

Abstract
The melanocortin receptor (MCR) subtype family is a member of the GPCR superfamily and each of them has a different pharmacological profile regarding the relative potency of the endogenous and synthetic melanocortin peptides. Substitution of Trp with DNaI (2′) in -γ-MSH resulted in the loss of binding affinity and potency at hMC4R. However, the molecular mechanism of this ligand selectivity is unclear. In this study, we utilized chimeric receptors and site-directed mutagenesis approaches to investigate the molecular basis of MC4R responsible for peptide [Pro5, DNaI (2′)8]-γ-MSH selectivity. Cassette substitutions of the second, third, fourth, fifth, and sixth TM of the human MC4R (hMC4R) with the homologous regions of hMC1R were constructed and the binding affinity of peptide [Pro5, DNaI (2′)8]-γ-MSH at these chimeric receptors was evaluated. Our results indicate that the cassette substitutions of TM2, TM3, TM4 and TM5 of hMC4R with homologous regions of the hMC1R did not significantly increase peptide [Pro5, DNaI (2′)8]-γ-MSH binding affinity and potency but substitution of the TM6 of the hMC4R with the same region of the hMC1R significantly enhances [Pro5, DNaI (2′)8]-γ-MSH binding affinity and potency. Further site-directed mutagenesis study indicates that four amino acid residues, Phe267, Tyr268, Ile269 and Ser270, in TM6 of the hMC4R may play an important role in [Pro5, DNaI (2′)-γ-MSH selective activity at MC4R.
Keywords: MC4R, γ-MSH, MC1R, Obesity, GPCR, Agonist
1. Introduction
The melanocortin system plays an important role in the regulation of animal physiology and pathophysiology [1–3]. This system consists of melanocortin peptides, melanocortin receptors (MCRs) and endogenous antagonists (agouti and agouti-related protein). The initial transcript of the pro-opiomelanocortin (POMC) gene is a large hormone precursor post-translationally processed into a number of biologically active peptides. These peptides include α-, β-, and γ-melanocyte stimulating hormone (MSH), adrenocorticotropic hormone (ACTH), the lipotropins, and β-endorphin. Melanocortin receptor subtype family is a member of the GPCR superfamily and each subtype has different physiological functions [4–6]. Each of the melanocortin receptor subtypes has a different pattern of tissue expression in the central nervous system and peripheral tissue and each receptor also has its own signature profile regarding the relative potency of the endogenous and synthetic melanocortin peptides. The MC1R is mainly expressed in melanocytes and monocytes and plays an important role in pigmentation and anti-inflammatory reactions [6]. The MC2R is mainly expressed in the adrenal gland and involved in adrenal steroidogenesis and adipocyte lipogenesis [7]. The MC3R and MC4R are expressed in the hypothalamus and play an important role in the regulation of food intake and body weight control [8,9]. The MC5R is expressed in the exocrine gland and is involved in the regulation of the exocrine gland function [4], Agouti and AGRP are endogenous antagonists of melanocortin receptors [10,11].
Energy homeostasis is a highly regulated process involving interacting signals between a variety of anorexigenic and orexigenic peptides, proteins and signaling molecules [12–15]. Genetic studies have highlighted the importance of the central melanocortin system in the control of mammalian energy homeostasis in mice and humans [16,17]. Mutations of the pro-opiomelanocortin gene, MC3R andMC4R gene result in an obese phenotype [18–20]. MC3R and MC4R are expressed in hypothalamic nuclei known to play different role in control of body weight [21–24]. MC3R knockout mice had normal food intake with increased body weight and body fat [23,24]. MC4R knockout mice were hyperphagia with increased body weight and fat mass [23]. Mutations of MC3R and MC4R have also been identified in human obese patients [25–29]. Mutations of hMC4R gene have been identified the most common cause of monogenic obesity because they are detected in almost 5–6% children with early onset severe obesity [30]. Over-expression of the melanocortin antagonists, agouti and AGRP, in transgenic animals also possesses an obese phenotype [31–34].
The melanocortin receptors consist of a single polypeptide featuring seven α-helical transmembrane domains (TMs), an extracellular N-terminus, three extracellular loops, three intracellular loops and an intracellular C-terminus. Many structural features conserved in other G-protein-coupled receptors are found in the melanocortin receptors; the consensus N-linked glycosylation sites near the amino terminus, a palmitoylation site in the COOH-terminal tail, and sites for phosphorylation in the first and third intracellular domains and in the COOH-terminal tail [5,7,21]. All melanocortin receptors contain the conserved DRY motif at the C-terminal end of TM3 and contain a C-terminal cysteine that may function as a fatty acid acylation site. However, the melanocortin receptors lack several features found in most G-protein-coupled receptors; one, or two, cysteine residues in the first and second extracellular loops and prolines found in the fourth and fifth TMs.
All melanocortin peptide agonists contain a His-(L/D)Pbe-Arg-Trp consensus sequence that is considered the “message” sequence, responsible for selectivity and activation of the melanocortin receptors [35,36]. The bioactive conformation of agonists peptides involves a β-turn on the His-(L/D)Phe-Arg-Trp motif [35,36] which is likely to serve as an organizing scaffold to orient the side chains of the message sequence in the best orientation for interaction with the receptor. Extensive structure-function studies on α-MSH have created many new, more potent, and enzyme-resistant analogs of the melanocortin peptides which include synthetic agonists NDP-MSH [Nle4, DPhe7]-α-MSH and Melanotan II (MTII) and antagonists such as SHU9119 and HS204 [37–39]. Recently, it is reported that substitution of a bulky hydrophobic amino acid in Trp8 of the γ-MSH with β-(2-naphthyl)-D-alanine (DNaI) ([Pro5, DNaI(2′)8]-γ-MSH) significantly decreased agonist selectivity at MC4R [40]. However, the molecular basis of MC4R responsible for [Pro5, DNaI (2′)8]-γ-MSH selectivity is unknown. In order to gain insights into the molecular determinants of the hMC4R responsible for [Pro5, DNaI (2′)8]-γ-MSH selectivity, we have utilized chimeric receptor and site-directed mutagenesis approaches to investigate the role of the TMs of MC4R in [Pro5, DNaI (2′)8]-γ-MSH selectivity. The chimeric receptors and single amino acid mutated receptor of the MC4R were constructed and tested. Our results suggest that four amino acid residues of TM6 of MC4R are crucial for [Pro5, DNaI (2′)8]-γ-MSH selectivity at MC4R.
2. Methods and materials
2.1. Reagents
[Nle4-D-Phe7]-MSH (NDP-MSH) was purchased from Peninsula Laboratories (Belmont, CA). 3-Isobutyl-methylxanthine (IBMX) from Sigma, and [125I] NDP-MSH from PerkinElmer Life Sciences (Boston, MA). The HEK-293 cell line was purchased from ATCC (Manassas, VA) and DMEM, lipofectamine from Life Technologies (Rockville MD). [Pro5, DNaI (2′)8]-γ-MSH was synthesized using method previous reported [40].
