

Abstract
Mitochondrial (mt) DNA biogenesis is critical to cardiac contractility. DNA polymerase gamma (pol γ) replicates mtDNA, whereas thymidine kinase 2 (TK2) monophosphorylates pyrimidines intramitochondrially. Point mutations in POLG and TK2 result in clinical diseases associated with mtDNA depletion and organ dysfunction. Pyrimidine analogs (NRTIs) inhibit Pol γ and mtDNA replication. Cardiac “dominant negative” murine transgenes (TGs; Pol γ Y955G, and TK2 H121N or I212N) defined the role of each in the heart. mtDNA abundance, histopathological features, histochemistry, mitochondrial protein abundance, morphometry, and echocardiography were determined for TGs in “2 × 2” studies with or without pyrimidine analogs. Cardiac mtDNA abundance decreased in Y955C TGs (∼50%) but increased in H121N and I212N TGs (20-70%). Succinate dehydrogenase (SDH) increased in hearts of all mutants. Ultrastructural changes occurred in Y955C and H121N TGs. Histopathology demonstrated hypertrophy in H121N, LV dilation in I212N, and both hypertrophy and dilation in Y955C TGs. Antiretrovirals increased LV mass (≈50%) for all three TGs which combined with dilation indicates cardiomyopathy. Taken together, these studies demonstrate three manifestations of cardiac dysfunction that depend on the nature of the specific mutation and antiretroviral treatment. Mutations in genes for mtDNA biogenesis increase risk for defective mtDNA replication, leading to LV hypertrophy.
Keywords: Cardiomyopathy, Murine model, mtDNA, NRTI, Toxicity
Introduction
Alteration of mitochondrial replication pathways and imbalance of precursor nucleotide pools for mitochondrial (mt) DNA replication can result in diminished mitochondrial biogenesis and concomitantly decreased energy output. POLG is the human nuclear gene that encodes the catalytic subunit of DNA polymerase gamma (pol γ), the replicase for mtDNA [1, 2]. Y955C, a POLG point mutation, is the commonest and most severe autosomal dominant mutation. Y955C Pol γ causes human chronic progressive external ophthalmoplegia (CPEO), an inherited disorder characterized by mtDNA depletion and mtDNA mutations [3].
Like Pol γ, thymidine kinase 2 (TK2) is a nuclear encoded enzyme. TK2 is a mitochondrial pyrimidine deoxynucleoside salvage enzyme involved in pyrimidine nucleoside phosphorylation and mtDNA precursor synthesis in mitotically-quiescent cells [4]. TK2 performs the critical first intra-mitochondrial phosphorylation of pyrimidines. The monophosphorylated product of TK2 (dTMP) is the initial phosphorylated precursor of dTTP used for mtDNA replication by Pol γ. Theoretically, TK2 phosphorylates native nucleosides as well as pyrimidine nucleoside analogs, such as zidovudine (AZT), creating pyrimidine NRTI-MPs in the latter case. TK2 mutations are one cause of “inherited mtDNA depletion syndromes” [5] (MDS; MIM 251880), a heterogeneous group of inherited disorders that share reduced mtDNA copy number and dysfunction of diverse organs. Specific point mutations of TK2, H121N, and I212N, exhibit severe mtDNA depletion [6].
Nucleoside reverse transcriptase inhibitors (NRTIs) for AIDS are nucleoside analogs that effectively block HIV reverse transcriptase. Depletion of mtDNA in vivo in some tissues also results from pharmacological administration of nucleoside analogs through inhibition of Pol γ [7, 8]. In particular, pyrimidine triphosphates (e.g. AZT-TP or stavudine, d4T-TP) can inhibit mitochondrial replication in cardiac myocytes [9]. Mitochondrial toxicity from NRTIs warrants careful monitoring for organ dysfunction in patients undergoing antiretroviral treatments (reviewed in [10-12]).
Present studies employed transgenic mice (TG) that over-expressed homologous point mutants of known human TK2 (H121N or I212N) or Pol γ in the murine heart using the α-MyHC promoter [9, 13, 14]. Previously published data demonstated decreased mtDNA, oxidative stress, and cardiomyopathy (CM) in the murine Y955C TG [15]. Altered mtDNA abundance was found in a murine native TK2 overexpressor TG [16]. Studies here characterize the combined impact of point mutations of Pol γ and TK2 and of NRTIs on cardiac mtDNA abundance, mitochondrial ultrastructure, and mitochondrial enzyme histochemistry.
Materials and Methods
Generation of α-MyHC Promoter-Driven Mutant Transgenic Mice (TG)
Established methods were employed as described previously [13] and applied to the TK2 cDNA constructs from Wang and Eriksson [17]. Briefly, murine homologs of the human TK2 mutants were genetically engineered to yield amino acid substitution of HIS 121 -> ASN (H121N), and ILE 212 ->ASN (I212N). Generation of cardiac-targeted Pol γ Y955C TG was previously described [15].
Treatment Protocols
Procedures complied with Emory IACUC and NIH guidelines. Antiretroviral drugs were from the manufacturers (compliments of Raymond Schinazi, VA Medical Center, Decatur, GA and Emory Center for AIDS Research Pharmacology Core). WT and TG littermates of both gender (male and female), were age-matched (8-12 weeks) at the start of the treatment regimen. Food and water were provided ad libitum. Dosing was done by daily gavage (morning, 0.25 ml/day) at doses that resemble human therapy. Mice received either vehicle control (1% carboxymethylcellulose) or a combination antiretroviral treatment (HAART) consisting of zidovudine (AZT; 0.22 mg/day), lamivudine (3TC; 0.11 mg/day) and indinavir (IDV; 0.9 mg/day). Morbidity and mortality from the procedure were negligible. At day 35, final weights and echocardiographic measurements were made, animals were terminated. and samples retrieved and stored for DNA extraction and analysis.
Genotyping
For the two TK2 mutants (H121N and I212N), the presence of the transgene was detected in the founders and their offspring using Southern blotting and real-time PCR essentially as reported in the past [13, 18]. Pol γ Y955C TGs were routinely verified as described previously [15].
TK2 Gene Copy Analysis
At least two lines were established for each of the targeted over-expressing mutant TK2 TGs. To determine the relative copy number in each line, the level of TK2 transgene was analyzed semi-quantitatively from murine tail DNA extracts using real-time PCR and Light Cycler TaqMan Master kit. Target genes were amplified using specific primers for TK2 (forward: 5′-TACGAGGAGTGGCTGGTCA-3′ and reverse: 5′-GTTGTGGTCAGCCTCAATCA-3′ and Universal ProbeLibrary probe #33; Roche Diagnostics Corp.), and a “housekeeping” gene, GAPDH (forward: 5′-GATGCTACAAGCAGGCCTTT-3′ and reverse: 5′-GCAGAAAGCAAGGGCAAA-3′, and Universal ProbeLibrary probe #4; Roche Diagnostics Corp., IN). DNA amplification was performed using a LightCycler (LC) 480 (Roche Diagnostics Corp.) on individual tissues extracted from at least six mice within each line. Relative copy number was normalized to TK2 (single copy gene) from wild-type (WT).
Mitochondrial DNA (mtDNA) and Nuclear DNA (nDNA) Quantitation in Heart Tissue Using Real-Time PCR
Methods employed were modifications used by others [19] as reported [15, 16]. Briefly, total DNA was extracted from heart tissue (∼10 mg wet weight) using a MagNA Pure System and reagents (Roche Life Sciences, Indianapolis, IN). DNA sequences for primers and probes used for quantitation of mitochondrial and nuclear DNA are described previously [15, 20]. Real-Time PCR was performed in duplicate for each amplicon. Amplification was performed using LC 480 (Roche). Standard DNA curves for quantitation of the LC products were employed. PCR products of mtDNA and nDNA were quantified using the corresponding external standard.
