

Abstract
Excitotoxic cell death is one of the precipitating events in the development of temporal lobe epilepsy. Of particular prominence is the loss of GABAergic hilar neurons. Although the molecular mechanisms responsible for the selective vulnerability of these cells are not well understood, activation of the extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) pathway has been implicated in neuroprotective responses to excitotoxicity in other neuronal populations. Here, we report that high levels of the striatal-enriched protein tyrosine phosphatase (STEP), a key regulator of ERK/MAPK signaling, are found in vulnerable somatostatin-immunoreactive hilar interneurons. Under both control conditions and after pilocarpine-induced status epilepticus (SE), ERK/MAPK activation was repressed in STEP-immunoreactive hilar neurons. This contrasts with robust SE-induced ERK/MAPK activation in the granule cell layer of the dentate gyrus, a cell region that does not express STEP. During pilocarpine-induced SE, in vivo disruption of STEP activity allowed activation of the MAPK pathway, leading to immediate-early gene expression and significant rescue from cell death. Thus, STEP increases the sensitivity of neurons to SE-induced excitotoxicity by specifically blocking a latent neuroprotective response initiated by the MAPK pathway. These findings identify a key set of signaling events that render somatostatinergic hilar interneurons vulnerable to SE-induced cell death.
Keywords: STEP, ERK/MAPK, cell death, apoptosis, hippocampus, seizure, somatostatin
Introduction
Status epilepticus (SE) triggers profound and lasting alterations in hippocampal physiology that can lead to the development of temporal lobe epilepsy (TLE) (Schwartzkroin, 1997; Scharfman, 2000; Cossart et al., 2005; Ribak and Dashtipour, 2005). One prominent pathophysiological effect of SE is a reduction in the number of GABAergic interneurons (Sloviter, 1987; Obenaus et al., 1993; Houser and Esclapez, 1996). Susceptibility to SE-induced cell death has been shown to vary among phenotypically distinct subgroups of hilar interneurons. Of note is the marked decrease in somatostatin-positive interneurons both in patients with TLE (de Lanerolle et al., 1989; Robbins et al., 1991) and in animal models of TLE (Sloviter, 1987; Sloviter, 1991; Buckmaster and Dudek, 1997; Buckmaster and Jongen-Rélo, 1999). For example, postmortem analysis has revealed that the number of somatostatin-positive neurons is reduced by 80% in individuals with TLE (Robbins et al., 1991), and, in the kainic acid model of TLE, somatostatin-positive hilar interneurons account for >80% of the GABAergic cells that were lost (Buckmaster and Jongen-Rélo, 1999). Given these observations, insight into the molecular mechanism responsible for the subclass-specific vulnerability of interneurons to SE-induced cell death may provide a deeper understanding of temporal lobe epileptogenesis.
Information regarding the molecular events underlying cell type-specific vulnerability may be gained by the analysis of inherent neuroprotective signaling responses actuated by excitotoxic stimuli. Along these lines, accumulating evidence shows that the p42/44 mitogen-activated protein kinase (MAPK) pathway plays an important role in neuronal cell survival. For example, the MAPK pathway mediates the anti-apoptotic effects of BDNF (Hetman et al., 1999). Likewise, the MAPK pathway stimulates preconditioning-mediated neuroprotection (Jiang et al., 2005; Lee et al., 2005), whereas disruption of MAPK signaling has been shown to precede cell death (Xia et al., 1995). Given the stringent, cell type-specific regulation of this pathway, these data raise the possibility that vulnerability to SE-induced cell death may, in part, depend on the capacity of a cell to elicit a MAPK pathway-dependent neuroprotective response.
Here, we examined the relationship between SE-induced MAPK pathway activation and neuronal survival in the dentate gyrus of the mouse. A key finding was that SE stimulated acute and robust MAPK pathway activation in SE-resistant granule cells but not in vulnerable populations of hilar interneurons. Our data reveal that the vulnerable cells express high levels of striatal-enriched protein tyrosine phosphatase (STEP), which is a key regulator of the extracellular signal-regulated kinase (ERK) pathway. (Lombroso et al., 1991). SE-induced MAPK activation within these neurons was repressed by STEP, leading to cell death. Blocking STEP activity by injecting FK506 allowed ERK to be activated and conferred protection against excitotoxicity. Collectively, these data suggest that STEP plays an important role in SE-induced somatostatinergic hilar interneuron cell death.
Materials and Methods
Pilocarpine-induced SE.
SE was induced in male C57BL/6 mice (7–8 weeks of age; Harlan, Indianapolis, IN) by the intraperitoneal injection of pilocarpine. Animals were initially given intraperitoneal injections of 1 mg/kg atropine methyl nitrate 30 min before receiving injections of pilocarpine (325 mg/kg diluted in physiological saline; Sigma, St. Louis, MO). SE was defined as a continuous motor seizure of stage 4 (rearing and falling), stage 5 (loss of balance, continuous rearing and falling), or stage 6 (severe tonic–clonic seizures) (Racine, 1972). Sham control animals were given injections of atropine methyl nitrate, followed by physiological saline 30 min later. Animals were killed at the indicated times. To block calcineurin activation, FK506 (1 mg/kg diluted in 20% DMSO and 80% physiological saline; Sigma) was injected intraperitoneally 15 min after pilocarpine injection. For all experiments, we only used mice that exhibited SE for a minimum of 2 h.
Immunohistochemistry and immunofluorescence.
Mice were perfused transcardially with cold saline, followed by 4% paraformaldehyde in PBS (10 mm, pH 7.4) under ketamine/xylazine anesthesia. Brains were postfixed in 4% paraformaldehyde for 4 h at 4°C and cryoprotected with 30% sucrose solution in PBS. Coronal sections (40 μm) through the dorsal hippocampus were prepared using a freezing microtome.
For immunohistochemistry, sections were washed with PBS and incubated in 0.3% hydrogen peroxide/PBS for 20 min to eliminate endogenous peroxidase activity. After several washes with PBS, sections were blocked with 10% normal goat serum in PBS, followed by overnight incubation at 4°C with one of the following antibodies: mouse anti-STEP antibody (1:2000; Novus Biologicals, Littleton, CO), rabbit anti-phospho-ERK (pERK) antibody (1:1000; Cell Signaling Technology, Beverly, MA), mouse anti-JunB antibody (1:2000; Santa Cruz Biochemicals, Santa Cruz, CA), rabbit anti-FosB antibody (1:2000; Santa Cruz Biochemicals), mouse anti-His antibody (1:1000; Cell Signaling Technology), rabbit anti-glutamic acid decarboxylase 65/67 (GAD65/67) antibody (1:1000; Sigma), or rabbit anti-somatostatin antibody (1:1000; Peninsula Lab, Belmont, CA). Sections were then processed using the ABC staining method (Vector Laboratories, Burlingame, CA). Nickel-intensified diaminobenzidine (DAB; Vector Laboratories) was used to visualize the signal. Photomicrographs were captured using a 16-bit digital camera (Micromax YHS 1300; Princeton Instruments, Trenton, NJ) mounted on a Leica (Nussloch, Germany) DM IRB microscope.