2.2. Characterization of the pharmacological profile of the peptide [Pro5, DNaI (2′)8]- γ-MSR
2.2.1. Construction o/the melanocortin chimeric receptors
The amino acid sequences of the human (h) MC1R and human (h) MC4R were examined by hydrophobicity plot (Genetics Computer Group, Inc., Madison, WI) and manually by comparing their sequences to a previously published alignment of seven transmembrane G-protein coupled receptor α-helices [41]. The chimeric receptors utilized in these studies are schematically diagramed in Fig. 1. The chimeric receptors were constructed by polymerase chain reaction (PCR) using Pfu polymerase (Stratagene, La Jolla, CA) [42]. The human MC4R served as template. During an initial round of PCR, partial-length receptor fragments were generated. The sequence of one of the PCR primer oligonucleotides consisted of the transmembrane domain of interest coupled to a portion of the extracellular domain required to form the chimeric receptor. The second oligonucleotide primer consisted of either the 5′ or 3′ end of the MC4R. Receptor fragments were separated by agarose gel electrophoresis and used in a second round of PCR in which full-length chimeric receptor constructs were assembled by cycling the appropriate fragments together for 10 cycles prior to adding both 5′ and 3′ receptor primers. The chimeric receptors were subcloned into the eukaryotic expression vector pcDNA 3.1 (Invitrogen, Carlsbad, CA). The entire coding region of the chimeric receptors was sequenced to confirm that the desired sequences will be present and that no sequence errors had been introduced by University of Alabama at Birmingham Sequence Core. The hMC4R/hMC1R (TM2) chimera encoded amino acids Met 1 to Ile83 and Ile103 to Tyr332 of hMC4R and amino acids Cys78 to Val97 of hMC1R. The hMC4R/hMC1R (TM3) chimera encoded amino acids Met 1 to Ile121 and Ile143 to Tyr332 of hMC4R and amino acids Asp117 to Ala137 of hMC1R. The hMC4R/hMC1R (TM4) chimera encoded amino acids Met 1 to Cys172 and Ser188 to Tyr332 of hMC4R and amino acids Ile168 to Tyr182 of hMC1R. The hMC4R/hMC1R (TM5) chimera encoded amino acids Met 1 to val193 and Leu231 to Tyr332 of hMC4R and amino acids leu189 to Val205 of hMC1R. The hMC4R/hMC1R (TM6) chimera encoded amino acids Met 1 to Ile251 and Cys271 to Tyr332 of hMC4R and amino acids Gly248 to Leu266 of hMC1R. The chimeric receptors were then subcloned into the eukaryotic expression vector pCDNA 3.1 (Invitrogen; Carlsbad, CA).
Fig. 1.
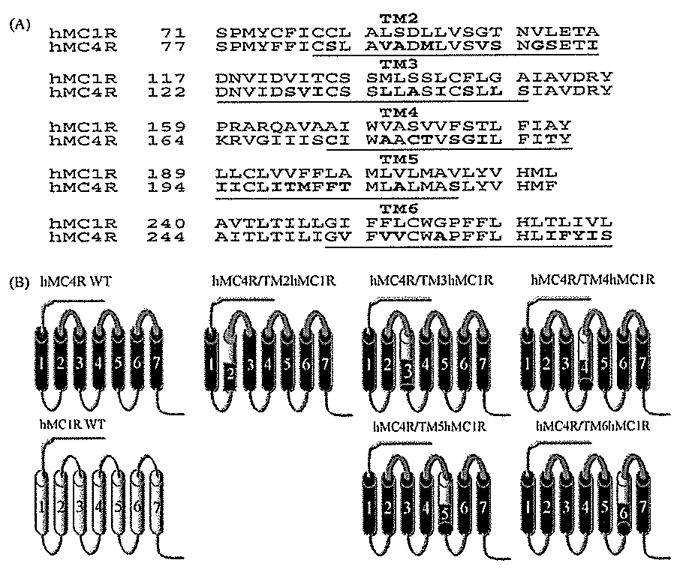
The strategy of making chimeric receptors. Panel A represents the receptor sequences of the hMC4R and hMC1R. Panel B schematically depicts the seven transmembrane structures of the “wild type” (WT) MC4R (drawn with heavy lines) and MC1R (drawn with thin lines) and the structure of the chimeric MC4R with the substituted TMs of the MC1R. Bold font represents the dissimilar residues.
2.2.2. Cell culture and transfection
The HEK-293 cell line was utilized in this study. The cells were cultured in DMEM medium containing 10% bovine fetal serum and HEPES. Cells at 80% confluence were washed twice, and the receptor constructs were transfected into cells using lipofectamine (Life Technologies, Rockville MD). The permanently transfected clonal cell lines were selected by resistance to the neomycin analogue G418 [43].
2.2.3. Binding assays
Binding experiments were performed using the conditions previously described [44], Briefly, after removal of the media, cells were incubated with non-radioligand [DPhe6]-γ-MSH or [Pro5, DNaI (2′)8]-γ-MSH from 10−10 to 10−6 M in 0.5 mL MEM containing 0.2% BSA and 2 × 105 cpm of 125I-NDP-MSH for 1 h. The binding reactions were terminated by removing the media and washing the cells twice with MEM containing 0.2% BSA, The cells were then lysed with 0.2N NaOH, and the radioactivity in the lysate was quantified in an analytical gamma counter (PerkinElmer, Shelton, CT). Nonspecific binding was determined by measuring the amount of 125I-label bound on the cells in the presence of excess 10−6 M unlabeled ligand. Specific binding was calculated by subtracting nonspecifically bound radioactivity from total bound radioactivity. Binding data are reported as IC50.
2.2.4. cAMP assay
Cellular cAMP generation was measured using a competitive binding assay kit (TRK 432, Amersham, Arlington Heights, IL). Briefly, cell culture media was removed, and cells were incubated with 0.5 mL Earle’s Balanced Salt Solution (EBSS), containing the melanocortin agonist [DPhe6]-γ-MSH or [Pro5, DNaI(2′)8]-γ-MSH (10−10–10−6 M), for 1 h at 37 °C in the presence of 10−3 M isobutylmethylxanthine. The reaction was stopped by adding ice-cold 100% ethanol (500 μl/well). The cells in each well were scraped, transferred to a 1.5-mL tube, and centrifuged for 10 min at 1900 × g, and the supernatant was evaporated in a 55 °C water bath with pre-purified nitrogen gas. cAMP content was measured as previously described, according to instructions accompanying the assay kit [45].
2.2.5. Receptor expression by using FACs
To determine whether the receptor proteins are expressed at the cell surface, we generated a chimeric hMC4R with the N terminal fusion to the FLAG (Flag-hMC4R). The FLAG protein is an eight amino acid peptide (Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys) and FLAG-tagged receptor expression vectors have been widely used to purify receptor protein and to determine the protein expression [47]. Cells transfected with the chimeric receptors were harvested using 0.2% EDTA and washed twice with phosphate buffer saline (PBS). Aliquots of 3 × 106 cells were centrifuged and fixed with 3% paraformaldehyde in PBS (pH 7.4). The cells were incubated with 50 μL of 10 μg/ml murine anti-FLAG M1 monoclonal antibody (Sigma, Catalog No. 316) in incubation buffer for 45 min. Under this condition the primary antibody binds only to receptors located at the cell surface. The cells were collected by centrifugation and washed three times with incubation buffer. The cell pellets were suspended in 100 μL of incubation buffer containing CY™3-conjugated Affinity Pure Donkey Anti-Mouse Ig G (ImmunoResearch Lab, Inc., West Grove, PA) and incubated at room temperature for 30 min. Flow cytometry was performed on a fluorescence-activated cell sorter (Becton Dickinson FACStar plus six parameter cytometer/sorter with a dual Argon ion laser, San Jose, CA) [46]. The results were analyzed using the software CellQuest (Beckton-Dickinson Immunocytometry Systems, San Jose, CA).
2.3. Statistical analysis
Each experiment was performed in duplicate three separate times. The mean value of the dose–response data of binding and cAMP production was fit to a sigmoid curve with a variable slope factor using non-linear squares regression analysis (Graphpad Prism, Graphpad Software, San Diego, CA}. Data are expressed as mean ± S.E. Significant difference was assessed by one-way ANOVA with P < 0.05 considered to be statistically significant.