Enzyme Histochemistry for Succinate Dehydrogenase (SDH)
Succinate dehydrogenase histochemistry is reliable for visualization of normal mitochondria and for identification of disrupted mitochondrial biogenesis. Measurement of SDH, a substrate-specific mitochondrial oxidative enzyme, served as a benchmark for the utilization of a metabolic intermediary in the Kreb’s cycle. Specifically, the reduction of a tetrazolium salt to an insoluble, dark blue colored formazon product correlates to the level of type I skeletal muscle fibers, or actively proliferating mitochondria, which are associated with most genetic and acquired mtDNA defects. Specific methods for histochemistry were adapted from standard protocols with minor modifications [21]. The relative intensity of the immunhistochemistry staining for SDH was semi-quantitated using a scoring scheme from 0 (control) up to +4 for maximum level using light microscopy (20× magnification).
Fine Structure Pathological Evaluations with Transmission Electron Microscopy (EM) Analysis of Mitochondrial Damage of Mutant TGs
Ultrastructure (N=12) was evaluated with transmission EM using methods employed regularly in the laboratory [22]. Sections (0.5 μ) were cut with glass knives and stained with Toluidine Blue for orientation. Ultrathin (900 Å) sections were cut with a diamond knife, stained with uranyl acetate and lead citrate and viewed on a Philips Morgagni EM201 microscope and images electronically captured at two different magnifications (14,000×, and 22,300×). Each EM image was reviewed independently by two investigators. Representative image for final collage was selected from careful review of a minimum of 12 captured EM images/mouse in a total group of 2-4 randomly selected mice from each cohort. Parameters included presence of structurally abnormal mitochondria, increased numbers of mitochondrial profiles per field, intra-mitochondrial lamellar bodies, abnormal cristae density, cristae reduplication, mitochondrial swelling, and intra-mitochondrial paracrystals [23].
Quantitative Mitochondrial Volumes
Representative images from EM cardiac sections were quantitatively analyzed for changes in mitochondrial volumes in TGs (with or without treatment) from WTs using Image J software (available from the NIH, http://rsb.info.nih.gov/ij/). From each cohort, four mice were randomly selected for EM imaging. For each mouse, 3-5 images were taken. Using the Image J software, all intact mitochondria were selected and measurements were collectively determined. The respective mitochondrial volumes were subsequently calculated by multiplying the measured area by the thickness of the prepared tissue slide (90nm).
Immunoblotting of Mitochondrial Complex I
Methods employed resembled those used by us recently [13] and applied to isolated cardiac mitochondria obtained from TG and WT hearts. Slight modifications were made to adjust the protocol to an infrared imaging, Odyssey 2-color detection system (LiCor Biosciences, Lincoln, NE) utilizing their specific LiCor PVDF membrane, proprietary Odyssey blocking buffer, primary antibodies including anti-oxphos Complex I 20 kDa subunit mouse IgG2b monoclonal 21Cll (lnvitrogen Corp., Carlsbad, CA) and rabbit pAb VCAD1/Porin (Abcam Inc., Cambridge, MA) followed by secondary antibodies goat anti-mouse IRDye 680 CW and goat anti-rabbit IRDye 800 CW (LI-COR Biosciences). Resultant blots were scanned using Odyssey system to determine relative quantitation of Complex I for each sample. Since differences in protein abundance can be found between male and female mice, groups were separated based by gender [24, 25].
Preparation and Selection of Whole-Mount Heart Sections
Since overall cardiac size can vary between males and females [26], we used only male mice with maximal LV dilation, as determined by echocardiograph, for representative whole mount sections for each cohort. Hearts were bi-valved with a razor blade, processed, embedded in paraffin, and stained with Masson’s trichome (MT) and Hematoxylin and Eosin (H&E). Images were photographed at 10× and 20× magnification. Change in cavity % were determined from measuring cavity area and wall area together with total area for each selected image using Image J software (as described above for mitochondrial volumes).
Echocardiography (ECHO) in Mutant TGs and WT
LV mass was quantified and normalized using ECHO in age- and gender-matched (littermates) WT and transgenes (TGs) as reported previously [22, 27]. Echocardiography was performed at the end of the studies. Methods used were described previously. Briefly, mice were anesthetized with Avertin (0.25 mg/g of body weight) and two-dimensionally targeted M-mode images were generally taken from the short axis (at the level of the largest LV diameter) by using a 15 MHz transducer (Acuson SequoiaTM). M-mode measurements of heart rate (HR), left ventricle end diastolic dimensions (LVEDD), left ventricle end systolic dimensions (LVESD), anterior wall thickness (AWTH), and posterior wall thickness (PWTH) were obtained from the original tracings using the leading-edge convention of the American Society of Echocardiography and by using the steepest continuous endocardial echoes. Left ventricle mass (LV Mass) was computed using the formula: (1) LV Mass=1.05*((LVEDD+AWTH+pWTH)3-(L VEDD)3). Fractional shortening (FS) was calculated by using the formula: (2) FS = ((LVEDD-LVESD)/LVEDD)*100
Statistical Analysis
Resultant values were expressed as the ratio of mean values for mtDNA to the mean values of nDNA × 10-3. Resultant values expressed as mean ± standard error, normalized to vehicle-treated WT mean (set at 1.0). A value of P < 0.05 was considered statistically significant. Echocardiographic data between TG cohorts with vehicle or AZT-HAART treatments and WTs were compared by one-way ANOVA with Newman-Keuls Multiple Comparison Test.
Results
General
Targeted transgenesis of murine TK2 mutants (H121N and I212N) was accomplished in at least two lines each. Transgenesis of pups was verified by Southern blot (Fig. 1). .Animals bred true for six generations before experimental use. Overall, no gross phenotype was identified in the H121N TGs or littermates over 1 year. However, one line of mutant TK2 (I212N-B) was runted compared to age-matched littermates (not shown), and did not survive long enough for experimental treatments. No changes in cognitive behavior, growth, maturation, breeding behavior, or Mendelian distribution were found in any other line. Data in Table 1 compares relative gene copy of the cardiac-targeted mutant TK2 TG lines in both H121N and I212N. Copies ranged from 1.5- to 4.0-fold of WT (single copy). Pol γ Y955C lines were previously determined [15].
Fig. 1.
Transgenic mutant TK2 lines. Southern blot analysis confirmed TK2 mutants (+), WT (-), and control (C). Gels document transgenesis in each of two mutant TK2 lines (H121N and I212N)
Table 1.
Mutant murine TK2 gene copy multiples
| Line | Fold increase above WT* | Operational genotype |
|---|---|---|
| I212N—A | 2 | Low |
| I212N—B | 4 | High |
| WT | 1 | Single copy |
| H121N—A | 3 | Medium |
| H121N—B | 4 | High |
| H121N—C | 1.5 | Low |
Relative copy number normalized to TK2 WT (single copy gene)
Due to the large number of TGs, one line for each mutant TK2 TG (I212N and H121N) was chosen for experiments. The low gene copy lines for each mutant TG were selected (1.8- and 1.2-fold above WT, respectively) to obviate potential artifact from higher TG expression. Kaplan-Meier survival curves were similar for both the H121N and I212N TGs and WT FVB/n (not shown).
mtDNA/nDNA Abundance
Within the timeframe of the study (8-12 week old mice at initiation and 5 weeks treatment duration), I212N and H121N TGs exhibited an unexpected finding. Cardiac mtDNA/nDNA ratios in vehicle-treated TK2 mutant TGs increased (∼50%) compared to that of WT littermates (P < 0.01; Fig. 2a, b). AZT-based HAART treatment (5 week duration) had no measurable effect on WT mtDNA/nDNA. Combined antiretroviral treatment of I212N TGs resulted in no change in mtDNA abundance from vehicle-treated I212N TGs, yet increased compared to treated WTs (P < 0.01). Thus, the I212N mutant phenotype impacted mtDNA abundance but AZT-HAART did not amplify those effects. Vehicle-treated H121N TGs also resulted in an increase in mtDNA abundance compared to WT littermates. In contrast to I212N, AZT-HAART treated H121N TGs had no change in mtDNA/nDNA ratio from vehicle- or AZT-HAART treated WT cohorts. As a result, vehicle-treated H121N TGs exhibited a higher mtDNA/nDNA ratio than the other three cohorts, including treated H121N TGs (P < 0.01). This result suggests a “mutant specific” difference between I212N and H121N TGs. Vehicle-treated H121N TGs increase mtDNA abundance (like I212N), but when combined with AZT-HAART their mtDNA/nDNA ratio has no increase from WT littermates (unlike I212N). Overall, I212N and H121N TGs (at least vehicle-treated) appear to induce paradoxical increases in mtDNA abundance, suggesting a compensational response or pathological mitochondrial proliferation response [28].