For immunofluorescent labeling, sections were washed with PBS and blocked with 10% normal goat serum in PBS, followed by overnight incubation with anti-STEP, anti-pERK, anti-GAD65/67, anti-FosB, and/or anti-somatostatin antibody. After several washes, sections were incubated with secondary antibodies conjugated with Alexa 488 and/or Alexa 594 (1:1000; Invitrogen, San Diego, CA) for 2 h at room temperature and mounted with Gelmount (Biomedia, Foster City, CA). Fluorescent images were captured using a Zeiss (Oberkochen, Germany) 510 Meta confocal microscope (2-μm-thick optical section).
Fluoro-Jade B staining.
Sections (40 μm) were initially mounted, rehydrated with 70% ethanol, washed with distilled water, and incubated in 0.06% potassium permanganate solution for 10 min. Next, the sections were incubated in 0.004% Fluoro-Jade B (Millipore, Bedford, MA) solution containing 0.1% glacial acetic acid for 20 min at room temperature, washed, and mounted with distrene plasticizer xylene (DPX; Electron Microscopy Sciences, Fort Washington, PA). For Fluoro-Jade B double labeling, sections were initially immunostained with mouse anti-STEP antibody (1:2000), followed by an anti-mouse antibody conjugated with Alexa 633. Sections were then incubated with 0.006% potassium permanganate solution for 5 min, then incubated in 0.004% Fluoro-Jade B solution for 20 min, and finally mounted with DPX.
Cresyl violet staining.
In brief, sections were mounted onto gelatin-coated slides and dried. After rehydrating in a graded alcohol series followed by water, sections were stained with 0.3% cresyl violet solution. After destaining with 95% ethanol containing 0.1% glacial acetic acid, sections were dehydrated and mounted with Permount.
Analysis of DNA breaks.
DNA strand breaks, a marker of apoptosis (Kondratyev and Gale, 2001), were analyzed by labeling the 3′-OH ends with terminal deoxynucleotidyl transferase (TdT) using the ApopTag Plus Fluorescein In Situ Apoptosis detection kit (Millipore). For this study, tissue was fixed by transcardial perfusion (as described above) 3 d after SE. The sections (30 μm) were processed following the manufacturer's instructions.
Microinjection of TAT-STEP or TAT-myc control peptide.
Mice were placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA) under ketamine/xylazine anesthesia. Guide cannulas (24 gauge) were located in the primary motor cortex [stereotaxic coordinates: anteroposterior (AP), −1.64 mm; mediolateral, +1.00 mm; dorsoventral, −0.50 mm]. Ten days later, 0.5 μl of TAT-STEP (100 nm) or TAT-myc (100 nm) was infused using an injector cannula (30 gauge). Pilocarpine was injected 1.5 h later.
Western blot.
Mice were killed, and hippocampi were rapidly dissected and stored at −80°C until used. Hippocampi were resuspended and sonicated in radioimmunoprecipitation assay buffer [50 mm Tris-HCl, 1% NP-40, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm sodium vanadate, and 1 mm NaF-containing protease inhibitor mixture (Roche Diagnostics, Rotkreuz, Switzerland)], and supernatants were isolated by centrifugation (13,000 × g) for 10 min. Immediately before loading, lysates (5 μg/lane) were diluted in 6× gel-loading buffer and electrophoresed on a 12% SDS polyacrylamide gel. Samples were transblotted onto polyvinylidene fluoride membrane (Immobilon P; Millipore), blocked, and incubated with the mouse anti-STEP antibody (1:4000 final dilution, in blocking solution) at 4°C overnight. Membranes were washed and incubated (2 h at room temperature) with horseradish peroxidase (HRP)-conjugated secondary antibody (1:2000; PerkinElmer Life Sciences, Norwalk, CT). The signal was visualized by using a Renaissance chemiluminescent HRP substrate (PerkinElmer Life Sciences).
Tissue culture.
Hippocampi were isolated from embryonic day 19–20 Sprague Dawley rat pups, and the tissue was processed as described by Lee et al. (2005). Cells were plated at a density of 5 × 105 cells/cm2, and cultures were maintained at 37°C and 5% CO2 in a Napco (Winchester, VA) 6100 incubator. Experiments were performed after 10–12 d in vitro.
Reporter gene assays.
Primary hippocampal neurons were plated in a 12-well dish and transfected (4 μg of DNA per well) after 8–10 d in culture with Lipofectamine 2000 (Invitrogen). The following constructs were used: pEGFP-N3 (500 ng/well), pcDNA3 STEP (500 ng/well), pcDNA3.1 (empty vector, 500 ng/well), GAL4-ELK (1.5 μg/well), and E1B-luciferase (1.5 μg/well). Cells were assayed 36 h after transfection, and luciferase activity was analyzed using the method described by Ford and Leach (1998).
Identification of cell death in cultured neurons.
Primary cultured hippocampal neurons were transfected as described above with the following constructs: pEGFP-N3 (500 ng) and either pcDNA3 STEP (3.5 μg) or pcDNA3.1 (empty vector, 3.5 μg). Two days after transfection, cells were stimulated with NMDA (50 μm) for 15 min (Lee et al., 2005), and cell viability was quantified 8 h later using the DNA stain Hoechst 33342 (1 μg/ml; Invitrogen). Immunolabeling for enhanced green fluorescent protein (EGFP) was used to identify transfected cells (Lee et al., 2005). A 16-bit digital camera (Micromax YHS 1300) connected to an inverted epifluorescence microscope (DM IRB; Leica) was used to capture photomicrographs. For the quantification of apoptotic cell death, the viability of transfected EGFP-positive neurons was determined by dividing the number of transfected neurons with fragmented or condensed nuclei (dead or dying) by the number of transfected neurons for each condition. We examined five coverslips for each treatment and quantified cell death in randomly selected fields. Significance was determined by using the two-tailed Student's t test.
Cell quantitation.
Photomicrographs were captured at 40× magnification using a 16-bit digital camera, and quantitation was performed using MetaMorph software (Universal Imaging, West Chester, PA). Cells between the upper and lower blades of the dentate gyrus that were not within area CA3 were defined as hilar. The total number of Fluoro-Jade B-, STEP-, JunB-, FosB- and cresyl violet-positive cells was counted bilaterally in each animal from two dorsal hippocampal sections separated by a 200 μm interval (stereotaxic coordinate AP, approximately −1.60 to −2.00), and the number of cells was given as mean ± SEM from six mice in each group. For the counting of cresyl violet-positive cells, we excluded cells in the subgranular zone because the distinction between granule cells and hilar cells could not be made unambiguously. With respect to STEP-positive cell quantitation, we found STEP-expressing cells in the subgranular zone, and these cells appeared to be lost after SE. Therefore, we decided to perform a more inclusive count of STEP-expressing cells, which included the subgranular zone. Thus, the density of cresyl violet cells was slightly lower than the STEP-positive cell density in Figure 3. Cell counts were analyzed statistically using Student's t test, and significance was accepted for p < 0.05.
Figure 3.