3. Results
3.1. Peptide [DPhe6]-γ-MSH and [Pro5, DNaI (2′]8-γ-MSH binding and activity at MC4R and MC1R
Peptide [DPhe6]-γ-MSH and [Pro5, DNaI (2′)8]-γ-MSH are γ-MSH analogues (Table 1). However, the binding affinities and potencies of these two analogues at MC1R and MC4R are different [40]. To determine [DPhe6]-γ-MSH and [Pro5, DNaI (2′)8]-γ-MSH binding affinity at wild type hMC4R and hMC1R (hMC4R-WT and hMC1R-WT), the ability of [DPhe6]-γ-MSH and [Pro5, DNaI (2′)8-γ-MSH to displace 125I NDP-MSH binding at hMC4R-WT and hMC1R-WT were first examined as shown in Fig. 2. [DPhe6]-γ-MSH dose-dependency displaces 125I NDP-MSH binding at the hMC1R WT and hMC4R WT (Fig. 2A). To examine the ability of [DPhe6]-γ-MSH to activate hMC1R or MC4R, [DPhe6]-γ-MSH stimulated cAMP production was determined. Consistent with binding data, peptide [DPhe6]-γ-MSH dose-dependently increased cAMP generation at the hMC1R-WT and hMC4R WT (Fig. 2B). [Pro5, DNaI (2′)8]-γ-MSH dose-dependently displaces 125I NDP-MSH binding at the hMC1R WT and hMC4R-WT but binding affinity is significantly decreased at hMC4R (Fig, 2C). To examine the ability of [Pro5, DNaI (2′)8]-γ-MSH to activate hMC1R or hMC4R, [Pro5, DNaI (2′)8-γ-MSH stimulated cAMP production was determined. Consistent with binding data, peptide [Pro5, DNaI (2′)8]-γ-MSH dose-dependently increased cAMP generation at the hMC1R-WT but its potency was significantly reduced at hMC4R. [Pro5, DNaI (2′)8]-γ-MSH becomes a partial agonist at hMC4R (Fig. 2D), Our results indicate that [DPhe6]-γ-MSH is a non selective agonist for the MC1R and MC4R but [Pro5, DNaI (2′)8]-γ-MSH becomes a partial agonist at the MC4R which is consistent with the previous report [40]. Their Ki and EC50 are shown in Table 2.
Table 1.
Sequences of γ-MSH, [DPhe6]-γ-MSH, and [Pro5, DNaI(2′)8]-γ-MSH
| Sequences of γ-MSH analogues | |
|---|---|
| γ-MSH | H-Tyr-Val-Nle-Gly-Pro-Phe-Arg-Trp-Asp-Arg-Phe-Gly-NH2 |
| [DPhe6]-γ-MSH | H-Tyr-Val-Nle-Gly-Pro-DPhe-Arg-Trp-Asp-Arg-Phe-Gly-NH2 |
| [Pro5, DNaI(2′)8]-γ-MSH | H-Tyr-Val-Nle-Gly-Pro-Phe-Arg-DNaI(2′)-Asp-Arg-Phe-Gly-NH2 |
The bold letter s represent key residues for receptor binding and signaling
Fig. 2.
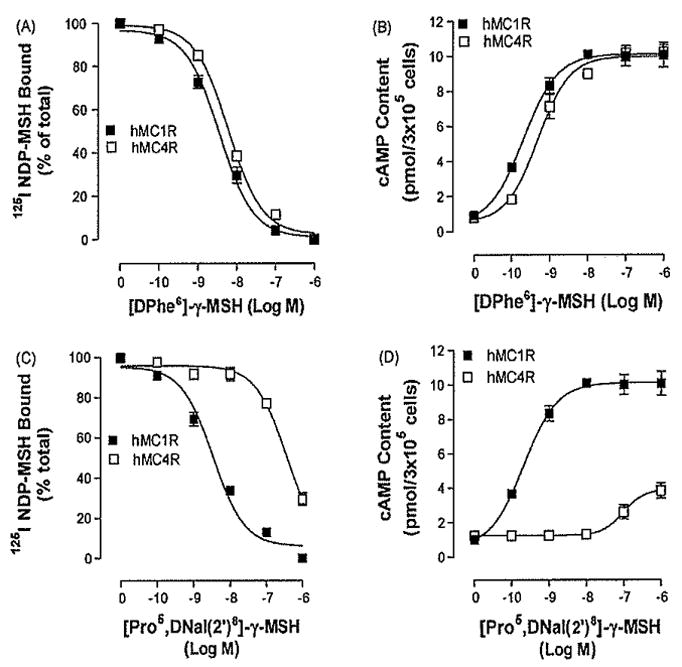
Binding affinities and potencies of peptide [DPhe6]-γ-MSH and [Pro5, DNaI (2′)8]-γ-MSH and NDP-MSH at the wild type hMC4R and hMC1R. Panel A depicts the binding affinity of [DPhe6]-γ-MSH as determined by inhibition of 125I NDP-MSH binding. Panel B represents the ability of [DPhe6]-γ-MSH to stimulate the production of intracellular cAMP. Panel C depicts the binding affinity of [Pro5, DNaI (2′)8]-γ-MSH as determined by inhibition of 125I NDP-MSH binding. Panel D represents the ability of [Pro5, DNaI (2′)8]-γ-MSH to stimulate the production of intracellular cAMP. Data points represent the mean ± S.E.M. of at least three independent experiments.
Table 2.
Effect of unlabled ligands on 125I-NDP-MSH binding, at the HEK cells transfected with the chimeric hMC4 receptors. All determinations were on cells transfected with wild type or chimeric receptors The receptor expression was determined by FLAG-tagged receptor expression and expressed as % of wild type receptor protein. Ki values for [DPhe6]-γ-MSH and [Pro5, DNaI(2′)8]-γ-were determined from displacement of 125I-NDP-MSH binding, as described under “Section 2”. The data shown are mean ± S.E.M of at least three independant experiments.
| Receptor expression(%WT) |
125I-NDP-MSHKi (nM) |
cAMP production EC50 (NM) |
|||
|---|---|---|---|---|---|
| [DPhe6]-γ-MSH | [Pro5, DNaI(2′)8]-γ-MSH | [DPhe6]-γ-MSH | [Pro5, DNaI(2′)8]-γ-MSH | ||
| hMC4RWT | 100 | 4.1±0.2 | 477±19.4 | 35± | partial agonist |
| hMC4R/TM2 hMC1R | 98±12.2 | 3.7±0.4 | 459±11.5 | 2.9±0.3 | partial agonist |
| hMC4R/TM3 hMC1R | 92±15.7 | 3.9±0.5 | 506±8.3 | 3.3±0.5 | partial agonist |
| hMC4R/TM4 hMC1R | 97.11.2± | 4.8±0.6 | 421±12.4 | 3.1±0.3 | partial agonist |
| hMC4R/TM5 hMC1R | 94±16.4 | 2.9±0.2 | 569±13.2 | 1.7±0.4 | partial agonist |
| hMC4R/TM6 hMC1R | 96±12.2 | 3.9±0.4 | 79±9.7* | 3.2±0.3 | 116±65* |
| hMC1RWT | 100 | 2.5±0.2 | 15.9±3.8 | 2.3±0.5 | 231±8.2 |
p < 0.05
3.2. Effects of substitutions of the transmembrane domain of the hMC4R with the corresponding regions of the hMC1R on [DPhe6]-γ-MSH and (Pro5, DNaI (2′)8]-γ-MSH specific bindings and signaling
To investigate the molecular determinant of hMC4R responsible for [Pro5, DNaI (2′)8]-γ-MSH selectivity, a domain-exchange strategy was used to localize regions of the hMC4R responsible for peptide [Pro5, DNaI (2′)8]-γ-MSH activity. [Pro5, DNaI (2′)8]-γ-MSH is a full agonist at hMC1R but partial agonist at MC4R. The advantage of using the hMC4R as a template is that replacement of hMC4R TMs with the corresponding region of hMC1R may increase [Pro5, DNaI (2′)8]-γ-MSH binding affinity and potency if the specific TM of the MC1R is involved in [Pro5, DNaI (2′)8]-γ-MSH selectivity. Cassette substitutions of the second, third, fourth, fifth, and sixth TMs of the hMC4R with homologous regions of the hMC1R were constructed. The first, and seventh TMs were not chosen for investigation because our previous data suggested that 1TM and 7TM of melanocortin receptors were not important in ligand binding [43,44,48]. In order to reduce the possibility of the receptor tertiary structure alternation by the entire TM domain exchange, upper half TM region of the hMC4R was replaced with the corresponding region of the hMC1R (Fig. 1).