Fig. 2.
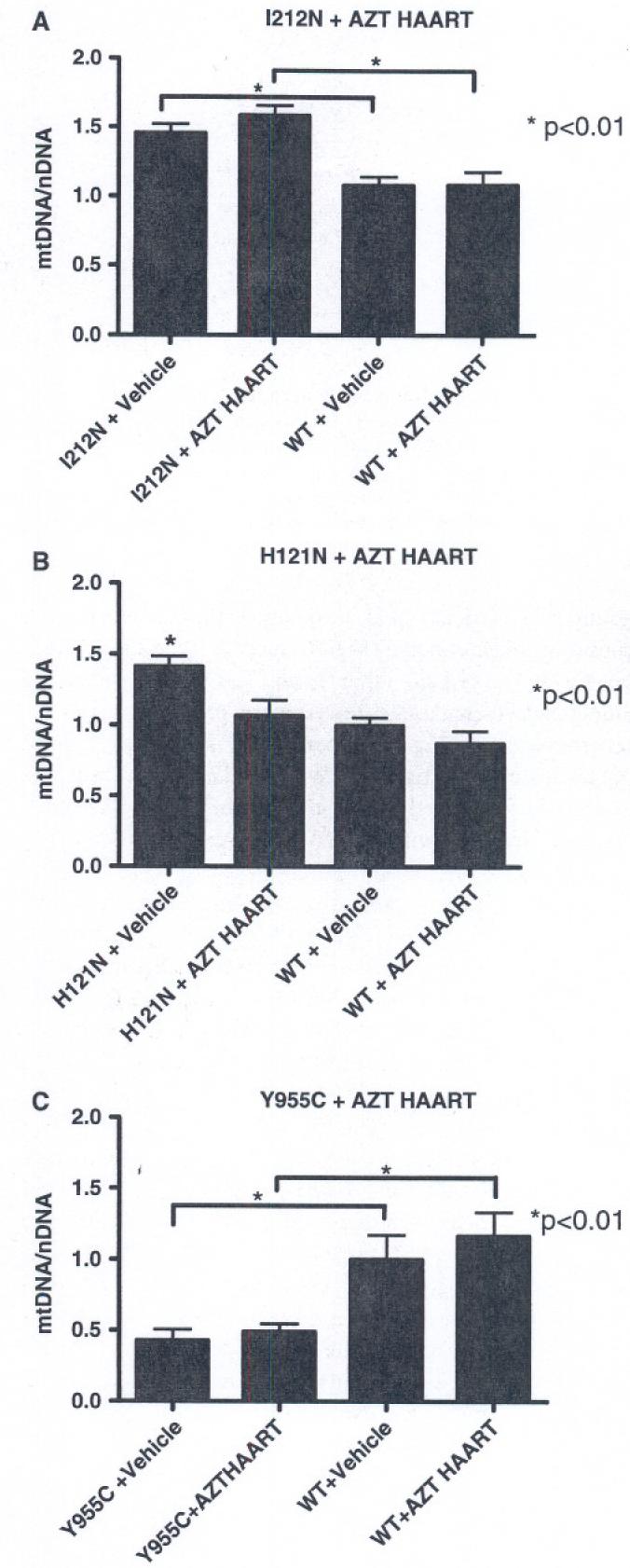
Real-time PCR mtDNA/nDNA ratios with NRTI treatment: TGs and WT cohorts were treated with combination antiretroviral (AZT, 3TC, IDV; HAART) or vehicle control for 35d. Tissue samples were analyzed using real-time PCR. (a) I212N TGs with vehicle- or AZT-HAART treatment had increased mtDNA/nDNA ratios compared to WT littermates. (b) Vehicle-treated H121N TGs also had increased mtDNA/nDNA ratio while AZT-HAART TGs have little change compared to WTs. (c) Vehicle- or AZT-HAART treated Y955C TGs had decreased mtDNA abundance compared to WTs
In contrast to the TK2 mutant TG phenotypes, mtDNA abundance in vehicle-treated Y955C TGs significantly decreased (∼50%) compared to that of vehicle-treated WT (Fig. 2c). Treated Y955C TGs had no additional change from vehicle-treated Y955C TGs, but again had a ratio that was significantly decreased from vehicle or AZT-HAART treated WTs (P < 0.01). Transgenes of the mutant Pol γ gene Y955C profoundly influenced mtDNA replication. This included decreased mtDNA abundance without further impact of AZT-HAART (at 5 weeks duration). Interestingly the response to NRTI regimens in each of these TGs differs.
SDH Enzyme Histochemistry
SDH histochemistry, as previously described [20], was used to evaluate dysfunctional mitochondrial biogenesis. Using a semi-quantitative scoring scheme with vehicle-treated WTs at 0, relative darker blue staining was graded up to +4. Heart sections from all vehicle-treated I212N, H121N, or Y955C TGs exhibited a stronger blue staining than vehicle-treated WT, with I212N TGs having the darkest stain (+3) while H121N and Y955C TGs both had +2 level of blue intensity (Fig. 3, upper panels). Antiretroviral treatment increased SDH signal in both the I212N (+4) and Y955C (+3) TGs as well as the WT (+1), whereas NRTI-treated H121N TGs had less intense SDH signal (+1) compared to respective vehicle-treated littermates (Fig. 3, lower panels compared upper panels). Relative SDH enzyme activities for each TG cohort appear to correlate with mtDNA abundance phenotypes following vehicle or AZT-HAART treatments.
Fig. 3.

SDH histochemical staining in cardiac tissues from both TGs and WTs: Frozen cardiac tissues from representative I212N, H121N, and Y955C TGs and WTs, with or without antiretroviral treatment (HAART). Representative stained tissues for vehicle-treated (upper panels) and AZT-HAART (lower panels) are shown. Increase in dark blue staining is indicative of increase in SDH enzyme activity. Darker blue staining occurs in all TG cohorts including WT following AZT-HAART treatment, with the exception of H121N TG. The relative intensity of the immunohistochemistry staining for SDH using light microscopy (20S magnification) is indicated from a relative score of 0 (vehicle-treated WT) to +4
Ultrastructural (EM) Features of Mitochondria in TG Hearts
EM is an established, semi-quantitative method of analysis for NRTI toxicity [13, 22] and for mitochondrial volume [29]. EM was used by other investigators to analyze mitochondrial profiles in mouse pup cardiomyocytes following NRTI treatment of the dam [30]. EM was used extensively by us in other studies [13, 31]. EM data here supported biochemical and molecular findings (above) and ECHO findings (below). Vehicle-treated WTs demonstrated normal cardiac mitochondria with regularly arranged spherical, oval, elongated mitochondria with densely packed christae (Fig. 4, upper left). AZT-HAART treated WTs, on the other hand, had slightly enlarged mitochondria containing fewer more prominent and discernable cristae (Fig. 4, upper right), suggesting a mild NRTI-treatment effect on WTs. I212N TGs revealed essentially no ultrastructural changes in the cardiac mitochondria with or without HAART treatments (data not shown).
Fig. 4.

