the HMG-CoA reductase pathway, was added to VIC cultures. Addition of mevalonate represents ‘blocking’ simvastatin’s action by reinitiating the HMG-CoA reductase pathway. However, Figure 6a shows that addition of mevalonate to simvastatin-treated cultures on TCPS did not reverse the nodule-inhibiting actions of simvastatin, indicating that nodule inhibition is not dependent on the HMG-CoA reductase pathway. Similar results were obtained on FB and LN surfaces (data not shown).
Figure 6.
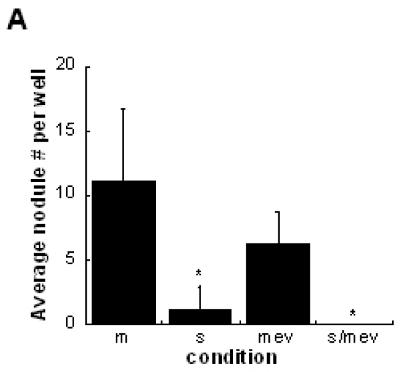
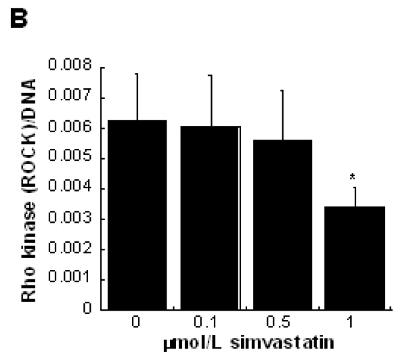
(A) Mevalonate (‘mev’) was added to VIC cultures to directly bypass statin (‘s’) inhibition of the HMG-CoA reductase pathway. * p < 0.0001 vs. untreated control (‘m’). (B) Rho kinase (ROCK) levels were evaluated in VICs cultured in 3-D collagen gels with increasing concentrations of simvastatin. * p < 0.03 vs. untreated control.
Quantification of Rho kinase (ROCK), an enzyme in the Rho contraction pathway, confirmed a positive correlation between VIC contraction and ROCK. The results in Figure 6b show that increased concentrations of simvastatin led to decreased ROCK expression. Because decreased ROCK expression generally correlates with decreased contraction, these results are complementary to the 3-D dose-dependence in Figure 1b, wherein simvastatin treatment decreased VIC contractility.
Discussion
Currently, few studies have investigated simvastatin treatment of VICs, and this work extends those initial findings 23, 24 through a more in-depth in vitro study involving multiple simvastatin dosages and treatment regimens, as well as several different nodule-conducive environments. This work is notable in its exploration of multiple types of culture environments, findings of statin-induced nodule regression and accompanying morphological analysis, and investigation of potential mechanisms.
We have shown that VICs respond to simvastatin treatment in a dose-dependent manner, both in 2-D and 3-D environments. Application of increasing concentrations of simvastatin resulted in decreased formation of nodules, with simvastatin treatment most effective on laminin-coated surfaces, followed by fibrin-coated surfaces, and finally, least effective on TCPS. The dose-dependent effect of simvastatin on nodule formation was consistent with results obtained for aortic valve myofibroblasts (AVMFs) on TCPS 23, 24. However, it is interesting to note that all previous studies of statins applied to VIC cultures were done exclusively on TCPS 23, 24, a culture condition which we found to be the least responsive to simvastatin treatment in terms of nodule reduction.
Culturing cells on TCPS alone cannot paint an accurate picture of cell behavior, as cells in their native environment are surrounded by ECM proteins that continuously modulate cellular responses. The ECM components studied in the present investigation were not intended to mimic native conditions; rather, they were selected for their ability to stimulate nodule formation in VIC cultures. However, use of these ECM molecules did illustrate an important point with respect to HMG-CoA reductase inhibitor efficacy: the composition of the culture substrate can greatly alter the response of VICs to simvastatin treatment. The significant substrate-dependence of simvastatin efficacy highlights the importance of performing such experiments in varied culture environments and provides motivation for future evaluations of pharmacological efficacy to be executed in more bioinspired environments.