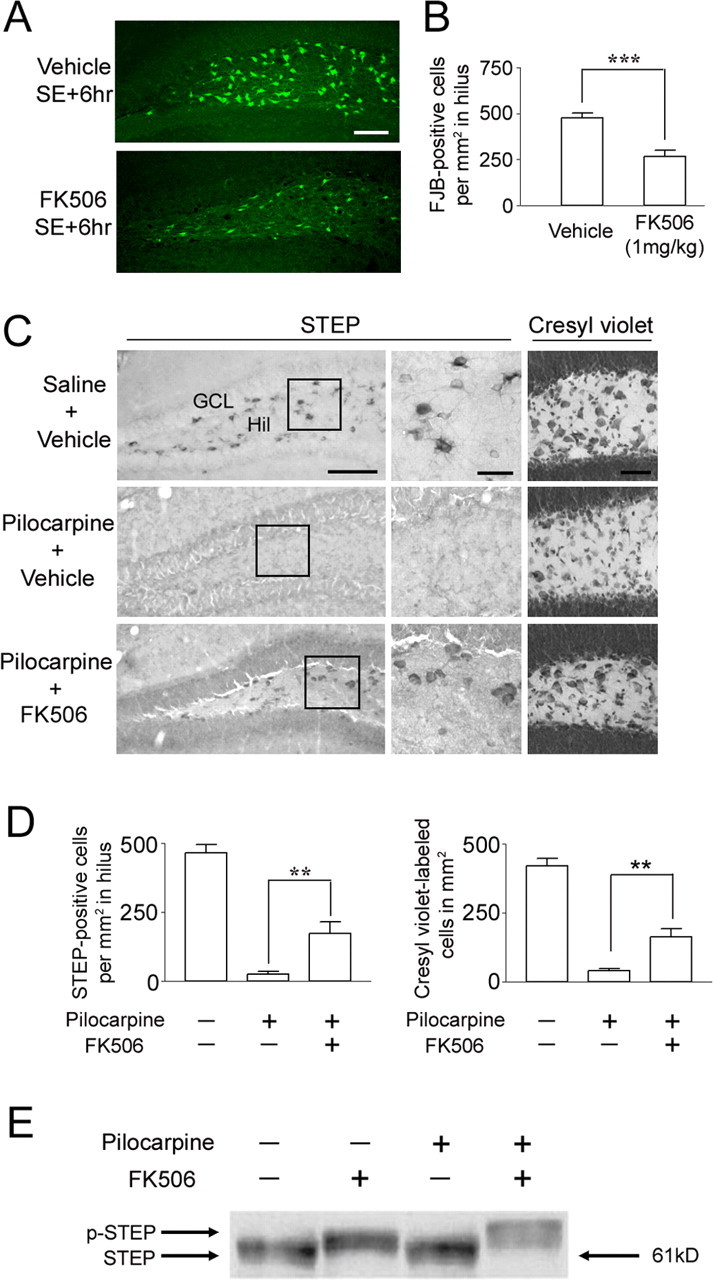
FK506 attenuates SE-induced cell death in the hilus. FK506 (1 mg/kg) or vehicle (20% DMSO in saline) was injected intraperitoneally 15 min after pilocarpine injection. Mice were killed 6 h after SE, and cell death was identified by Fluoro-Jade B staining. A, Representative images reveal Fluoro-Jade B-positive cells in the hilus. Scale bar, 100 μm. B, Cell counts of Fluoro-Jade B (FJB)-positive cells reveal that FK506 significantly reduced SE-induced cell death. ***p < 0.001. C, FK506 conferred lasting protection to STEP-expressing hilar (Hil) interneurons. Representative images of STEP immunolabeling in the dentate gyrus from animals killed 7 d after SE are shown. Note the large number of STEP-positive cells in the hilus of mice given injections of FK506 (bottom). In contrast, in the absence of FK506 treatment, STEP-positive cells were not observed in SE-induced mice (middle). Cresyl violet staining (right) is consistent with STEP immunostaining; FK506 attenuated SE-induced cell loss in the hilus. Scale bars: low-magnification images, 100 μm; high-magnification images, 25 μm. D, Quantitation of STEP- and cresyl violet-positive cells 7 d after pilocarpine-induced SE. **p < 0.01. E, Western blotting was used to detect the active (dephosphorylated) and inactive (phosphorylated) form of STEP (p-STEP). Under both control conditions (lane 1) and after pilocarpine injection (lane 3) (15 min after SE onset), STEP migrated in its lower-molecular-weight (dephosphorylated) active form. After FK506 (1 mg/kg) injection alone (lane 2) or with pilocarpine (lane 4), STEP migrated in its higher-molecular-weight (phosphorylated) inactive form. Error bars indicate SEM.
Results
STEP is expressed in hilar interneurons
To examine STEP expression in the hippocampus, coronal mouse brain sections were immunohistochemically processed with an anti-STEP antibody. As shown in Figure 1A, relatively high levels of STEP were observed in both the deep hilus and in the border area between the hilus and granule cell layer (GCL). Quantitative analysis of STEP expression in the deep hilus and hilar border area can be found in supplemental Figure 1 (available at www.jneurosci.org as supplemental material). A high-magnification micrograph revealed strong STEP expression in the cytoplasm and cellular processes. This subcellular staining pattern is consistent with our previous observations in the rat striatum (Boulanger et al., 1995). Limited STEP expression was detected throughout the CA1 and CA3 regions, and relatively high levels were observed in CA2 (data not shown) (Boulanger et al., 1995). Expression was not detected in the GCL (Fig. 1A). Double labeling for STEP and GAD65/67 revealed that STEP was highly expressed in GABAergic interneurons of the hilus (Fig. 1B). Within the deep hilus, both the morphology and distribution of STEP-positive interneurons is consistent with that of somatostatin-expressing hilar interneurons (Buckmaster et al., 2002). Indeed, double labeling for STEP and somatostatin revealed that almost all somatostatin-positive hilar cells (>99%, 302 somatostatin-positive cells were examined from six mice) strongly express STEP (Fig. 1C). However, a subset of STEP-positive hilar cells (23%, 393 cells were examined from six mice) did not coexpress somatostatin. A number of these somatostatin-negative cells were positive for the putative mossy cell marker glutamate receptor 2/3 (GluR2/3; data not shown). However, this finding was complicated when we identified a subset of GluR2/3-positive cells that also express GAD6 (36%, 213 cells were observed). STEP was also strongly expressed in somatostatin-positive interneurons in the stratum oriens layer of CA1 (Fig. 1C). Few STEP-positive cells were found in stratum radiatum and the molecular layer, where virtually no somatostatin-immunoreactive cells are found (Esclapez and Houser, 1995).
Figure 1.