Electron photomicrographs of mitochondria from cardiac myocytes of H121N and Y955C TG compared to WT: Shown are representative images for each cohort selected from minimum of 12 captured EM images/ mouse from 2-4 randomly selected mice from each cohort. AZT-HAART treated WTs have slightly enlarged mitochondria containing fewer more prominent and discernable cristae compared to vehicle-treated WTs (upper right and left panels, respectively). Spherical or oval mitochondria in the vehicle-treated and AZT-HAART treated H121N TGs appear to be accompanied by an occasional irregular shaped mitochondrion (middle panels). Vehicle-treated and AZT-HAART treated Y955C TGs have profound increase in mitochondrial number, smaller size, and irregular shape and structure with substantially reduced numbers of prominent and often truncated cristae (lower panels). (original magnification 26,000×. Marker indicates 1μ)
In H121N TGs, spherical or oval mitochondria in the vehicle-treated and NRTI-treated cohorts appear to be accompanied by an occasional irregular shaped mitochondrion (Fig. 4, middle panels). Changes between the AZT-HAART treated and vehicle-treated H121N TGs are subtle. AZT-HAART treated H121N TGs may have more irregular-shaped and some smaller mitochondria, which is concordant with decreased mtDNA abundance in AZT-treated verses vehicle-treated H121N TGs (Fig. 2). Likewise, vehicle-treated and NRTI-treated Y955C TGs had profound increase in mitochondrial number, smaller size, and irregular shape and structure with substantially reduced numbers of prominent and often truncated cristae compared to WTs (Fig. 4, lower panels). While AZT-treated Y955C TGs have some differences compared to vehicle-treated Y955C TGs, it is difficult to assess the increased severity or significance. No significant changes in sarcomeres occurred in any of the cohorts.
To confirm those pathologic impressions, changes in mitochondrial volumes were calculated from EMs for each cohort using Image J software. Relative volumes of mitochondria from comparable EM images were measured for H121N and Y955C cohorts and WT littermates with or without treatment. Mitochondrial volumes did not change for I212N TGs (data not shown). In contrast, mitochondrial volumes increased for both H121N and Y955C TGs compared to WT in hearts of vehicle-treated mice (Fig. 5, P < 0.05). In addition, mitochondrial volume in hearts from antiretroviral treated H121N TGs decreased back to WT levels. This was statistically different from vehicle-treated H121N TGs. As previously mentioned, it may be reasonable to suggest that the H121N TG and antiretroviral treatment counteracted each other and resulted in a mitochondrial volume in treated H121N TGs that more closely resembled that of the vehicle-treated WT. Antiretroviral treatment of Y955C TGs did not change mitochondrial volumes compared to that of vehicle-treated Y955C TGs.
Fig. 5.
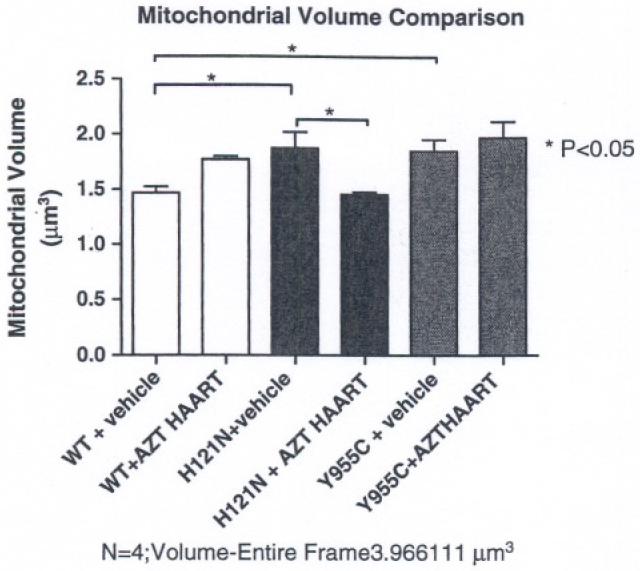
Comparison of mitochondrial volumes of H121N and Y955C TG and WT cardiac sections: Relative mitochondrial/total field section volumes were determined on EM sections. H121N transgenesis alone resulted in increased volume over WT while antiretroviral treatment in H121N TGs was lower than treated WTs. Treated Y955C TGs had a higher mitochondrial volume than vehicle-treated WTs, but were no different from treated WTs
Mitochondrial 20-kd Subunit of Complex I Immunoblot Quantitation
Abundance of polypeptides encoded by mtDNA (specifically those elements of complex I of the electron transport chain) was determined by immunoblot analysis of cardiac mitochondria isolated from TG and WT hearts with or without AZT-HAART treatment using specific infrared probes (anti-OxPhos Complex I, 20-kD mitochondrial-encoded subunit mouse IgG2b monoclonal and rabbit pAB VDAC1/Porin). Detection of the 20-kD subunit of complex I from mitochondrial samples was normalized to porin signals and then converted to arbitrary units for comparison (Fig. 6).
Fig. 6.

Steady state abundance of 20-kd mitochondrial subunit of complex I polypeptide by Western blot in H121N and Y955C TGs with AZT-HAART. Infrared scans were performed using antibody probes to the 20-kd (mitochondrial-encoded) subunit of complex I using the Odyssey infrared imaging system. Band signal intensities were normalized to porin control on the same blot. (a) H121N and Y955C TGs (males) with vehicle or AZT-HAART had slight increase (non-statistical) in Complex I/ porin ratio compared to WTs. (b) Vehicle- and AZT-HAART treated (5 weeks) Y955C TGs (females) had a decrease in steady state abundance of 20-kd mitochondrial subunit compared to H121N TGs and WT littermates
Using this approach, AZT-based therapy caused no detectable change in steady state abundance of the 20-kD mitochondrial-encoded subunit of Complex I in mitochondria samples of I212N TG (data not shown) or H121N TG (Fig. 6a and b, males and females, respectively). Likewise, no change was detected in male samples from Y955C TGs with vehicle or AZT-HAART treatment (Fig. 6a). However, female Y955C TG samples (vehicle or AZT-HAART treated) demonstmted a significant decrease (P < 0.05) in the steady state abundance of the 20-kD mitochondrial-encoded subunit of Complex I compared to their WT littermates (Fig. 6b). Of note, significant differences related to gender (although considered throughout the study) were only seen in this parameter.
Cardiac Whole-Mount Histological Comparisons
Histologic analyses of whole-mount heart sections stained with Masson Trichrome were used to determine cardiac gross pathological changes in ventricular walls and heart cavities of all TGs compared to WT (Fig. 7). H121N TGs demonstrated evidence of LV hypertrophy with a decreased cavity % (7.7%) compared to WT (15.0%). I212N TGs exhibited LV dilation with an increased cavity % (18.2%) compared to WT. These pathological results suggested that cardiac performance may be altered in both the H121N and I212N TGs, but in pathophysiologica1ly different ways. Y955C TG cavity % (20.9%) had the greatest change suggesting both hypertrophic and dilated compared to WT.
Fig. 7.

Histologic analysis of sectioned hearts: H121N TGs demonstrated evidence of LV hypertrophy with decreased cavity % (7.7%), whereas the I212N TGs exhibited LV dilation with increased cavity % (18.2%) compared to WT (15.1%). Y955C TGs had the largest change in cavity % (20.9%) suggesting both hypertrophy and dilation. (original magnification 2×; Masson Trichrome)
Echocardiographic (ECHO) Evaluation of LV Mass in Hearts of TGs With or Without Combined Antiretroviral Therapy
An increase in LV mass or LV chamber cavity volume may be considered characteristic of cardiac dysfunction. Echocardiographic data from hearts of all three TGs with and without antiretroviral treatment were notably different from WT (5 weeks; Fig. 8). Three distinct outcomes resulted from the three different mutant TGs.
Fig. 8.