To determine whether chimeric receptor proteins are expressed at the cell surface and to quantify receptor expression level, the antigenic epitope FLAG sequence was inserted into the NH2 terminus of hMC4R-WT or chimeric receptors using polymerase chain reaction [47]. Our results indicate that the FLAG signal was detected by FACs at hMC4R-WT and chimeric receptors. The expression levels of all chimeric receptors were not significantly different from that of wild type receptor (Table 2).
To determine whether the receptor domain exchange alters receptor function [DPhe6]-γ-MSH binding affinity and potency were evaluated at these chimeric receptors. Our results indicate that unlabelled [DPhe6]-γ-MSH dose-dependently displaces 125I-NDP-MSH binding at these chimeric receptors and all chimeric receptors possess high [DPhe6]-γ-MSH binding affinity (Figs. 3 and 4Figs. 3A and 4A). Consistent with the binding data, our results also indicate that [DPhe6]-γ-MSH dose dependently increased cAMP generation at these chimeric receptors (Figs.3B and 4B), The domain exchange did not significantly alter [DPhe6]-γ-MSH potency and suggest that the tertiary structures of the chimeric receptors were not grossly disrupted and that the normal function of the receptor was retained. Their Ki and EC50 values are shown in Table 2. NDP-MSH has similar pharmacological profile at these chimeric receptors compared to that of [DPhe6]- γ-MSH (data are not shown).
Fig. 3.
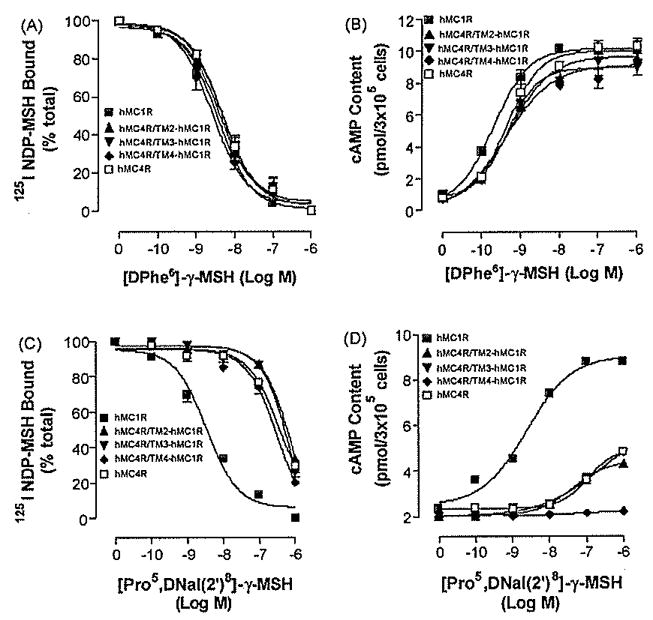
Binding affinities and potencies of [DPhe6]-γ-MSH and [Pro5, DNaI (2′)a]-γ-MSH at hMC4R/TM2hMC1R, hMC4R/TM3hMC1R and hMC4R/TM3hMC1R. Panel A depicts the binding affinity of [DPhe6]-γ-MSH as determined by inhibition of 125INDP-MSH binding at the chimeric receptors. Panel B represents the ability of [DPhe6]-γ-MSH to stimulate the production of intracellular cAMP at the chimeric receptors. Panel G depicts the binding affinity of [Pro5, DNaI (2′)8]-γ-MSH as determined by inhibition of 125INDP-MSH binding at the chimeric receptors. Panel D represents the ability of [Pro5, DNaI (2′)8]-γ-MSH to stimulate the production of intracellular cAMP at the chimeric receptors. Data points represent the mean ± S.E.M. of at least three independent experiments.
Fig. 4.
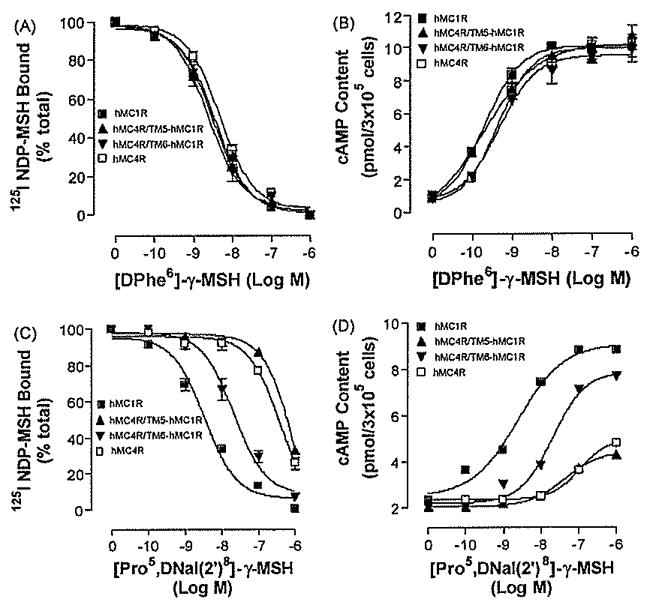
Binding affinities and potencies of [DPhe6]-γ-MSH and [Pro5, DNaI(2′)8]- γ-MSH at hMC4R/TM5hMC1R and hMC4R/TM6hMC1R. Panel A depicts the binding affinity of [DPhe6]- γ-MSH as determined by inhibition of 125INDP-MSH binding at the chimeric receptors. Panel B represents the ability of [DPhe6]-γ-MSH to stimulate the production of intracellular cAMP at the chimeric receptors. Panel C depicts the binding affinity of [Pro5, DNaI (2′)8]- γ-MSH as determined by inhibition of 125I NDP-MSH binding at the chimeric receptors. Panel D represents the ability of [Pro5, DNaI (2′)8]- γ-MSH to stimulate the production of intracellular cAMP at the chimeric receptors. Data points represent the mean ± S.E.M. of at least three independent experiments.