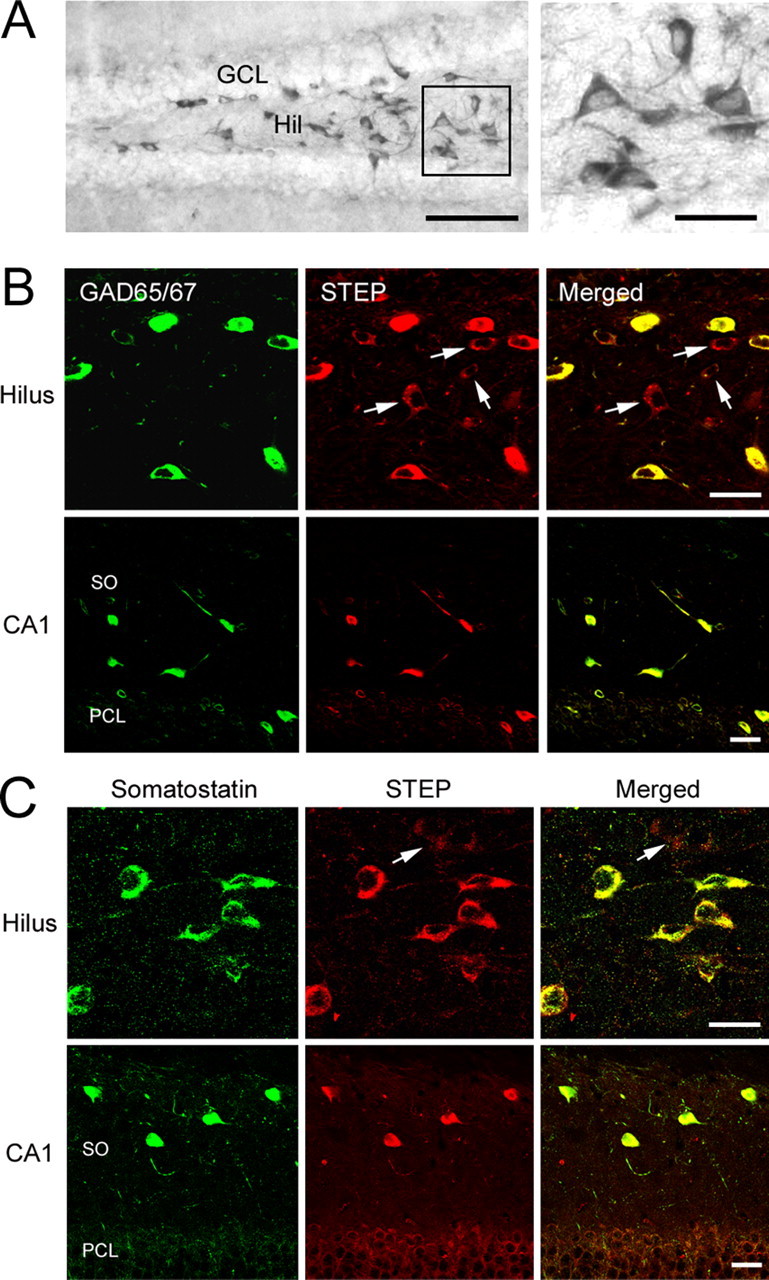
STEP is expressed in GABAergic hilar interneurons. A, DAB-based immunohistochemical labeling revealed marked STEP expression in the hilus (Hil). Note the lack of STEP staining in the GCL. The boxed region is magnified and shown on the right. B, Top row, Immunofluorescent double labeling for STEP and GAD65/67 revealed that STEP is expressed in GABAergic hilar interneurons. Bottom row, STEP is also expressed in GAD65/67-positive neurons in the stratum oriens of CA1. Arrows indicate cells expressing STEP but not GAD65/67. C, Double labeling shows that somatostatinergic interneurons express STEP in the hilus (top row) as well as in the stratum oriens of CA1 (bottom row). Arrow denotes a somatostatin-negative cell weakly expressing STEP. Scale bars: A, 100 μm (low-magnification image) and 25 μm (high-magnification image); B, C, 20 μm. SO, Stratum oriens; PCL, pyramidal cell layer.
Degeneration of hilar neurons expressing STEP after pilocarpine-induced SE
The muscarinic acetylcholine receptor agonist pilocarpine is commonly used to elicit SE in rodent models of TLE. The loss of hilar interneurons is a well characterized result of pilocarpine-induced SE and can be detected at 6 h after SE (Borges et al., 2003). To assess whether STEP-positive hilar interneurons are vulnerable to SE, mice (C57BL/6) were given injections of pilocarpine and killed 6 h after SE onset, and hippocampal brain sections were processed with Fluoro-Jade B, a sensitive histochemical stain for dying cells. At this time point, Fluoro-Jade B-labeled cells were specifically observed in the hilus (Fig. 2A). This contrasts with the GCL, where Fluoro-Jade B staining was not detected.
Figure 2.
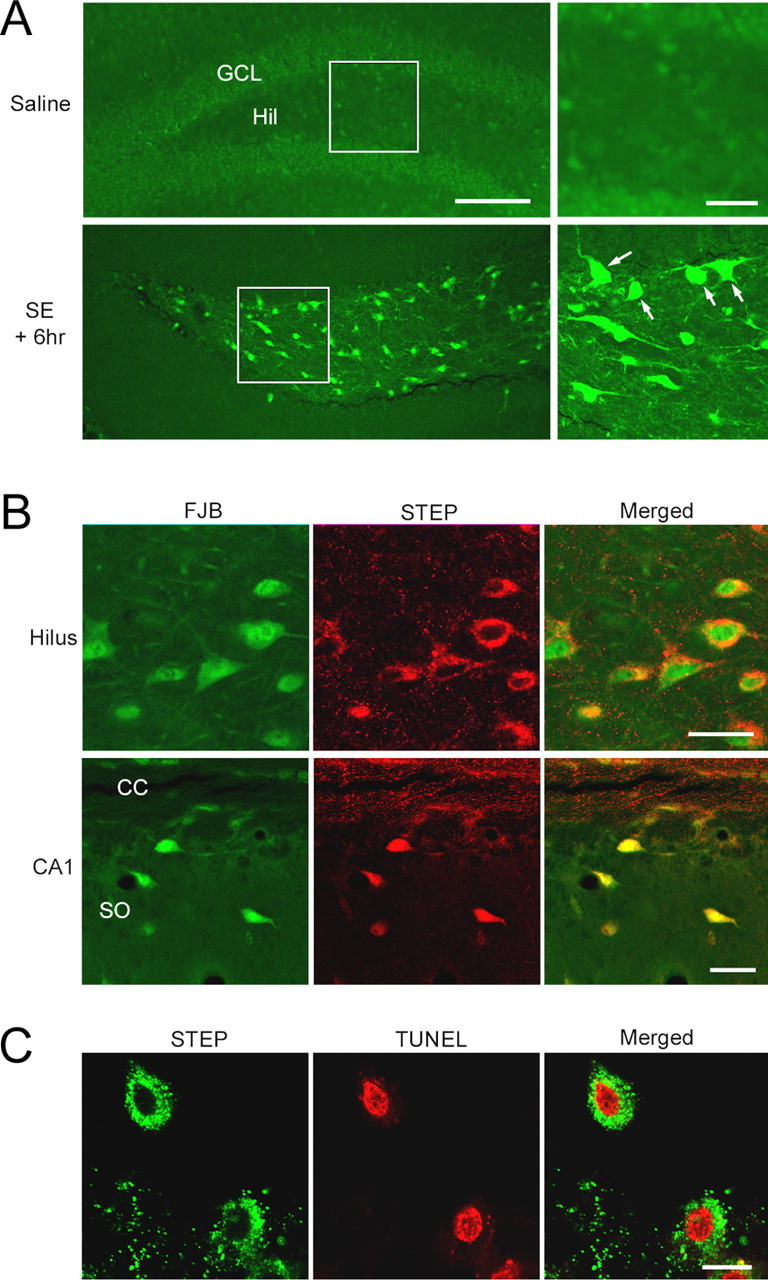
Pilocarpine-induced SE elicits cell death in the hilus. A, Mice were given injections of either pilocarpine (325 mg/kg, i.p.) or saline (control), and brain sections were stained with Fluoro-Jade B, a fluorescent marker of dead and dying cells. Mice were killed 6 h after SE onset. Fluoro-Jade B labeling was observed specifically in the hilus (Hil). In contrast, cell death was not observed in the GCL. Boxed regions are shown magnified on the right. Arrows denote dead/dying cells. B, Double labeling revealed that Fluoro-Jade B (FJB) -labeled cells in the hilus express STEP. In stratum oriens of CA1, cell death was specifically observed in STEP-positive cells. CC, Corpus callosum; SO, stratum oriens. Scale bars, 20 μm. C, Results of double labeling in the hilus for STEP and TUNEL, a marker of apoptotic cell death, were consistent with the Fluoro-Jade B staining data; TUNEL-labeled cells were STEP positive. For TUNEL labeling, animals were killed 3 d after SE. Scale bars: A, 100 μm (low-magnification image) and 25 μm (high-magnification image); B, 20 μm; C, 10 μm.