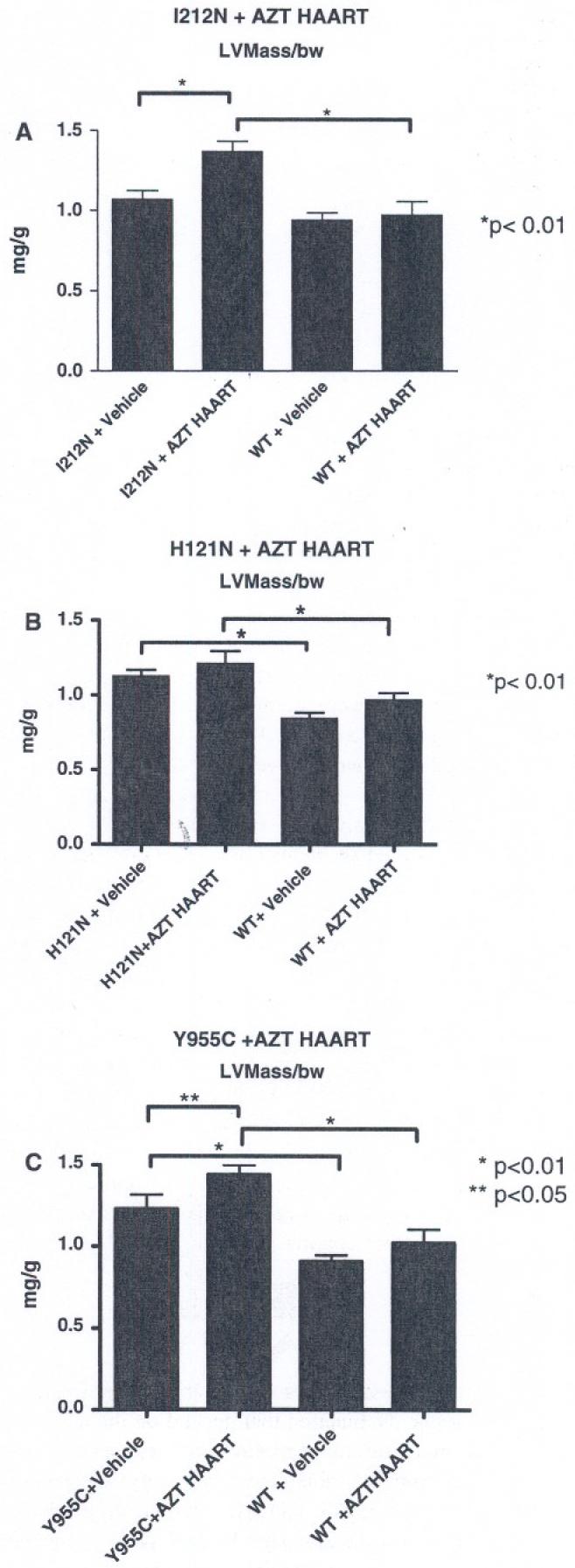
Quantitative analyses of ECHO images: LV mass was calculated in a blinded fashion, code was broken, and data tabulated. (a) I212N TGs only had an increase in LV mass following combination antiretroviral treatment. (b) H121N TGs without anti-retrovira1 treatment (HAART) had significant increase (∼25-50%) in LV mass from WT. (c) Y955C TGs also demonstrated an increase in LV mass (both vehicle and AZT-HAART treated) with an apparent additive impact in hearts from treated Y955C TGs. Data were normalized to body weight (mg/g) and plotted as mean ± SEM
In the absence of antiretroviral treatment, I212N TG hearts exhibited no ECHO phenotype (normalized LV mass; mg/g body weight) compared to LV mass found in hearts from WT cohorts (Fig. 8a). This result parallels (in some ways) clinical findings in which patients with mutations in TK2 do not exhibit a substantial cardiac phenotype [5]. Moreover, treatment with antiretroviral NRTI combinations had no impact on LV mass in WT. LVmass increased in I212N TG hearts following antiretroviral treatment (P < 0.05). It is reasonable to conclude that a combination of expression of the mutant I212N and antiretroviral treatment together resulted in cardiac dysfunction.
ECHO studies for H121N TGs hearts without treatment exhibited approximately 50% increase in LV mass compared to respective controls (Fig. 8b). These data supported a conclusion that compensation occurred in H121N TGs induced by the expression of the mutant TK2 (H121N). In contrast, I212N TGs required both mutant TK2 (I212N) expression and antiretroviral treatment to elicit pathophysiologic changes.
A third outcome occurred in hearts from Y955C TGs. Here, vehicle-treated Y955C TG exhibited increased LV mass compared to WT (Fig. 8c). Antiretroviral treatment increased the LV mass of hearts in Y955C TGs to a level greater than seen for vehicle-treated Y955C TGs (P < 0.05), suggesting an additive effect. Thus, Y955C TGs alone, or the combination of mutant expression with antiretroviral treatment resulted in an increased (or even additive) LV mass, respectively.
Cardiac functional parameters were further analyzed (summarized in Table 2). Left ventricle end diastolic (LVEDD) and systolic (LVESD) dimension levels for all vehicle-treated TGs were higher than in vehicle-treated WTs (P < 0.05). In contrast, FS and HR levels for all vehicle-treated TGs were lower than in vehicle-treated WTs.
Table 2.
Cardiac functional parameters by echocardiography
| I212N (N = 12) | I212 + HAART (N = 13) | H121N (N = 7) | H121N+HAART (N = 7) | Y955C (N = 13) | Y955C+HAART (N = 14) | WT (N = 38) | WT+HAART (N = 36) | |
|---|---|---|---|---|---|---|---|---|
| LVEDD (mm) | 3.52 ± 0.04 | 3.87 ± 0.07 | 3.31 ± 0.09 | 3.43 ± 0.15ns | 3.25 ± 0.08 | 3.80 ± 0.08 | 2.99 ± 0.05 | 3.15 ± 0.06 |
| LVESD (mm) | 1.67 ± 0.04 | 2.07 ± 0.05 | 1.57 ± 0.08 | 1.77 ± 0.12ns | 1.63 ± 0.09 | 2.15 ± 0.10 | 1.25 ± 0.04 | 1.43 ± 0.06 |
| FS (%) | 52.64 ± 0.83 | 46.44 ± 0.97 | 52.62 ± 1.64 | 48.37 ± 2.28ns | 50.01 ± 1.8 | 43.32 ± 1.6 | 58.05 ± 0.98 | 55.03 ± 1.08 |
| HR (bpm) | 336 ± 31 | 358 ± 8 | 342 ± 12 | 356 ± 18 | 312 ± 8 | 273 ± 17 | 383 ± 8 | 390 ± 12 |
WT cohorts were pooled from each 2 × 2 TG and normalized. Data presented are means +/- SEM for each functional parameter. Abbreviations include: left ventricular end diastolic dimension (LVEDD), left ventricular end systolic dimension (LVESD), fractional shortening (FS), and heart rate (HR). ns = no statistical difference between vehicle treated and AZT-treated
AZT-HAART treatment of WTs compared to vehicle-treated WTs resulted in increased LVEDD, LVESD, and HR while FS decreased (all P < 0.05). Similar trends occurred in H121N, I212N, and Y955C TGs treated with AZT-HAART. AZT-HAART treated I212N and Y955C TGs were statistically different from vehicle-treated littermates as well as AZT-HAART WTs (P < 0.05). AZT-HAART treated H121N TGs were not statistically different from their vehicle-treated littermates for LVEDD, LVESD, and FS (P > 0.05). Notable exception in overall patterns was found in Y955C TGs which had a decreased HR following AZT-HAART compared to vehicle-treated Y955C TGs. In addition, AZT-treated verses vehicle-treated Y955C TGs exhibited the largest changes in all parameters compared to all other cohorts.
Discussion
Taken together, these studies demonstrate three manifestations of cardiac dysfunction that depend on the nature of the specific mutation and antiretroviral treatment. I212N alone impacts mtDNA abundance, but only when combined with AZT-HAART increases cardiac hypertrophy while H121N is just the opposite; H121N alone is capable of increasing cardiac hypertrophy and increasing mtDNA abundance but with AZT-HAART, the mtDNA abundance remains unchanged from WTs. Y955C impacts both mtDNA abundance and cardiac function without AZT-HAART treatment and the deleterious effects are amplified by antiretrovirals (Table 3).