After confirming that substitution of the receptor domains did not alter receptor tertiary structure and function, we then investigated which region of the hMC4R is responsible for [Pro5, DNaI (2′)8]- γ-MSH selective binding and potency, HEK cells expressing these chimeric receptors were incubated with labeled 1251 NDP-MSH and various concentrations of unlabelled [Pro5, DNaI (2′)8]- γ-MSH and its binding affinity was assessed. Our results indicate that substitutions of TM2, TM3, TM4 and TM5 of the hMC4R with the corresponding regions of the hMC1R (hMC4R/TM2hMC1R, hMC4R/TM3hMC1R, hMC4R/TM4hMC1R and hMC4R/TM5hMC1R) did not significantly increase peptide [Pro5, DNaI (2′)8]-γ-MSH binding affinity (Figs. 3C and 4C). However, substitution of the hMC4R TM6 with the corresponding region of the hMC1R (hMC4R/TM6hMC1R) significantly increased peptide [Pro5, DNaI (2′)8]- γ-MSH binding affinity (Fig. 4C). The IC50 values of [Pro5, DNaI (2′)8] -γ-MSH are summarized in Table 2. To determine whether the substitutions of the hMC4Rwith the corresponding regions of the hMC1R will also increase [Pro5, DNaI (2′)8]- γ-MSH potency [Pro5, DNaI (2′)8]- γ-MSH stimulated cAMP production were determined. Our results indicate that hMC4R/TM2hMC1R, hMC4R/TM3hMC1R, hMC4R/TM4hMC1R and hMC4R/TM5hMC1R did not significantly increase [Pro5, DNaI (2′)8]- γ-MSH potency (Fig. 3D and 4D). However, substitutions of the hMC4R TM6 with the corresponding regions of the hMC1R (hMC4R/TM5hMC1R) did significantly increase peptide [Pro5, DNaI (2′)8]- γ-MSH potency which is consistent with the binding results (Fig. 4D). Their Ki, and EC50 are shown in Table 2.
3.3. Mutations of non-conserved amino acid residues in TM6 of the hMC4R for peptide [Pro5, DNaI (2′)8]- γ-MSH binding and signaling
Substitution of TM6 of the hMC4R with the corresponding region of the hMC1R significantly increases peptide [Pro5, DNaI (2′)8]- γ-MSH binding affinity and potency suggest that non-conserved amino acid residues of the TM6 of the hMC4R may be involved in peptide [Pro5, DNaI (2′)8]- γ-MSH specific binding. Five amino acid residues of the TM6 are identified to be non-conserved between MC4R and MC1R. To determine whether these amino acid residues are involved in peptide [Pro5, DNaI (2′)8]- γ-MSH specific binding and activity, these residues were individually mutated with the corresponding amino acid residues of hMC1R and evaluated. Our results indicate that these mutant receptors were expressed at cell surface and the expression levels showed no significant variation compared with that of wild type receptor (Table 3), The binding affinities and potencies of DPhe-γ-MSH at these mutated receptors are similar to that of hMC4R (Fig. 5A and B). The binding affinity of [Pro5, DNaI (2′)8]- γ-MSH at mutations, Ile266Ala, Phe267Leu, and Ser270Leu was also similar to that of the hMC4R-WT but the binding affinity of [Pro5, DNaI (2′)8]- γ-MSH at mutations Tyr268Ile, Ile269Val was increased (Fig. 5C). Consistent with the binding results, the mutations, Ile266Ala, Phe267Leu, and Ser270Leu did not significantly increase [Pro5, DNaI (2′)8]- γ-MSH-mediated cAMP production but the mutations of Tyr268Ile, Ile269Val increased [Pro5, DNaI (2′)8]-γ-MSH potency (Fig. 5D). Their Ki and EC50 are shown in Table 3.
Table 3.
Mutations of the non-conserved residues in TM6 of the hMC4R on DNaI(2′)8-γ-MSH binding and CAMP production. All determinations were on cells transfected with wild type or mutated receptors. Ki values for [Pro5, DNaI(2′)8]-γ-MSH were determined from displacement of 125I-NDP-MSH binding, as described under “Section 2”. The data shown are mean ± S.E.M of at least three independant experiments.
| Receptor expression | [Pro5, DNaI(2′)8]-γ-MSH |
|||
|---|---|---|---|---|
| Ki (nM) | EC50(nM) | Emax(%) | ||
| hMC1RWT | 100 | 2.1±0.1 | 0.3±0.02 | 100 |
| I266T | 95±15.4 | 133.3±18.2 | Partial agonist | 33 |
| F267L | 96±12.2 | 169.3±21.2 | Partial agonist | 32 |
| Y268I | 94±3.5 | 154.2±11.1 | Partial agonist | 45 |
| I269V | 92±7.7 | 212.9±30.3 | Partial agonist | 51 |
| S270L | 93±8.1 | 124.3±12.5 | Partial agonist | 30 |
| hMC4-WT | 100 | 126.6±13.1 | Partial agonist | 35 |
Fig. 5.
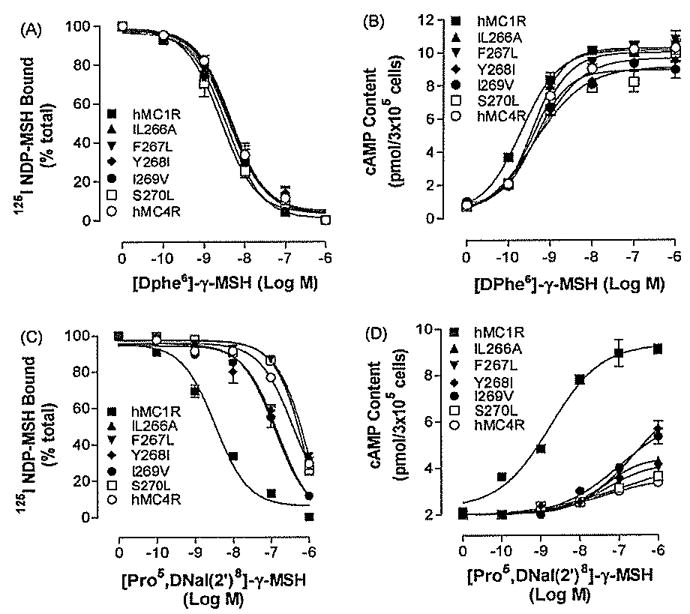
Binding affinities and potencies of [DPhe6]- γ-MSH and [Pro5, DNaI (2′)8]- γ-MSH at mutation of the non-conserved TM6 amino acid residues of hMC4R, Panel A depicts the binding affinity of [DPhe6]-γ-MSH as determined by inhibition of 125I NDP-MSH binding at the mutated receptors. Panel B represents the ability of [DPhe6]- γ-MSH to stimulate the production of intracellular cAMP at the mutated receptors. Pane C depicts the binding affinity of [Pro5, DNaI (2′)8]- γ-MSH as determined by inhibition of125I NDP-MSH binding at the mutated receptors. Panel D represents the ability of [Pro5, DNaI (2′)8]- γ-MSH to stimulate the production of intracellular cAMP at the mutated receptors. Data points represent the mean ± S.E.M. of at least three independent experiments.
Single mutation of hMC4R did not dramatically increase [Pro5, DNaI (2′)8]- γ-MSH binding affinity and potency, we therefore examined the combination of four amino acid mutations in TM6 on [Pro5, DNaI (2′)8]- γ-MSH selectivity. The multiple amino acid mutations, including Phe267Leu, Tyr268Ile, Ile269Val and Ser270Leu, were created and tested. Our results indicate that this multiple amino acid mutation of TM6 did result in an increase of [Pro5, DNaI (2′)8]- γ-MSH selectivity at MC4R. The binding affinity of [Pro5, DNaI (2′)8]- γ-MSH at this multiple mutations was significantly increased (Fig. 6A). Consistent with the binding results, this multiple mutations did significantly increase [Pro5, DNaI (2′)8]- γ-MSH-mediated cAMP production (Fig. 6B), suggesting that these four amino acid residues maybe involved in [Pro5, DNaI (2′)8]-γ-MSH selectivity.