The findings described herein reveal the unexpected, yet exciting, finding that simvastatin was capable of not only inhibiting the formation of nodules in VIC cultures, but also reducing nodule number after the nodules had formed. When TGF-β1 was applied for 5 days to induce nodule formation, simvastatin treatment was most effective at reversing nodule formation on laminin, followed by fibrin, and finally, least effective on TCPS — a trend consistent with that seen in Figure 1. The aforementioned nodule-regressive effects of simvastatin were noted only in conditions pre-treated with TGF-β1. Although unusual, these results are also consistent with those obtained with concurrent TGF-β1+statin treatment reported in Figure 1, where TGF-β1 appeared to enhance the nodule-inhibiting influence of simvastatin when compared against simvastatin alone. This relationship between TGF-β1 and simvastatin is intriguing— across several experiments, administration of TGF-β1 appeared to sensitize VICs to simvastatin, resulting in greater simvastatin efficacy. A potential mechanism for this occurrence is currently not known.
The authors recognize that there have not been clinical reports of nodule disappearance as reported herein. Thus, although we have documented clear evidence of nodule dissipation upon statin treatment of nodules, it is possible that these results cannot be directly translated to an in vivo environment. However, because in vivo studies of nodule-like calcification consist primarily of end-point analyses 25-27, it is also possible that in vivo nodule dissipation events may proceed undocumented due to limitations in the manner in which diseased valves can be analyzed. A recent in vivo study has utilized novel imaging technologies to monitor and visualize the earliest stages of arterial and valvular calcification, which may be comparable to nodule formation 28. This new technology appears very promising in its ability to clinically confirm the nodule regression reported herein, but because the technology is currently emerging, it has not yet been widely utilized. Thus, due to historical limitations in studying in vivo valve composition over time, the exact fate of calcific nodules during clinical studies of statins is not known. The clinical studies that have tracked the condition of a patient’s heart valve over time tend to examine stenosis, which is generally characterized by blood flow measurements, and may or may not be directly related to calcification.
Although previous studies have examined some effects of HMG-CoA reductase inhibitor treatment on other cell types 23, 29, 30, VICs are a unique cell type, both in their physiological function and their response to statin treatment. Unlike smooth muscle cells, which exhibit decreased proliferation with HMG-CoA reductase inhibitor treatment 30, VICs did not experience any changes in proliferation upon simvastatin treatment (p>0.4 for all comparisons, 3 simvastatin concentrations; data not shown). This result illustrates the uniqueness of VICs and the possibility that their response to treatment with HMG-CoA reductase inhibitors will be significantly different in comparison to other cell types. Additionally, previous HMG-CoA reductase inhibitor studies performed with other cell types did not investigate the possibility of a differential cellular response depending on the ECM environment, and the present effort uncovered significant differences in that regard.
Inhibition of the HMG-CoA reductase pathway is simvastatin’s predominant mechanistic action with respect to cholesterol reduction. However, addition of mevalonate to simvastatin-treated VIC cultures was not successful in reversing the nodule-suppressing actions of simvastatin, implying that simvastatin’s actions on nodule inhibition or regression in VIC cultures occured independently of simvastatin’s cholesterol-lowering function. These results are somewhat different than those documented by Wu et al 23, who found that addition of mevalonate to aortic valve myofibroblasts (AVMFs) did partially reverse simvastatin- and pravastatin-induced decreases in alkaline phosphatase (an indicator of calcification). However, AVMFs and VICs are slightly different cell populations, and the effects of simvastatin (and, by extension, any potential reversal by mevalonate) are likely to vary with cell type, as noted above.
In addition to inhibiting or regressing nodule formation, simvastatin also decreased VIC contractility in a dose-dependent manner. The increase in VIC contractility in conditions associated with increased nodule number is consistent with previous characterizations of diseased valves and calcified VIC cultures 22. Our results showed an inverse relationship between simvastatin concentration and ROCK concentration; decreased ROCK is consistent with the decreased contractility achieved with simvastatin administration. These results come together to suggest a possible mechanism for regulation of VIC nodule formation by simvastatin: simvastatin either directly or indirectly reduces the amount of ROCK, which consequently leads to decreased cell contraction, ultimately resulting in decreased levels of nodule formation in VIC cultures. Supplementary Figure 2 further underscores the importance of ROCK in regulating nodule formation in VIC cultures, as this experiment demonstrated that targeted inhibition of ROCK via H1152 drastically reduced nodule formation in VIC cultures, thereby providing a more direct link between ROCK activity and nodule formation. These reductions in VIC contractility were not accompanied by a decrease in the total amount of α-SMA in VIC cultures (Supplementary Figure 3), although these data do not account for possible changes in the presentation of α-SMA.