Table 3.
Summary of molecular and physiological phenotypes
| TG | mtDNA abundance | EM | LV Mass/bw |
|---|---|---|---|
| I212N (TK2) | ↑ (compensation) | No change from WT | TG alone: No change from WT TG + HAART: Increase LV Dilation |
| H121N (TK2) | ↑ (compensation) | ↑ cristae density | TG alone: Increase; TG + HAART: same as TG alone Hypertrophy |
| Y955C (pol γ) | ↓ (depletion) | ↑ cristae density, and ultrastructural damage | TG alone: Increase; TG + HAART: higher than TG alone Hypertrophy and LV dilation |
HAART: (combination antiretroviral treatment with AZT, 3TC and IDV for 35 days)
We systematically determined the effects of clinically relevant mutations (I212N, H121N, and Y955C) in two key mitochondrial enzymes (TK2 and Pol γ) involved in mitochondrial nucleotide homeostasis and mtDNA replication by using an approach based on over-expression of the mutant genes in vivo. Changes in mtDNA abundance, quantitative ultrastructure changes of mitochondria, and left ventricular functional changes were observed. All resulted from mtDNA replication defects at the level of DNA polymerase γ, thymidine kinase, or the nucleotide pool. It follows that disruption of mitochondrial replication impacts electron transport and contractile function, particularly in cardiac or skeletal muscle [32].
In a clinical context, mtDNA depletion is considered a shared hallmark of NRTI toxicity and of mutations in TK2 and Pol γ. Some studies suggest mtDNA abundance in peripheral blood cells is a surrogate marker for mitochondrial toxicity [10, 19], but this is not universally accepted and remains a challenge clinically. Unfortunately, mtDNA abundance in target organs can rarely be assessed directly in patients. In experiments here, steady state abundance of mtDNA was determined directly in cardiac samples from TGs and WTs with and without antiretroviral treatment (n ≥ 9 per cohort). All mtDNA/nDNA ratios were normalized to vehicle-treated WT cohorts.
mtDNA/nDNA ratios increased in hearts of both I212N and H121N TGs alone and in I212N TGs with AZT-HAART treatment. At first glance, this observed increased mtDNA abundance may appear counter-intuitive. Decreased mtDNA is considered characteristic of mitochondrial dysfunction associated with toxicity from long-term NRTI exposure [33]. Increased mtDNA could serve as an attempt by mitochondria to compensate for defective mtDNA replication. This effect occurs in some mitochondrial genetic disorders [34], in some HIV/AIDS patients treated with NRTIs [35], and in some of our own early in vitro studies in which mtRNA increased with short-term AZT exposure [36]. Some may argue that the mutants may be simply more efficient at making mtDNA. The enzyme activity of these two mutants, however, is significantly reduced from the native TK2 (discussed below) which suggests a compensatory response is a more likely explanation [28]. mtRNA was not quantitated in this study. In vitro data from Poirier and colleagues who demonstrated increased mtDNA following treatment with AZT in tissue culture [37] supports our explanation for increase in mtDNA abundance and has been described by others [38, 39] as a compensatory response [28].
H121N responds differently to NRTI treatment than I212N. With respect to mtDNA abundance, H121N demonstrated a suppression of mtDNA abundance following AZT-HAART compared to I212N. Reports in humans showed H121N mutant TK2 exhibited control Km values, but 2-fold lower Vmax values compared to WTTK2. I212N mutants showed less than 1% enzyme activity [40]. The Km values for each NRTI with these TK2 mutants have not been determined (to our knowledge). It may appear that H121N TG and AZT-based antiretroviral treatment counteracted each other with respect to mtDNA abundance. The resultant mtDNA abundance was similar to that of native WT treated with AZT-HAART. However, comparison of photmicrographs for H121N TGs following AZT-HAART demonstrated mitochondria that were distinctly different in appearance from WT mitochondria. In a follow-up experiment in which AZT was replaced with stavudine (d4T) in combination HAART regimen for 5 weeks duration, H121N TGs maintained an increased mtDNA abundance phenotype similar to vehicle-treated H121N TGs (data not shown). These results are concordant with previous studies that demonstrated alternative mitochondrial polypeptides generated in cardiac-targeted transgenic mice can interact differently with specific antiretrovirals [13]. This observation supports clinical findings in which a specific drug treatment (e.g. NRTI-based HAART) in one individual is safe and effective while in another results in toxicity or lack of efficacy; the recurring molecular dilemma that genetic background can influence response to drug treatment and the current focus of “individualized medicine”.
In contrast, mtDNA abundance decreased significantly in Y955C TGs, with or without AZT-HAART. These results suggest that specific mutations in target genes can impact mtDNA abundance in different ways. An increase or decrease in mtDNA abundance compared to WT serves as a biological marker for impaired mitochondrial biogenesis, compensation, and organ-specific pathogenesis.
Alternative phosphorylation pathways may circumvent dysfunctional TK2 [13], whereas no alternative exists for Pol γ, the only enzyme responsible for mtDNA replication in eukaryotes. Recent evidence supports the hypothesis that a p53-responsive R2 subunit of ribonucleotide reductase is present at constant low levels in both post-mitotic (such as cardiomyocytes) and proliferating cells. The potential existence of this p53R2 subunit in cardiomyocytes provides explanation for an alternative salvage pathway of deoxyribonucleosides in mitochondria [41].
It is also reasonable to suggest that transgenic manipulation has a potential biological effect downstream of the genetic target. This effect may be most evident in conditions where the selected transgene encodes an enzyme (or component of an enzyme complex) that generates precursors for a series of reactions.
NRTI-monophosphates (NRTI-MPs) also inhibit Pol γ in vitro, albeit not competitively [42]. Recent data from Bianchi’s group provided evidence for a rapid, highly selective import of dTMP into the mitochondria, and a 100-fold increased concentration of intra-mitochondrial dTMP [43]. The responsible dTMP importer has yet to be identified. If H121N moiety were proven to exist, intra-mitochondrial monophosphorylation (of dThd or NRTI) could be obviated, and an important mechanism to defend against NRTI mitochondrial toxicity in mitotically-quiescent tissues could be elucidated.
From a clinical genetics perspective, it remains unclear why the phenotypes of human TK2 mutations are tissue-selective and the same dilemma exists with NRTI tissue specific toxicity. It has been proposed that in unaffected tissues, pyrimidine phosphorylation may be rescued by cytoplasmic enzymes with overlapping substrate specificity [44]. In the case of skeletal muscle, several conditions could account for vulnerability to TK2 mutations. First, low expression of the previously described deoxyribonucleotide carrier (DNC), now thought to be the thiamine pyrophosphate transporter, was speculated to have an important role in transport of phosphorylated nucleotides into the mitochondria. DNC transporters may facilitate entry of diphosphate products from cytoplasmic thymidine phosphorylation and their intra-mitochondrial accummulation, bypassing TK2 requirement, although the true mitochondrial nucleotide transporter has yet to be identified [45]. Second, increased mtDNA abundance in muscle relative to other tissues, implies a high rate of synthesis with accompanying increase in vulnerability to defects in biogenesis. Lastly, decreased basal TK2 activity in muscle mitochondria [6] may impact mitochondrial biogenesis. Clinical findings together with recent in vivo murine studies suggest that the DNC transporter is incapable of rescuing mtDNA synthesis in post-mitotic cells, perhaps because TK1 expression and activity is minimal [5, 13, 43].
Disparate effects occurred with each respective TG, based on increased LV mass. In vehicle-treated I212N TK2 mutant TGs, no phenotypic changes in cardiac function occurred, based on changes in LV mass. However, in H121N TK2 mutant TGs LV mass statistically increased. Treatment with NRTIs in any of the three TGs caused cardiac dysfunction. Results from these studies suggest that genetic mutations (particularly in target gene products involved in the pathways of NRTI processing/phosphorylation) may have clinical implications on selection of antiretroviral therapies used to treat HIV/AIDS.