Fig. 6.
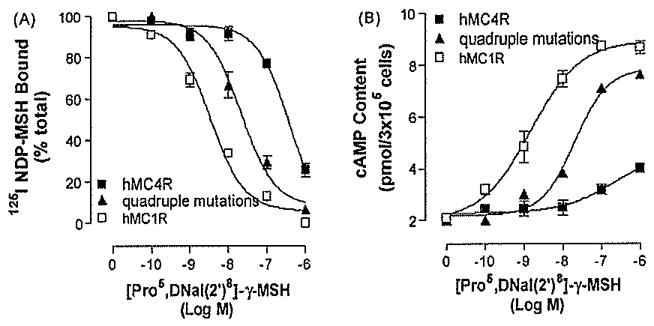
Binding affinity and potency of [Pro5, DNaI (2′)8]- γ-MSH at multiple mutations of hMC4R. Panel A depicts the binding affinity of [Pro5, DNaI (2′)8]- γ-MSH as determined by inhibition of 125I NDP-MSH binding at this mutated receptor. Panel B represents the ability of [Pro5, DNaI (2′)8]- γ-MSH to stimulate the production of intracellular cAMP at the mutated receptor. Data points represent the mean ± S.E.M. of at least three independent experiments.
4. Discussion
We have identified that several key amino acid residues of TM6 of MC4R are crucial for [Pro5, DNaI (2′)8]- γ-MSH selectivity at the hMC4R by utilizing chimeric receptors and site-directed mutagenesis.
Melanocortin agonists bind in a β-turn conformation that organizes the message sequence (His-L/DPhe-Arg-Trp) in an optimal arrangement for binding and activation of the receptors. Extensive pharmacological studies have demonstrated that the tripeptide D-Phe-Arg-Trp in NDP-MSH is important not only for ligand binding but also for receptor activation at MC1R, MC3R and MC4R [44,49,50]. When Phe, Arg and Trp in MSH were substituted with alanine, the peptide binding affinities and potencies were significantly decreased at these receptors [51–53]. However, substitution of DPhe in position 7 of MTII with a D-(2′-naphthyl)-D-alanine (D-NaI (2′)) (SHU9119) and Phe in position 6 of γ-MSH with DNaI(2′) switches peptide from agonist to antagonist at MC3R and MC4R but retain agonist activity at MC1R and MC5R [39,40]. This information provides an initial estimate of the main contacts between peptides and receptor.
In the absence of a crystal structure of the MC4R, free or in complex with a ligand, little is known about the exact orientation of MC4 agonists in their binding pocket. Homology models of GPCRs based on bovine rhodopsin in combination with site-directed mutagenesis have been successfully used in the past to provide a structural framework for both ligand binding and functional studies. Accumulated evidence also indicates that TM3 of hMC3R and hMC4R was crucial for DNaI (2′)7-SHU9119 ligand antagonist selectivity [50,54].
The Trp9 (super) residue of MSH was identified to be crucial for peptide binding affinity at hMC4R [52]. The Ala substitution resulted in a significant decrease of ligand binding affinity at hMC4R [44,55]. The bulky lipophilic amino acid derivative c-Hex Ala9 resulted in a large decrease in affinity but still retained efficacy [55], implying that the aromatic nature of Trp9-α-MSH is crucial for ligand binding affinity at hMC4R [44,50]. Pharmacological studies indicate that the core sequence His-Phe-Arg-Trp of γ-MSH is crucial for -γ-MSH binding affinity and potency although -γ-MSH has seven amino acid residues different from α-MSH. The last four amino acids in the C-terminal region of γ-MSH are not important for its biological activity and selectivity at melanocortin receptors [56], Substitution of the residue Trp8 of the -γ-MSH with D-(2′-naphthyl)-D-alanine (D-NaI (2′)8), [Pro5, DNaI (2′)8]- γ-MSH, significantly decreases its binding affinity and potency at MC4R [40], Pogozheva et al. have modeled both the active and inactive states of hMC4R and propose that TMs movement accompany MC4R activation [57], TM6 shifts outward and undergoes counterclockwise movement, changing the position of TM6 into the binding pocket. Several residues of the hMC4R have been identified to be involved in NDP-MSH-induced receptor activation [55,57,58], However, substitution of Trp with DNaI (2) in -γ-MSH [Pro5, DNaI (2′)8]- γ-MSH resulted in a significantly reduced MC4R activation but not for hMC1R. The aim of this study was to explore the molecular mechanism of MC4R responsible for this ligand selectivity using chimeric receptor and site-directed mutagenesis. The hMC4R and the hMC1R share a number of structural, functional and pharmacologic similarities. In the putative TM regions, the hMC4R and hMC1R show 67% identity. [Pro5, DNaI(2′)8]- γ-MSH is a potent agonist at MC1R but significantly decreased binding affinity and potency at MC4R. We speculate that the unique residues of the hMC4R might play an important role in this ligand selectivity. To determine which unique residue of the hMC4R is involved in this ligand selectivity, a chimeric receptor approach between hMC4R and hMC1R was utilized. We anticipate that the chimeric receptor using MC4R as a template will gain [Pro5, DNaI (2′)8]- γ-MSH binding affinity instead of using the chimeric receptor of the MC1R as a template losing peptide binding affinity. Our results demonstrate that the replacement of MC4R TM6 by MC1R TM6 results in increased binding and potency of [Pro5, DNaI (2′)8]- γ-MSH. This led us to the hypothesis that the D-(2′-naphthyl)-D-alanine (D-NaI (2′)8) in [Pro5, DNaI (2′)8]- γ-MSH may interfere in the TM6 shift outward and counterclockwise movement, physically hindering the con formational changes necessary to induce receptor activation. Comparison of the amino acid sequences of TM6 between the hMC4R and the hMC1R revealed that five amino acid residues are different. They are residues, Ile266, Phe267, Tyr268, Ile269 and Ser270, which could potentially confer affinity characteristics of [Pro5, DNaI (2′)8]- γ-MSH. We examined the roles of these residues on ligand selectivity using a site directed mutagenesis study. However, our results indicate that single amino acid substitution of these residues did not significantly increase ligand efficacy, suggesting that multiple amino acid residues of TM6 may be important for this ligand selectivity. We then make a multiple amino acid mutation, including Phe267, Tyr268, Ile269 and Ser270 in TM6. These receptor residues at hMC4R were replaced with the corresponding residues of hMC1R and tested. Our results support our hypothesis that these residues are crucial for [Pro5, DNaI (2′)8]- γ-MSH selectivity at MC4R. The replacements of these residues increased [Pro5, DNaI (2′)8]- γ-MSH selectivity at MC4R. The possible mechanism is that these mutations remove the steric hindrances of TM6 movement and induce receptor activation. However, substitution of TM6 of hMC4R with the corresponding region of hMC1R neither bring binding nor potency to the same level of hMC1R, suggesting that other areas close to this site may play a role and this is important for future potential investigation.
In conclusion, we have utilized chimeric receptor and site-directed mutagenesis approaches to identify critical amino acid residues of MC4R responsible for [Pro5, DNaI (2′)8]- γ-MSH selectivity and potency. We believe that identification of the molecular basis of hMC4R responsible for different ligand-mediated receptor binding and signaling will provide valuable information of the molecular mechanism of MC4R in obesity development and provide important information for rational design of the MC4R potent and selective agonist which can be used for obesity treatment in the future. Our results provide the first evidence that the TM6 of the hMC4R is critical for [Pro5, DNaI (2′)8]- γ-MSH selectivity and potency.