The effects of simvastatin with respect to regressing 2-D nodule formation were also somewhat mimicked in 3-D contraction assays. Application of simvastatin was able to partially reverse TGF-β1-induced cell contraction, but only when the simvastatin was applied very early in the contraction process — contraction reversal was no longer possible when simvastatin was applied at t=24 hours after collagen gel release. Interestingly, no such reversal was possible in the converse situation, where simvastatin-treated gels were switched to pro-contractile conditions. This result may imply that some long-term cell relaxation is imparted by simvastatin (even after delivery of simvastatin has been replaced with delivery of a pro-contractile factor), or that disruption of early contractile events in this assay is sufficient to prohibit any contraction from occurring.
This work clearly demonstrates that, in several 2-D and 3-D in vitro environments, simvastatin exerts a potent anti-nodule effect on cultures of valvular interstitial cells. Moreover, the efficacy of this treatment is highly dependent upon multiple factors, including the nature of the disease stimulus, the composition of the culture environment, and the timing of the statin treatment regimen. There are several implications of this work with respect to studying and treating calcific valvular disease. First, these findings stress the importance of investigating pharmaceutical interventions in in vitro environments that are more complex or biologically-inspired than traditional TCPS. Furthermore, our data suggest that a reduction in VIC contractility is a likely mechanism by which simvastatin reduces VIC nodule formation, and this finding may be further exploited to better elucidate the etiology of valvular disease as well as identify potential therapeutics. Statin-induced decreases in apoptosis may also play a role in nodule inhibition (Supplementary Figure 1), but neither apoptotic nor proliferation events were responsible for nodule regression.
The results obtained in this work upon combination of TGF-β1 with simvastatin also highlight an intriguing avenue for further studies. Ultimately, these results, combined with continued investigation of interactions between HMG-CoA reductase inhibitors and VICs, may help to shed more light upon the current controversy related to treatment of valvular disease with HMG-CoA reductase inhibitors and lead to the identification of appropriate treatment regimens to reduce valvular disease.
Supplementary Material
Acknowledgements and Funding Sources
(a) Funding for this work was provided by a National Science Foundation CAREER Award (CBET-0547374) to KSM and a National Institutes of Health Translational Cardiovascular Sciences Traineeship (T32-HL 007936-06) to ELM.
(b) The authors are grateful to Justin Turner (Zygo) for assistance with interferometry.
Footnotes
Condensed abstract
In vitro treatment of valvular interstitial cells with simvastatin revealed that simvastatin dramatically altered nodule morphology, but in a manner that depended upon the type of disease stimulus and the composition of the extracellular environment. The probable mechanism for these occurrences was simvastatin-induced downregulation of cell contractility.
References
- 1.Schoen FJ, Levy RJ. Calcification of tissue heart valve substitutes: progress toward understanding and prevention. Ann Thorac Surg. 2005;79:1072–1080. doi: 10.1016/j.athoracsur.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 2.Mohler ER, 3rd, Wang H, Medenilla E, Scott C. Effect of statin treatment on aortic valve and coronary artery calcification. J Heart Valve Dis. 2007;16:378–386. [PubMed] [Google Scholar]
- 3.Mohler ER., 3rd Mechanisms of aortic valve calcification. Am J Cardiol. 2004;94:1396–1402. doi: 10.1016/j.amjcard.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 4.Jian B, Narula N, Li QY, Mohler ER, 3rd, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–466. doi: 10.1016/s0003-4975(02)04312-6. [DOI] [PubMed] [Google Scholar]
- 5.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 6.Hinton RB, Jr., Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98:1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- 7.Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 8.Sacks FM, Pasternak RC, Gibson CM, Rosner B, Stone PH. Effect on coronary atherosclerosis of decrease in plasma cholesterol concentrations in normocholesterolaemic patients. Harvard Atherosclerosis Reversibility Project (HARP) Group. Lancet. 1994;344:1182–1186. doi: 10.1016/s0140-6736(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 9.Sacks FM, Moye LA, Davis BR, Cole TG, Rouleau JL, Nash DT, Pfeffer MA, Braunwald E. Relationship between plasma LDL concentrations during treatment with pravastatin and recurrent coronary events in the Cholesterol and Recurrent Events trial. Circulation. 1998;97:1446–1452. doi: 10.1161/01.cir.97.15.1446. [DOI] [PubMed] [Google Scholar]