In summary, this study utilized TGs that individually expressed pathogenetic point mutants of human TK2 or Pol γ in the murine heart to define defective mtDNA replication and cardiac dysfunction. Increased mtDNA abundance with I212N or H121N TG in a “dominant negative” approach indicated that monophosphorylation of nucleotides for mtDNA synthesis played a de facto role in cardiac mtDNA homeostasis. Alternatively, Y955C TG demonstrated an overwhelming suppression of mtDNA abundance that also leads to altered cardiac mtDNA homeostasis. Thymidine triphosphate abundance as a substrate for Pol γ remains crucial in both mtDNA biogenesis and cardiac function. A role for TK2 and Pol γ in heart disease is underscored by these in vivo experiments.
Acknowledgments
This work was supported by DHHS, NIH NHLBI R01 HL072707 (to WL). We thank Staffan Eriksson and Liya Wang (Department of Molecular Biosciences, SLU, Uppsala, Sweden) for helpful comments.
Contributor Information
James J. Kohler, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA
Seyed H. Hosseini, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA
Elgin Green, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Amy Hoying-Brandt, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Ioan Cucoranu, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Chad P. Haase, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA
Rodney Russ, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Jaya Srivastava, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Kristopher Ivey, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Tomika Ludaway, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Victor Kapoor, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Allison Abuin, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Alexsey Shapoval, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Robert Santoianni, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
Ann Saada, The Metabolic Disease Unit, Hadassah University Medical Centre, Jerusalem, Israel.
Orly Elpeleg, The Metabolic Disease Unit, Hadassah University Medical Centre, Jerusalem, Israel.
William Lewis, Department of Pathology, Emory University School of Medicine, 7126 Woodruff Memorial Building, 101 Woodruff Circle, Atlanta, GA 30322, USA.
References
- 1.Kagunim LS. DNA polymerase gamma, the mitochondrial replicase. Annual Review of Biochemistry. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- 2.Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase gamma in mitochondrial DNA replication and repair. Chemical Reviews. 2006;106:383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- 3.Horvath R, Hudson G, Ferrari G, Futterer N, Ahola S, Lamantea E, Prokisch H, Lochmuller H, McFarland R, Ramesh V, Klopstock T, Freisinger P, Salvi F, Mayr JA, Santer R, Tesarova M, Zeman J, Udd B, Taylor RW, Turnbull D, Hanna M, Fialho D, Suomalainen A, Zeviani M, Chinnery PF. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain. 2006;129:1674–1684. doi: 10.1093/brain/awl088. [DOI] [PubMed] [Google Scholar]
- 4.Eriksson S, Munch-Petersen B, Johansson K, Eklund H. Structure and function of cellular deoxyribonucleoside kinases. Cellular and Molecular Life Sciences. 2002;59:1327–1346. doi: 10.1007/s00018-002-8511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nature Genetics. 2001;29:342–344. doi: 10.1038/ng751. [DOI] [PubMed] [Google Scholar]
- 6.Saada A, Shaag A, Elpeleg O. mtDNA depletion myopathy: Elucidation of the tissue specificity in the mitochondrial thymidine kinase (TK2) deficiency. Molecular Genetics and Metabolism. 2003;79:1–5. doi: 10.1016/s1096-7192(03)00063-5. [DOI] [PubMed] [Google Scholar]
- 7.Vivet-Boudou V, Didierjean J, Isel C, Marquet R. Nucleoside and nucleotide inhibitors of HIV-1 replication. Cellular and Molecular Life Sciences. 2006;63:163–186. doi: 10.1007/s00018-005-5367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitsuya H, Weinhold KJ, Furman PA, St Clair MH, Lehrman SN, Gallo RC, Bolognesi D, Barry DW, Broder S. 3′-Azido-3′-deoxythymidine (BW A509U): An antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:7096–7100. doi: 10.1073/pnas.82.20.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis W. Nucleoside reverse transcriptase inhibitors, mitochondrial DNA and AIDS therapy. Antiviral Theraphy. 2005;10(Suppl 2):M13–M27. [PubMed] [Google Scholar]
- 10.Lewis W, Day BJ, Copeland WC. Mitochondrial toxicity of nrti antiviral drugs: An integrated cellular perspective. Nature Reviews. Drug Discovery. 2003;2:812–822. doi: 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- 11.Lewis W, Dalakas MC. Mitochondrial toxicity of antiviral drugs. Nature Medicine. 1995;1:417–422. doi: 10.1038/nm0595-417. [DOI] [PubMed] [Google Scholar]
- 12.Kohler JJ, Lewis W. A brief overview of mechanisms of mitochondrial toxicity from NRTIs. Environmental and Molecular Mutagenesis. 2007;48:166–172. doi: 10.1002/em.20223. [DOI] [PubMed] [Google Scholar]
- 13.Lewis W, Kohler JJ, Hosseini SH, Haase CP, Copeland WC, Bienstock RJ, Ludaway T, McNaught J, Russ R, Stuart T, Santoianni R. Antiretroviral nucleosides, deoxynucleotide carrier and mitochondrial DNA: Evidence supporting the DNA pol gamma hypothesis. Aids. 2006;20:675–684. doi: 10.1097/01.aids.0000216367.23325.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the alphamyosin heavy chain gene promoter in transgenic mice. Journal of Biological Chemistry. 1991;266:24613–24620. [PubMed] [Google Scholar]
- 15.Lewis W, Day BJ, Kohler JJ, Hosseini SH, Chan SS, Green EC, Haase CP, Keebaugh ES, Long R, Ludaway T, Russ R, Steltzer J, Tioleco N, Santoianni R, Copeland WC. Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Laboratory Investigation. 2007;87:326–335. doi: 10.1038/labinvest.3700523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hosseini SH, Kohler JJ, Haase CP, Tioleco N, Stuart T, Keebaugh E, Ludaway T, Russ R, Green E, Long R, Wang L, Eriksson S, Lewis W. Targeted transgenic over-expression of mitochondrial thymidine kinase (TK2) alters mitochondrial DNA (mtDNA) and mitochondrial polypeptide abundance: Transgenic TK2, mtDNA, and antiretrovirals. American Journal of Pathology. 2007;170:865–874. doi: 10.2353/ajpath.2007.060655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Eriksson S. Cloning and characterization of full-length mouse thymidine kinase 2: The N-terminal sequence directs import of the precursor protein into mitochondria. Biochemical Journal. 2000;351(Pt 2):469–476. [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis W, Miller YK, Haase CP, Ludaway T, McNaught J, Russ R, Steltzer J, Folpe A, Long R, Oshinski J. HIV viral protein R causes atrial cardiomyocyte mitosis, mesenchymal tumor, dysrhythmia, and heart failure. Laboratory Investigation. 2005;85:182–192. doi: 10.1038/labinvest.3700222. [DOI] [PubMed] [Google Scholar]
- 19.Cote HC, Yip B, Asselin JJ, Chan JW, Hogg RS, Harrigan PR, O’Shaughnessy MV, Montaner JS. Mitochondrial: Nuclear DNA ratios in peripheral blood cells from human immunodeficiency virus (HIV)-infected patients who received selected HIV antiretroviral drug regimens. Journal of Infectious Diseases. 2003;187:1972–1976. doi: 10.1086/375353. [DOI] [PubMed] [Google Scholar]