Acknowledgments
This work has been supported by NIH Grants R03 HD047312-01A1, 1 R03 HD058789-01 (Yang, Y-K) and U.S.P.H.S., N.I.H, grant DK 17420 (Hruby, V.J).
References
- 1.Adan RA, Hillebrand JJ, De Rijke C, Nijenhuis W, Vink T, Garner KM, et al. Melanocortin system and eating disorders. Ann N Y Acad Sci. 2003;994:267–74. doi: 10.1111/j.1749-6632.2003.tb03189.x. [DOI] [PubMed] [Google Scholar]
- 2.Yang YK, Harmon CM. Recent developments in our understanding of melanocortin system in the regulation of food intake. Obes Rev. 2003;4:239–48. doi: 10.1046/j.1467-789x.2003.00104.x. [DOI] [PubMed] [Google Scholar]
- 3.Cone RD. Studies on the physiological functions of the melanocortin system. Endocr Rev. 2006;27:736–49. doi: 10.1210/er.2006-0034. [DOI] [PubMed] [Google Scholar]
- 4.Gantz I, Shimoto Y, Konda Y, Miwa H, Dickinson CJ, Yamada T. Molecular cloning, expression, and characterization of a fifth melanocortin receptor. Biochem Biophys Res Commun. 1994;200:1214–20. doi: 10.1006/bbrc.1994.1580. [DOI] [PubMed] [Google Scholar]
- 5.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, et al. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem. 1993;268:15174–9. [PubMed] [Google Scholar]
- 6.Gantz I, Yamada T, Tashiro T, Konda Y, Shimoto Y, Miwa H, et al. Mapping of the gene encoding the melanocortin-1 (alpha-melanocyte stimulating hormone) receptor (MC1R) to human chromosome 16q24.3 by fluorescence in situ hybridisation. Genomics. 1994;19:394–5. doi: 10.1006/geno.1994.1080. [DOI] [PubMed] [Google Scholar]
- 7.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–51. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 8.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–8. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 9.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–41. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 10.Yang YK, Dickinson C, Lai YM, Li JY, Gantz I. Functional properties of an agouti signaling protein variant and characteristics of its cognate radioligand. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1877–86. doi: 10.1152/ajpregu.2001.281.6.R1877. [DOI] [PubMed] [Google Scholar]
- 11.Yang YK, Thompson DA, Dickinson CJ, Wilken J, Barsh GS, Kent SB, et al. Characterization of agouti-related protein binding to melanocortin receptors. Mol Endocrinol. 1999;13:148–55. doi: 10.1210/mend.13.1.0223. [DOI] [PubMed] [Google Scholar]
- 12.Woods SC, Schwartz MW, Baskin DG, Seeley RJ. Food intake and the regulation of body weight. Annu Rev Psychol. 2000;51:255–77. doi: 10.1146/annurev.psych.51.1.255. [DOI] [PubMed] [Google Scholar]
- 13.Woods SC, Seeley RJ. Adiposity signals and the control of energy homeostasis. Nutrition. 2000;16:894–902. doi: 10.1016/s0899-9007(00)00454-8. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 15.Benoit S, Schwartz M, Baskin D, Woods SC, Seeley RJ. CNS melanocortin system involvement in the regulation of food intake. Horm Behav. 2000;37:299–305. doi: 10.1006/hbeh.2000.1588. [DOI] [PubMed] [Google Scholar]
- 16.Fisher SL, Yagaloff KA, Burn P. Melanocortin and leptin signaling systems: central regulation of catabolic energy balance. J Recept Signal Transduct Res. 1999;19:203–16. doi: 10.3109/10799899909036646. [DOI] [PubMed] [Google Scholar]
- 17.Fisher SL, Yagaloff KA, Burn P. Melanocortin-4 receptor: a novel-signalling pathway involved in body weight regulation. Int J Obes Relat Metab Disord. 1999;23(Suppl 1):54–8. doi: 10.1038/sj.ijo.0800796. [DOI] [PubMed] [Google Scholar]
- 18.Zemel MB, Shi H. Pro-opiomelanocortin (POMC) deficiency and peripheral melanocortins in obesity. Nutr Rev. 2000;58:177–80. doi: 10.1111/j.1753-4887.2000.tb01857.x. [DOI] [PubMed] [Google Scholar]
- 19.Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe early onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155–7. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 20.Challis BG, Coll AP, Yeo GS, Pinnock SB, Dickson SL, Thresher RR, et al. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3–36) Proc Natl Acad Sci U S A. 2004;101:4695–700. doi: 10.1073/pnas.0306931101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, et al. Molecular cloning of a novel melanocortin receptor. J Biol Chem. 1993;268:8246–50. [PubMed] [Google Scholar]
- 22.Lembertas AV, Perusse L, Chagnon YC, Fisler JS, Warden CH, Purcell-Huynh DA, et al. Identification of an obesity quantitative trait locus on mouse chromosome 2 and evidence of linkage to body fat and insulin on the human homologous region 20q. J Clin Invest. 1997;100:1240–7. doi: 10.1172/JCI119637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, et al. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 24.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, et al. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–21. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 25.Tao YX. Functional characterization of novel melanocortin-3 receptor mutations identified from obese subjects. Biochim Biophys Acta. 2007;1772:1167–74. doi: 10.1016/j.bbadis.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 26.Santoro N, Perrone L, Cirillo G, Raimondo P, Amato A, Brienza C, et al. Effect of the melanocortin-3 receptor C17A and G241A variants on weight loss in childhood obesity. Am J Clin Nutr. 2007;85:950–3. doi: 10.1093/ajcn/85.4.950. [DOI] [PubMed] [Google Scholar]
- 27.Rutanen J, Pihlajamaki J, Vanttinen M, Salmenniemi U, Ruotsalainen E, Kuulasmaa T, et al. Single nucleotide polymorphisms of the melanocortin-3 receptor gene are associated with substrate oxidation and first-phase insulin secretion in offspring of type 2 diabetic subjects. J Clin Endocrinol Metab. 2007;92:1112–7. doi: 10.1210/jc.2006-1201. [DOI] [PubMed] [Google Scholar]
- 28.Lee YS, Poh LK, Kek BL, Loke KY. The role of melanocortin 3 receptor gene in childhood obesity. Diabetes. 2007;56:2622–30. doi: 10.2337/db07-0225. [DOI] [PubMed] [Google Scholar]
- 29.Feng N, Young SF, Aguilera G, Puricelli E, Adler-Wailes DC, Sebring NG, et al. Co-occurrence of two partially inactivating polymorphisms of MC3R is associated with pediatric-onset obesity. Diabetes. 2005;54:2663–7. doi: 10.2337/diabetes.54.9.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–95. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 31.Wilson BD, Ollmann MM, Barsh GS. The role of agouti-related protein in regulating body weight. Mol Med Today. 1999;5:250–6. doi: 10.1016/s1357-4310(99)01471-9. [DOI] [PubMed] [Google Scholar]
- 32.Wirth MM, Giraudo SQ. Agouti-related protein in the hypothalamic paraventricular nucleus: effect on feeding. Peptides. 2000;21:1369–75. doi: 10.1016/s0196-9781(00)00280-1. [DOI] [PubMed] [Google Scholar]