Double labeling with an anti-STEP antibody and Fluoro-Jade B revealed that 96% of Fluoro-Jade B-positive cells in the hilus were also STEP positive (268 cells were examined from three mice) (Fig. 2B). In addition, Fluoro-Jade B-positive cells in the stratum oriens, another region of the hippocampus where interneuron cell death occurs, were also STEP positive (Fig. 2B). TdT-mediated biotinylated UTP nick end labeling (TUNEL) staining was used to confirm that STEP-positive cells died after SE (Fig. 2C). Collectively, these data reveal that STEP-positive hilar interneurons are highly vulnerable to the excitotoxic effects of SE.
STEP inhibition decreases SE-induced hilar interneuron cell death
Accumulating evidence suggests that activation of the MAPK pathway is important for neuroprotection against several kinds of injury (Hetman and Xia, 2000). Because STEP is a negative regulator of the ERK/MAPK pathway, we tested whether inhibiting STEP would reduce hilar interneuron cell death caused by SE. To this end, mice were given injections of the calcineurin inhibitor FK506. Calcineurin is an upstream activator of STEP (Paul et al., 2003), and blocking calcineurin inhibits STEP activity. We chose FK506 to target calcineurin because there are no specific inhibitors of STEP. A 1 mg/kg intraperitoneal injection of FK506 has been shown to cross the blood–brain barrier and attenuate ischemia-induced neuropathology (Uchino et al., 2002; Furuichi et al., 2003; Noto et al., 2004). Systemic injection of FK506 (1 mg/kg) 15 min after pilocarpine injection (∼30 min before SE onset) significantly reduced cell death in the hilus 6 h after SE (Fig. 3A,B). The efficacy of FK506 was examined by using an immunoblotting assay that detects an activation state-dependent shift in the migration rate of STEP (Paul et al., 2003). This shift in migration has been ascribed to calcineurin-dependent dephosphorylation of STEP61 at Ser-221 (Paul et al., 2003). Hippocampal tissue was processed from control mice and from mice with SE (15 min after SE onset). Under both conditions, STEP61, the major STEP isoform in the hippocampus, migrated as a single band (Fig. 3E). The injection of FK506 (1 mg/kg) caused STEP to run as a higher-molecular-weight species. This shift in the migration rate of STEP indicates that FK506 blocks the activation of STEP in vivo and that STEP is constitutively active under control conditions.
We also examined the viability of STEP-expressing hilar interneurons and the neuroprotective effects of FK506 7 d after SE onset (Fig. 3C,D). Consistent with the Fluoro-Jade B staining pattern observed 6 h after SE, there was a total loss of STEP expression at the 7 d time point. In marked contrast, STEP-positive neurons were observed throughout the hilar region of mice given injections of FK506 before SE onset. Cresyl violet staining was also used to verify that FK506 protected hilar cells from SE-induced death (Fig. 3C,D). As noted in Materials and Methods, cresyl violet-positive cells were only counted within the deep hilus, whereas STEP-positive cells were counted in both the deep hilus and GCL border area. Thus, the density of cresyl violet-positive cells shown in Figure 3D was lower than that of STEP-positive cells. These data indicate that a single injection of FK506 did not simply delay SE-induced cell death but, rather, prevented the death of STEP-positive hilar interneurons. To determine whether the neuroprotective effects of FK506 were dependent on the expression of STEP, we compared the effects of FK506 on SE-induced cell death in the hilus and endopiriform nucleus. We selected the endopiriform nucleus because it does not express high levels of STEP. In contrast to the hilus, systemic injection of FK506 did not protect neurons of the endopiriform nucleus from SE-induced cell death (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This lack of FK506-mediated neuroprotection supports the hypothesis that FK506 attenuates cell death via a calcineurin/STEP-dependent mechanism. Consistent with this idea and a previous report (Moriwaki et al., 1998), FK506 had only modest effects on seizure latency and severity (Table 1). Importantly, we did not detect a difference in the severity of SE between two groups examined 3 h after SE. Likewise, animals from both groups showed continuous myoclonus for at least an additional 3 h or longer, although FK506-injected mice showed a slightly milder form of myoclonus at 6 h after SE. We also found that FK506 decreased the percentage of mice experiencing seizures (22% decline for stage 4–5 seizures and 11% for stage 6 seizures) and reduced mortality by 15%. An additional set of experiments in which FK506 was microinfused into the hilus is described below.
Table 1.
Summary of behavioral observations after pilocarpine injection
| Drug | Number of mice | Stage 4–5 seizures |
Stage 6 seizures |
Mortality (%) | ||
|---|---|---|---|---|---|---|
| Latency to onset (min) | Percentage | Latency to onset (min) | Percentage | |||
| Vehicle (20% DMSO) | 27 | 39.6 ± 13.1 | 92.6 | 49.2 ± 11.3 | 66.7 | 25.9 |
| FK506 (1 mg/kg) | 27 | 46.1 ± 14.4 | 70.4 | 58.6 ± 16.1 | 55.6 | 11.1 |
Data are represented as mean ± SD.
STEP and the MAPK cascade
The finding that blocking STEP activity enhanced neuronal survival suggested that FK506 treatment combined with SE stimulated an otherwise dormant MAPK pathway in the dentate gyrus. To test this possibility, we examined activation of ERK with an antibody against the active, dually phosphorylated form of ERK (pERK). Under control conditions, little pERK expression was observed in both the GCL and hilus (Fig. 4A). Pilocarpine triggered strong ERK activation in the GCL 15 min after SE onset. In contrast, SE did not stimulate pERK expression in the hilus, consistent with the role of STEP as a negative regulator of ERK activation.
Figure 4.

STEP and seizure-induced ERK activation. A, Immunolabeling for the activated form of ERK. Under both the control condition [Saline (Sal) + Vehicle (Veh), Panel 1] and 15 min after SE [Pilocarpine (Pilo) + Veh, Panel 2] onset, ERK activity was not observed in the hilus. In contrast, SE triggered a robust increase in ERK activation in the GCL. FK506 alone (1 mg/kg) triggered ERK activation in the hilus (Sal + FK506, Panel 3). ERK activation was also observed in the hilus after SE in mice given injections of FK506 (Pilo + FK506, Panel 4). The boxed regions are magnified images shown to the right of each panel. Arrows denote cells with marked pERK expression. B, Double labeling for pERK and STEP demonstrates that FK506 injection activated ERK signaling in two of the three STEP-positive hilar interneurons. Representative images were taken from animals killed 15 min after SE onset. C, Double labeling demonstrates that FK506 triggered the expression of pERK in somatostatin-positive cells of the hilus. Animals were killed 15 min after SE onset. Arrows denote somatostatinergic cells. Scale bars: A, 100 μm (low-magnification image) and 25 μm (high-magnification image); B, 10 μm; C, 25 μm.