- 10.Rosenson RS. Statins in atherosclerosis: lipid-lowering agents with antioxidant capabilities. Atherosclerosis. 2004;173:1–12. doi: 10.1016/S0021-9150(03)00239-9. [DOI] [PubMed] [Google Scholar]
- 11.Liebe V, Brueckmann M, Borggrefe M, Kaden JJ. Statin therapy of calcific aortic stenosis: hype or hope? Eur Heart J. 2006;27:773–778. doi: 10.1093/eurheartj/ehi697. [DOI] [PubMed] [Google Scholar]
- 12.Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–1719. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 13.Aronow WS, Ahn C, Kronzon I, Goldman ME. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. Am J Cardiol. 2001;88:693–695. doi: 10.1016/s0002-9149(01)01821-5. [DOI] [PubMed] [Google Scholar]
- 14.Novaro GM, Tiong IY, Pearce GL, Lauer MS, Sprecher DL, Griffin BP. Effect of hydroxymethylglutaryl coenzyme a reductase inhibitors on the progression of calcific aortic stenosis. Circulation. 2001;104:2205–2209. doi: 10.1161/hc4301.098249. [DOI] [PubMed] [Google Scholar]
- 15.Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G, Baumgartner H. Statins but not Angiotensin-Converting Enzyme Inhibitors Delay Progression of Aortic Stenosis. Circulation. 2004;110:1291–1295. doi: 10.1161/01.CIR.0000140723.15274.53. [DOI] [PubMed] [Google Scholar]
- 16.Antonini-Canterin F, Zuppiroli A, Popescu BA, Granata G, Cervesato E, Piazza R, Pavan D, Nicolosi GL. Effect of statins on the progression of bioprosthetic aortic valve degeneration. Am J Cardiol. 2003;92:1479–1482. doi: 10.1016/j.amjcard.2003.08.066. [DOI] [PubMed] [Google Scholar]
- 17.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. A Randomized Trial of Intensive Lipid-Lowering Therapy in Calcific Aortic Stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- 18.Houslay ES, Cowell SJ, Prescott RJ, Reid J, Burton J, Northridge DB, Boon NA, Newby DE. Progressive coronary calcification despite intensive lipid-lowering treatment: a randomised controlled trial. Heart. 2006;92:1207–1212. doi: 10.1136/hrt.2005.080929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson CM, Hanson MN, Helgeson SC. Porcine cardiac valvular subendothelial cells in culture: cell isolation and growth characteristics. J Mol Cell Cardiol. 1987;19:1185–1193. doi: 10.1016/s0022-2828(87)80529-1. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez KJ, Masters KS. Regulation of valvular interstitial cell mineralization by components of the extracellular matrix. J Biomed Mater Res. doi: 10.1002/jbm.a.32187. In Press. DOI: 10.1002/jbm.a.32187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng H, Rogers JD, Sweany AE, Dobrinska MR, Stein EA, Tate AC, Amin RD, Quan H. Influence of age and gender on the plasma profiles of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitory activity following multiple doses of lovastatin and simvastatin. Pharm Res. 1992;9:1629–1633. doi: 10.1023/a:1015828811865. [DOI] [PubMed] [Google Scholar]
- 22.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95:253–260. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- 23.Wu B, Elmariah S, Kaplan FS, Cheng G, Mohler ER., 3rd Paradoxical effects of statins on aortic valve myofibroblasts and osteoblasts: implications for end-stage valvular heart disease. Arterioscl Throm Vas. 2005;25:592–597. doi: 10.1161/01.ATV.0000154278.01871.64. [DOI] [PubMed] [Google Scholar]
- 24.Osman L, Yacoub MH, Latif N, Amrani M, Chester AH. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation. 2006;114:I547–552. doi: 10.1161/CIRCULATIONAHA.105.001115. [DOI] [PubMed] [Google Scholar]
- 25.Mohler ER, 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 26.Srivatsa SS, Harrity PJ, Maercklein PB, Kleppe L, Veinot J, Edwards WD, Johnson CM, Fitzpatrick LA. Increased cellular expression of matrix proteins that regulate mineralization is associated with calcification of native human and porcine xenograft bioprosthetic heart valves. J Clin Invest. 1997;99:996–1009. doi: 10.1172/JCI119265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]
- 29.Taylor ML, Wells BJ, Smolak MJ. Statins and cancer: a meta-analysis of case-control studies. Eur J Cancer Prev. 2008;17:259–268. doi: 10.1097/CEJ.0b013e3282b721fe. [DOI] [PubMed] [Google Scholar]
- 30.Turner NA, Midgley L, O’Regan DJ, Porter KE. Comparison of the efficacies of five different statins on inhibition of human saphenous vein smooth muscle cell proliferation and invasion. J Cardiovasc Pharmacol. 2007;50:458–461. doi: 10.1097/FJC.0b013e318123767f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.