- 20.Hosseini SH, Kohler JJ, Haase CP, Tioleco N, Stuart T, Keebaugh E, Ludaway T, Russ R, Green E, Long R, Wang L, Eriksson S, Lewis W. Targeted transgenic over-expression of mitochondrial thymidine kinase (TK2) alters mitochondrial DNA (mtDNA) and mitochondrial polypeptide abundance. American Journal of Pathology. 2007;170:865–874. doi: 10.2353/ajpath.2007.060655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sciacco M, Bonilla E. Cytochemistry and immunocytochemistry of mitochondria in tissue sections. Methods Enzymol. 1996;264:509–521. doi: 10.1016/s0076-6879(96)64045-2. [DOI] [PubMed] [Google Scholar]
- 22.Lewis W, Haase CP, Raidel SM, Russ RB, Sutliff RL, Hoit BD, Samarel AM. Combined antiretroviral therapy causes cardiomyopathy and elevates plasma lactate in transgenic AIDS mice. Laboratory Investigation. 2001;81:1527–1536. doi: 10.1038/labinvest.3780366. [DOI] [PubMed] [Google Scholar]
- 23.Dalakas MC, Illa I, Pezeshkpour GH, Laukaitis JP, Cohen B, Griffin JL. Mitochondrial myopathy caused by long-term zidovudine therapy. New England Journal of Medicine. 1990;322:1098–1105. doi: 10.1056/NEJM199004193221602. see comments. [DOI] [PubMed] [Google Scholar]
- 24.Golden KL, Marsh JD, Jiang Y. Testosterone regulates mRNA levels of calcium regulatory proteins in cardiac myocytes. Hormone and Metabolic Research. 2004;36:197–202. doi: 10.1055/s-2004-814445. [DOI] [PubMed] [Google Scholar]
- 25.Rosenkranz-Weiss P, Tomek RJ, Mathew J, Eghbali M. Gender-specific differences in expression of mRNAs for functional and structural proteins in rat ventricular myocardium. Journal of Molecular and Cellular Cardiology. 1994;26:261–270. doi: 10.1006/jmcc.1994.1029. [DOI] [PubMed] [Google Scholar]
- 26.Llamas B, Belanger S, Picard S, Deschepper CF. Cardiac mass and cardiomyocyte size are governed by different genetic loci on either autosomes or chromosome Y in recombinant inbred mice. Physiol Genomics. 2007;31:176–182. doi: 10.1152/physiolgenomics.00072.2007. [DOI] [PubMed] [Google Scholar]
- 27.Lewis W, Grupp IL, Grupp G, Hoit B, Morris R, Samarel AM, Bruggeman L, Klotman P. Cardiac dysfunction occurs in the HIV-1 transgenic mouse treated with zidovudine. Laboratory Investigation. 2000;80:187–197. doi: 10.1038/labinvest.3780022. [DOI] [PubMed] [Google Scholar]
- 28.Sebastiani M, Giordano C, Nediani C, Travaglini C, Borehi E, Zani M, Feccia M, Mancini M, Petrozza V, Cossarizza A, Gallo P, Taylor RW, d’Amati G. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. Journal of the American College of Cardiology. 2007;50:1362–1369. doi: 10.1016/j.jacc.2007.06.035. [DOI] [PubMed] [Google Scholar]
- 29.Barth E, Stammler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. Journal of Molecular and Cellular Cardiology. 1992;24:669–681. doi: 10.1016/0022-2828(92)93381-s. [DOI] [PubMed] [Google Scholar]
- 30.Bishop JB, Witt KL, Tice RR, Wolfe GW. Genetic damage detected in CD-I mouse pups exposed perinatally to 3′-azido-3′-deoxythymidine and dideoxyinosine via maternal dosing, nursing, and direct gavage. Environmental and Molecular Mutagenesis. 2004;43:3–9. doi: 10.1002/em.10210. [DOI] [PubMed] [Google Scholar]
- 31.Raidel SM, Haase C, Jansen NR, Russ RB, Sutliff RL, Velsor LW, Day BJ, Hoit BD, Samarel AM, Lewis W. Targeted myocardial transgenic expression of HIV Tat causes cardiomyopathy and mitochondrial damage. American Journal of Physiology. Heart and Circulatory Physiology. 2002;282:H1672–H1678. doi: 10.1152/ajpheart.00955.2001. [DOI] [PubMed] [Google Scholar]
- 32.Katz AM. Heart failure: A hemodynamic disorder complicated by maladaptive proliferative responses. Journal of cellular and molecular medicine. 2003;7:1–10. doi: 10.1111/j.1582-4934.2003.tb00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McComsey G, Lonergan JT. Mitochondrial dysfunction: Patient monitoring and toxicity management. Journal of Acquired Immune Deficiency Syndromes. 2004;37:S30–S35. doi: 10.1097/01.qai.0000137004.63376.27. [DOI] [PubMed] [Google Scholar]
- 34.Barthelemy C, Ogier de Baulny H, Diaz J, Cheval MA, Frachon P, Romero N, Goutieres F, Fardeau M, Lombes A. Late-onset mitochondrial DNA depletion: DNA copy number, multiple deletions, and compensation. Annals of Neurology. 2001;49:607–617. [PubMed] [Google Scholar]
- 35.Miro O, Lopez S, Rodriguez de la Concepcion M, Martinez E, Pedrol E, Garrabou G, Giralt M, Cardellach F, Gatell JM, Vilarroya F, Casademont J. Upregulatory mechanisms compensate for mitochondrial DNA depletion in asymptomatic individuals receiving stavudine plus didanosine. Journal of Acquired Immune Deficiency Syndromes. 2004;37:1550–1555. doi: 10.1097/00126334-200412150-00002. [DOI] [PubMed] [Google Scholar]
- 36.d’Amati G, Lewis W. Zidovudine causes early increases in mitochondrial ribonucleic acid abundance and induces ultrastructural changes in cultured mouse muscle cells. Laboratory Investigation. 1994;71:879–884. [PubMed] [Google Scholar]
- 37.Divi RL, Haverkos KJ, Humsi JA, Shockley ME, Tharnire C, Nagashima K, Olivero OA, Poirier MC. Morphological and molecular course of mitochondrial pathology in cultured human cells exposed long-term to Zidovudine. Environmental and Molecular Mutagenesis. 2007;48(34):179–189. doi: 10.1002/em.20245. [DOI] [PubMed] [Google Scholar]
- 38.Abu-Amero KK, Bosley TM. Increased relative mitochondrial DNA content in leucocytes of patients with NAION. British Journal of Ophthalmology. 2006;90:823–825. doi: 10.1136/bjo.2006.090332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang WW, Masayesva B, Zahurak M, Carvalho AL, Rosenbaum E, Mambo E, Zhou S, Minhas K, Benoit N, Westra WH, Alberg A, Sidransky D, Koch W, Califano J. Increased mitochondrial DNA content in saliva associated with head and neck cancer. Clinical Cancer Research. 2005;11:2486–2491. doi: 10.1158/1078-0432.CCR-04-2147. [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Saada A, Eriksson S. Kinetic properties of mutant human thymidine kinase 2 suggest a mechanism for mitochondrial DNA depletion myopathy. Journal of Biological Chemistry. 2003;278:6963–6968. doi: 10.1074/jbc.M206143200. [DOI] [PubMed] [Google Scholar]
- 41.Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chretien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, Rotig A. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (P53R2), causes severe mitochondrial DNA depletion. Nature Genetics. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 42.Lim SE, Copeland WC. Differential incorporation and removal of antiviral deoxynucleotides by human DNA polymerase gamma. Journal of Biological Chemistry. 2001;276:23616–23623. doi: 10.1074/jbc.M101114200. [DOI] [PubMed] [Google Scholar]
- 43.Ferraro P, Nicolosi L, Bernardi P, Reichard P, Bianchi V. Mitochondrial deoxynucleotide pool sizes in mouse liver and evidence for a transport mechanism for thymidine monophosphate. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18586–18591. doi: 10.1073/pnas.0609020103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saada A. Deoxyribonucleotides and disorders of mitochondrial DNA integrity. DNA and Cell Biology. 2004;23:797–806. doi: 10.1089/dna.2004.23.797. [DOI] [PubMed] [Google Scholar]
- 45.Iacobazzi V, Ventura M, Fiermonte G, Prezioso G, Rocchi M, Palmieri F. Genomic organization and mapping of the gene (SLC25A19) encoding the human mitochondrial deoxynucleotide carrier (DNC) Cytogenetics and Cell Genetics. 2001;93:40–42. doi: 10.1159/000056945. [DOI] [PubMed] [Google Scholar]