Next, we tested whether STEP inhibits ERK activation in the hilus. Initially, in a control experiment, we tested whether blocking STEP activity (in the absence of SE) would trigger ERK activation. To this end, mice were given injections of FK506 (1 mg/kg) and killed 15 min later. FK506 injection alone triggered ERK activation in the hilus but not in the GCL (Fig. 4A, Panel 3). Given that STEP is expressed specifically in hilar cells and is tonically active, this result indicates that STEP is a potent negative regulator of ERK activation under control, unstimulated conditions. In the hilus, the level of ERK activation after SE and FK506 injection appeared to be greater than the level of ERK activation resulting from FK506 injection alone (Fig. 4A, Panel 4), indicating that SE stimulates ERK activation. To determine whether ERK activation occurred within STEP-positive neurons, hippocampal sections from mice that received injections of FK506 and pilocarpine were double labeled for STEP and pERK. Figure 4B shows that SE induced ERK phosphorylation in STEP-positive cells. Furthermore, double immunolabeling revealed that FK506 elicited pERK expression within somatostatin-positive hilar interneurons (Fig. 4C). These results reveal that STEP functions as a potent negative regulator of the MAPK pathway in the hilus.
MAPK signaling has been shown to exert a neuroprotective response via a transcriptionally dependent mechanism (Criswell et al., 2005). To assess whether disruption of STEP stimulates the expression of MAPK-regulated genes, we monitored the induction of FosB and JunB (Coffer et al., 1994; Inoue et al., 2004). In control mice, JunB and FosB expression was low in the hilus and the GCL (Fig. 5A). At 1.5 h after SE onset, robust JunB and FosB expression was observed in the GCL, whereas little expression was observed in the hilus. In contrast, FK506 injection significantly increased the number of JunB- and FosB-positive cells in the hilus (p < 0.004 for JunB and p < 0.009 for FosB) (Fig. 5C). To verify that gene expression occurred in STEP-positive interneurons, tissue was double labeled for FosB and STEP. As shown in Figure 5B, marked FosB expression was observed in STEP-positive hilar interneurons. These data raise the possibility that the disruption of STEP activity results in the emergence of a latent, neuroprotective, MAPK-dependent transcriptional response in hilar interneurons.
Figure 5.

Blocking STEP activation triggers immediate-early gene expression. A, SE stimulated FosB (left) and JunB (right) expression in the GCL but not in the hilus. FK506 injection (1 mg/kg, 15 min after pilocarpine injection) significantly increased SE-induced FosB and JunB expression in the hilus. Mice were killed 1.5 h after SE onset. Boxed regions are magnified and shown on the right. Arrows denote cells with immediate-early gene expression. B, Double labeling with FosB and STEP revealed that immediate-early gene expression occurred in STEP-positive hilar cells. C, Quantitative analysis of FosB- or JunB-positive cells in the hilus under the different treatment conditions. Data from six animals were examined for each condition and expressed as the mean ± SEM. **p < 0.01. Error bars indicate SEM. Scale bars: A, 100 μm (low-magnification image) and 25 μm (high-magnification image); B, 10 μm; C, 25 μm.
Ectopic STEP increases cell death
To more rigorously test the role of STEP as a regulator of SE-induced cell death, we used an enzymatically inactive mutant of STEP46 (TAT-STEP), in which the essential cysteine in the catalytic domain was converted to a serine (Pelkey et al., 2002; Paul et al., 2007). A TAT-peptide sequence was added to the N terminus of the construct to allow STEP to penetrate cells (Green and Loewenstein, 1988), and myc and His epitopes were added to allow TAT-STEP to be distinguished from endogenous STEP. TAT-STEP still binds to ERK but cannot dephosphorylate it. Thus, TAT-STEP functions by sequestering ERK in the cytosol and, in turn, disrupting signaling via the MAPK cascade (Paul et al., 2007). In the motor cortex, TAT-STEP infusion (100 nm, 0.5 μl) effectively reduced nuclear translocation of pERK after SE (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). This contrasts with nuclear translocation of pERK in the contralateral cortex. In addition, TAT-STEP did not significantly alter the phosphatase activity of wild-type STEP (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). Because TAT-STEP blocks the nuclear translocation of pERK and thereby prevents the activation of a putative neuroprotective transcriptional response, we reasoned that misexpression of TAT-STEP might increase vulnerability to SE-induced injury and cell death. To this end, we infused TAT-STEP (100 nm, 0.5 μl) into the motor cortex. This brain region was chosen because we found that a relatively small percentage of cells normally die in this area after SE. Thus, given the low level of SE-induced cell death in the motor cortex, a potential STEP-dependent increase in cell vulnerability should be readily detected. Initially, the expression pattern of TAT-STEP was examined 8 h after microinfusion. Immunofluorescence using an anti-His antibody confirmed that TAT-STEP was taken up in the cortical region surrounding the site of injection (Fig. 6A).
Figure 6.
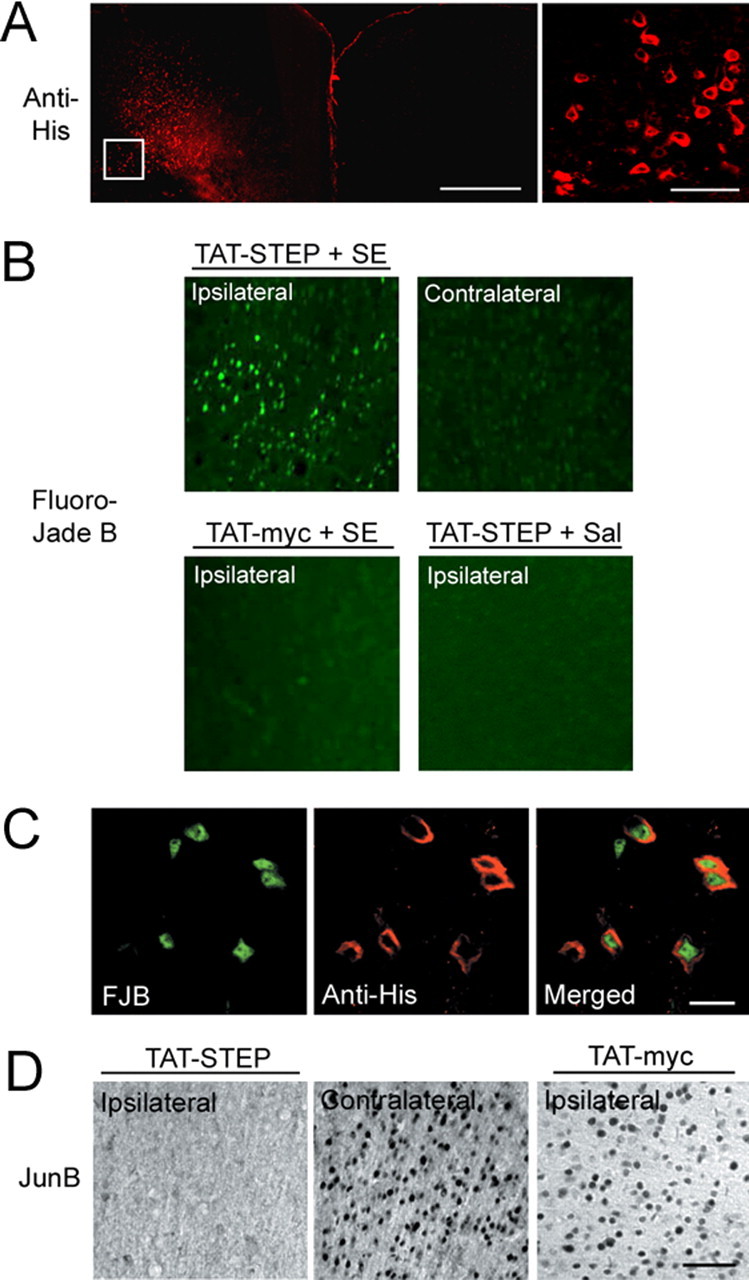
TAT-STEP increases SE-induced cell death and decreases immediate-early gene expression in the cortex. In A–C, His-tagged TAT-STEP was microinjected into the motor cortex via an indwelling guide cannula. SE was induced 2 h later, and mice were killed 6 h after SE onset. A, TAT-STEP was visualized with an anti-His antibody, followed by Alexa 488-conjugated secondary antibody (color-coded red for clarity). The high-magnification image (taken from the boxed region in the left panel) identifies individual cells that had taken up TAT-STEP. B, Cell death was determined using Fluoro-Jade B staining. Compared with the contralateral hemisphere or with the injection of TAT-myc, TAT-STEP increased SE-induced cell death. In saline (Sal)-injected mice, TAT-STEP did not induce cell death. C, Double labeling for Fluoro-Jade B (FJB; left) and TAT-STEP (anti-His; middle) revealed that SE-induced death occurred in TAT-STEP-expressing cells. D, Representative data showing that SE-induced JunB expression was decreased in the TAT-STEP-infused hemisphere compared with the contralateral hemisphere or to TAT-myc infusion. Animals were killed 2 h after SE onset. Scale bars: A, 500 μm (low-magnification image) and 50 μm (high-magnification image); B, 200 μm (low-magnification image) and 50 μm (high-magnification image); B, 10 μm; C, 10 μm; D, 50 μm.