- 33.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, et al. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–8. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 34.Butler AA, Cone RD. The melanocortin receptors: lessons from knockout models. Neuropeptides. 2002;36:77–84. doi: 10.1054/npep.2002.0890. [DOI] [PubMed] [Google Scholar]
- 35.Holder JR, Haskell-Luevano C. Melanocortin ligands: 30 years of structure-activity relationship (SAR) studies. Med Res Rev. 2004;24:325–56. doi: 10.1002/med.10064. [DOI] [PubMed] [Google Scholar]
- 36.Irani BG, Holder JR, Todorovic A, Wilczynski AM, Joseph CG, Wilson KR, et al. Progress in the development of melanocortin receptor selective ligands. Curr Pharmaceut Des. 2004;10:3443–79. doi: 10.2174/1381612043382891. [DOI] [PubMed] [Google Scholar]
- 37.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, et al. 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone: a highly potent alpha-melanotropin with ultralong biological activity. Proc Natl Acad Sci U S A. 1980;77:5754–8. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sawyer TK, Hruby VJ, Darman PS, Hadley ME. [half-Cys4, half-Cys10]-alpha-Melanocyte-stimulating hormone: a cyclic alpha-melanotropin exhibiting superagonist biological activity. Proc Natl Acad Sci U S A. 1982;79:1751–5. doi: 10.1073/pnas.79.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schioth HB, Muceniece R, Mutulis F, Prusis P, Lindeberg G, Sharma SD, et al. Selectivity of cyclic [D-NaI7] and [D-Phe7] substituted MSH analogues for the melanocortin receptor subtypes. Peptides. 1997;18:1009–13. doi: 10.1016/s0196-9781(97)00079-x. [DOI] [PubMed] [Google Scholar]
- 40.Cai M, Mayorov AV, Cabello C, Stankova M, Trivedi D, Hruby VJ. Novel 3D pharmacophore of alpha-MSH/gamma-MSH hybrids leads to selective human MC1R and MC3R analogues. J Med Chem. 2005;48:1839–48. doi: 10.1021/jm049579s. [DOI] [PubMed] [Google Scholar]
- 41.Baldwin JM. The probable arrangement of the helices in G protein-coupled receptors. EMBO J. 1993;12:1693–703. doi: 10.1002/j.1460-2075.1993.tb05814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang YK, Dickinson CJ, Zeng Q, Li JY, Thompson DA, Gantz I. Contribution of melanocortin receptor exoloops to agouti-related protein binding. J Biol Chem. 1999;274:14100–6. doi: 10.1074/jbc.274.20.14100. [DOI] [PubMed] [Google Scholar]
- 43.Yang Y, Dickinson C, Haskell-Luevano C, Gantz I. Molecular basis for the interaction of [Nle4, D-Phe7]-melanocyte stimulating hormone with the human melanocortin-1 receptor. J Biol Chem. 1997;272:23000–l. doi: 10.1074/jbc.272.37.23000. [DOI] [PubMed] [Google Scholar]
- 44.Yang YK, Fong TM, Dickinson CJ, Mao C, Li JY, Tota MR, et al. Molecular determinants of ligand binding to the human melanocortin-4 receptor. Biochemistry. 2000;39:14900–11. doi: 10.1021/bi001684q. [DOI] [PubMed] [Google Scholar]
- 45.Yang YK, Ollmann MM, Wilson BD, Dickinson C, Yamada T, Barsh GS, et al. Effects of recombinant agouti-signaling protein on melanocortin action. Mol Endocrinol. 1997;11:274–80. doi: 10.1210/mend.11.3.9898. [DOI] [PubMed] [Google Scholar]
- 46.Conrad DM, Hanniman EA, Watson CL, Mader JS, Hoskin DW. Ryanodine receptor signaling is required for anti-CD3-induced T cell proliferation, interleukin-2 synthesis, and interleukin-2 receptor signaling. J Cell Biochem. 2004;92:387–99. doi: 10.1002/jcb.20064. [DOI] [PubMed] [Google Scholar]
- 47.Mizoue LS, Bazan JF, Johnson EC, Handel TM. Solution structure and dynamics of the CX3C chemokine domain of fractalkine and its interaction with an N-terminal fragment of CX3CR1. Biochemistry. 1999;38:1402–14. doi: 10.1021/bi9820614. [DOI] [PubMed] [Google Scholar]
- 48.Yang X, Wang Z, Dong W, Ling L, Yang H, Chen R. Modeling and docking of the three-dimensional structure of the human melanocortin 4 receptor. J Protein Chem. 2003;22:335–44. doi: 10.1023/a:1025386022852. [DOI] [PubMed] [Google Scholar]
- 49.Haskell-Luevano C, Hendrata S, North C, Sawyer TK, Hadley ME, Hruby VJ, et al. Discovery of prototype peptidomimetic agonists at the human melanocortin receptors MC1R and MC4R. J Med Chem. 1997;40:2133–9. doi: 10.1021/jm960840h. [DOI] [PubMed] [Google Scholar]
- 50.Chen M, Aprahamian CJ, Celik A, Georgeson KE, Garvey WT, Harmon CM, et al. Molecular characterization of human melanocortin-3 receptor ligand–receptor interaction. Biochemistry. 2006;45:1128–37. doi: 10.1021/bi0521792. [DOI] [PubMed] [Google Scholar]
- 51.Grieco P, Balse-Srinivasan P, Han G, Weinberg D, MacNeil T, Van der Ploeg LH, et al. Synthesis and biological evaluation on hMC3, hMC4 and hMC5 receptors of gamma-MSH analogs substituted with L-alanine. J Pept Res. 2002;59:203–10. doi: 10.1034/j.1399-3011.2002.01966.x. [DOI] [PubMed] [Google Scholar]
- 52.Sahm UG, Olivier GW, Branch SK, Moss SH, Pouton CW. Synthesis and biological evaluation of alpha-MSH analogues substituted with alanine. Peptides. 1994;15:1297–302. doi: 10.1016/0196-9781(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 53.Bednarek MA, Silva MV, Arison B, MacNeil T, Kalyani RN, Huang RR, et al. Structure–function studies on the cyclic peptide MT-II, lactam derivative of alpha-melanotropin. Peptides. 1999;20:401–9. doi: 10.1016/s0196-9781(99)00048-0. [DOI] [PubMed] [Google Scholar]
- 54.Yang Y, Chen M, Lai Y, Gantz I, Georgeson KE, Harmon CM. Molecular determinants of human melanocortin-4 receptor responsible for antagonist SHU9119 selective activity. J Biol Chem. 2002;277:20328–35. doi: 10.1074/jbc.M201343200. [DOI] [PubMed] [Google Scholar]
- 55.Fleck BA, Ling N, Chen C. Substituted NDP-MSH peptides paired with mutant melanocortin-4 receptors demonstrate the role of transmembrane 6 in receptor activation. Biochemistry. 2007;46:10473–8. doi: 10.1021/bi700406k. [DOI] [PubMed] [Google Scholar]
- 56.Castrucci AM, Hadley ME, Sawyer TK, Wilkes EC, al-Obeidi F, Staples DJ, et al. Alpha-melanotropin: the minimal active sequence in the lizard skin bioassay. Gen Comp Endocrinol. 1989;73:157–63. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 57.Pogozheva ID, Chai BX, Lomize AL, Fong TM, Weinberg DH, Nargund RP, et al. Interactions of human melanocortin 4 receptor with nonpeptide and peptide agonists. Biochemistry. 2005;44:11329–41. doi: 10.1021/bi0501840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hruby VJ, Lu D, Sharma SD, Castrucci AL, Kesterson RA, al-Obeidi FA, et al. Cyclic lactam alpha-melanotropin analogues of Ac-Nle4-cyclo[Asp5, D-Phe7, Lys10] alpha-melanocyte-stimulating hormone-(4–10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–61. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]