TAT-STEP-positive cells were highly vulnerable to pilocarpine-induced SE. Double labeling for TAT-STEP and Fluoro-Jade B revealed a marked increase in dead and dying cells in the infused hemisphere (Fig. 6C). This contrasts with the rather low level of Fluoro-Jade B labeling observed in the contralateral control hemisphere (Fig. 6B). Consistent with the effects of endogenous STEP in the hilus, TAT-STEP blocked the expression of JunB in the cortex (Fig. 6D). Importantly, in the absence of SE, microinjection of TAT-STEP did not induce cell death in the motor cortex. Likewise, the injection of the control peptide TAT-myc (100 nm, 0.5 μl) alone did not increase SE-induced cell death. These findings are consistent with the work of Aarts et al. (2002) that reported that nanomolar concentrations of TAT-fusion proteins were not toxic. However, in slice culture, Prendergast et al. (2002) did observe TAT-mediated toxicity.
We next examined whether injection of TAT-STEP into the hilus would block the neuroprotective effects of FK506. Hence, if FK506 is conferring neuroprotection via the inhibition of calcineurin-mediated STEP activation, then ectopic TAT-STEP should override this effect and increase SE-induced cell death. In this experiment, TAT-STEP was infused into the hilus, and 1.5 h later, FK506 was infused into the hilus, followed by an intraperitoneal injection of pilocarpine. We provide representative and quantitative data showing that hilar cells were transduced with TAT-STEP and that TAT-STEP blocked the neuroprotective effects of FK506 (supplemental Figs. 4, 5, available at www.jneurosci.org as supplemental material). In addition, infusion of the MEK1/2 (MAP/ERK kinase 1/2) inhibitor U0126 (1,4-diamino-2,3-dicyano-1,4-bis[2-amino-phenylthio]butadiene) blocked the neuroprotective effects of FK506 (supplemental Figs. 4, 5, available at www.jneurosci.org as supplemental material). Differences in SE onset and duration were not detected across any of the microinfusion experimental groups. Collectively, these data provide a link between STEP, the MAPK pathway, and SE-induced hilar interneuron cell death.
STEP overexpression increases cell death after NMDA stimulation
Last, we examined the effect of STEP on excitotoxic cell death in primary hippocampal neuronal cultures. For this study, we isolated neurons from embryonic day 20 rat pups and maintained them in culture for 10–12 d. The cultures were then transfected with EGFP (transfection marker) and either wild-type STEP or empty vector (pcDNA). Forty-eight hours later, cells were stimulated with NMDA (50 μm for 15 min), and the effects on cell viability were assayed 8 h later using Hoechst labeling (Fig. 7A). Relative to control vector-transfected neurons, STEP overexpression significantly increased the excitotoxicity of NMDA (Fig. 7B).
Figure 7.

STEP increases neuronal vulnerability to excitotoxic cell death. A, Hippocampal neurons isolated from embryonic rat pups were transfected with EGFP expression vector and either a STEP expression vector or an empty expression vector (pcDNA). Two days after transfection, cells were stimulated with NMDA (50 μm) for 15 min, and cell viability was quantified 8 h later using Hoechst staining. A total of 326 pcDNA- and 366 STEP-transfected cells were examined. Arrows identify representative transfected cells that are dying (condensed nuclei); arrowheads denote healthy cells. Scale bar, 50 μm. B, Quantitation of the percentage of transfected neurons undergoing cell death. Relative to control vector transfection, transfection with STEP increased the toxic effects of NMDA (***p < 0.001, significantly different from control vector transfection). C, STEP attenuates MAPK-dependent gene expression. Neurons were transfected with Gal4-ELK and E1B-luciferase reporter and either the STEP expression vector or empty expression vector (pcDNA). Cells were stimulated (15 min) with NMDA (20 μm) or TPA (1 μm) and lysed 6 h later. Both NMDA and TPA stimulated a significant increase in MAPK-dependent reporter gene expression. STEP overexpression blocked both NMDA- and TPA-mediated gene expression. Pretreatment (30 min) with FK506 (1 μm) blocked the repressive effects of STEP. The values are means ± SEM of quadruplicate determinations and are expressed as fold stimulation normalized to the unstimulated pcDNA control, which was set equal to one. ***p < 0.001, significant difference relative to control buffer treatment.
To determine whether STEP overexpression blocks MAPK-dependent transcription, neurons were transfected with the MAPK reporter constructs GAL4-ELK/E1B (Chung et al., 1999) and stimulated with phorbol ester 12-O-tetradecanoyl-phorbol-13-acetate (TPA; 1 μm for 30 min) or NMDA (20 μm for 15 min). Cotransfection of STEP blocked the capacity of TPA and NMDA to stimulate MAPK-dependent transcription. Importantly, pretreatment (15 min) with FK506 (1 μm) blocked STEP repression of TPA- and NMDA-mediated transcription (Fig. 7C). These data demonstrate that STEP acts as a potent negative regulator of the MAPK pathway and confirm the in vivo results showing that STEP increases neuronal vulnerability to potentially excitotoxic stimuli.
Discussion
Here, we have identified a central role for STEP in SE-induced, somatostatinergic hilar interneuron cell death. Principal among our findings is that the high level of tonic STEP activity in hilar interneurons represses a potentially neuroprotective ERK/MAPK signaling cascade. The connection between STEP and increased vulnerability to excitotoxic death was supported by the results of the ectopic STEP expression assays and the data showing that the disruption of STEP activity with FK506 is neuroprotective. Together, these data delineate a set of signaling events that render hilar interneurons highly vulnerable to SE-induced cell excitotoxicity.
Anatomical studies have shown that hilar interneurons receive excitatory input from granule cells and the perforant pathway (Leranth et al., 1990; Buckmaster and Schwartzkroin, 1995; Acsády et al., 1998) and project axon collaterals to granule cell apical dendrites in the outer molecular layer (Leranth et al., 1990; Han et al., 1993). This feedback circuit from the hilus to the GCL has been suggested to function as a negative regulator of GCL excitability (Leranth et al., 1990; Han et al., 1993). Recently, it was reported that granule cells of epileptic rats had <50% of the level of evoked, monosynaptic IPSP conductance and GABAA receptor-mediated spontaneous and miniature IPSCs compared with controls (Kobayashi and Buckmaster, 2003). In the case of epileptic patients, reduced evoked IPSPs in granule cells were also reported (Williamson et al., 1999). Thus, the selective loss of inhibitory input to the GCL has been suggested to enhance hippocampal excitability. However, it should also be noted that a number of studies have found that inhibitory drive to the GCL is enhanced in animal models of TLE (Tuff et al., 1983; Haas et al., 1996; Buckmaster and Dudek, 1997).
A striking finding of our study was the marked increase in pERK expression in the GCL 15 min after pilocarpine-induced SE but the lack of pERK induction in the hilus. Given that hilar interneurons receive excitatory synaptic input from the GCL mossy fiber collaterals and that pERK is induced in CA3, another target of the mossy fibers, it was our prediction that SE would induce robust MAPK pathway activation. This lack of induction is likely not the result of a weak excitatory input to the hilus. Rather, neuroanatomical studies have shown that hilar cells receive robust glutamatergic input from the GCL (Leranth et al., 1990; Acsády et al., 1998). These findings suggested that there is a negative regulatory mechanism that blocks activity-dependent MAPK stimulation in the hilus.
The identification of STEP in hilar neurons provides the mechanistic link needed to explain the lack of SE-induced MAPK pathway activation. STEP, which was initially characterized in the striatum, is also expressed in a number of other brain regions, including the hippocampus, nucleus accumbens, and septal nucleus (Boulanger et al., 1995). However, our study is the first to examine STEP in the hilus. STEP expression was found in the cytoplasm and cell processes of hilar interneurons. Double labeling for somatostatin revealed that STEP was enriched in somatostatinergic hilar interneurons. As noted above, this subclass of interneurons is highly vulnerable to SE-induced excitotoxicity (Robbins et al., 1991; Buckmaster and Jongen-Rélo, 1999). We found that acute SE-induced cell death was correlated with STEP expression in the hilus. However, it is important to state that the effects of STEP on excitotoxicity are context specific. Along these lines, area CA2 expresses high levels of STEP (Boulanger et al., 1995); yet, unlike the hilus, it is relatively resistant to excitotoxic cell death (Zhang et al., 1997; El Bahh et al., 1999). Mechanistically, it is not known why CA2 neurons are resistant, although numerous studies have shown that CA2 pyramidal neurons are enriched in an array of receptors and trophic factors (Tucker et al., 1993; Ochiishi et al., 1999; Lein et al., 2005) that may affect excitability and/or provide trophic support and thus decrease susceptibility to excitotoxic stimuli. Likewise, divergence also holds at the level of synaptic afferents (Haglund et al., 1984; Köhler et al., 1985). Thus, for CA2 neurons, the expression of STEP may not supplant an inherent level of excitability, synaptic input, or trophic support, which are all key regulators that contribute to vulnerability. In addition, SE-induced cell death occurs in cell populations that do not have high levels of STEP, such as the GCL (Sloviter et al., 1996; Roux et al., 1999).
Given our data showing the correlation between STEP expression and cell death, it is tempting to hypothesize that STEP is a determinant of whether a neuron is acutely sensitive to excitotoxicity. In line with this idea, overexpression of STEP in cultured hippocampal neurons increased NMDA-induced cell toxicity, and in vivo membrane- permeable TAT-STEP increased the vulnerability of cortical neurons to SE-induced cell death. The connection between STEP and excitotoxicity was further examined by using the calcineurin antagonist FK506. Previously, Paul et al. (2003) reported that calcineurin-dependent dephosphorylation of STEP leads to its binding to and dephosphorylation of ERK. By blocking the activation of STEP, FK506 led to a significant decrease in SE-induced hilar interneuron cell death. The specificity of FK506-mediated neuroprotection was assessed by comparing cell death in two vulnerable brain regions: the hilus, which expresses high levels of STEP, and the endopiriform nucleus, which has limited STEP expression. In contrast to the neuroprotective effects conferred to hilar cells, the level of endopiriform cell death was unaffected by systemic injection of FK506. Together, these data support the idea that FK506 confers neuroprotection via a calcineurin/STEP-dependent mechanism.
The association between disrupted STEP activation and attenuated cell death suggests that a latent MAPK-dependent response was engaged in hilar interneurons. Indeed, we found that the MAPK pathway was activated in the hilus after FK506 injection. This effect of FK506 was specific to hilar cells; FK506 did not stimulate ERK activation in the GCL. Given that STEP is expressed in the hilus, but not in the GCL, these data indicate the effect of FK506 was the specific result of STEP inhibition. It should be noted that FK506 has been shown to have neuroprotective effects in a number of brain injury models (Butcher et al., 1997; Singleton et al., 2001). However, the mechanism by which FK506 provides protection has not been established. Given the results reported here, it will be interesting to assess whether alterations in STEP/MAPK signaling may be a common mechanism by which FK506 affords neuroprotection.
Western analysis of hippocampi from control or pilocarpine-treated mice revealed that STEP was active even under control conditions. As a depolarization/calcium-activated phosphatase, this finding suggests that hilar interneurons receive a tonic level of excitatory activity that is sufficient to stimulate STEP. In line with this finding, a number of studies have shown that the perforant pathway and GCL, two principal inputs to hilar interneurons, have spontaneous firing rates of 7.12 ± 9.0 Hz and 0.05–0.13 Hz, respectively (Quirk et al., 1992; Howard et al., 1998; Vogt and Regehr, 2001), and thus may provide the excitatory stimulus necessary to drive tonic STEP activation. The chronic repression of ERK activation would likely have profound effects on the gene profile of hilar interneurons under both normal and SE conditions. Along these lines, we found that blocking STEP activity allowed SE to stimulate the expression of the ERK-regulated genes FosB and JunB. In addition, cAMP response element-binding protein-dependent transcription was also derepressed by disruption of STEP activity (Y.-S. Choi and K. Obrietan, unpublished observations).
The MAPK pathway consists of the kinases Raf, Mek, and ERK. With a stimulus of sufficient intensity and duration, the activated form of ERK translocates to the nucleus and stimulates transcription factors such as ELK-1 (Treisman, 1996; Cobb, 1999; Grewal et al., 1999). In addition, ERK targets a number of kinases such as MSK1/2 and RSK2 (Deak et al., 1998; McDonald et al., 1998; Soloaga et al., 2003) that stimulate gene transactivation. The induction of MAPK-dependent transcription has been shown to confer neuroprotection against excitotoxic stimuli. For example, BDNF-dependent neuroprotection is dependent on the MAPK cascade (Hetman et al., 1999). Interestingly, BDNF is released in an activity-dependent manner (Goodman et al., 1996), thus raising the possibility that potentially neuroprotective effects of SE-induced BDNF release may not be imparted to STEP-positive cells as a result of repressed MAPK activation. Furthermore ERK activation may afford protection against c-Jun N-terminal kinase (JNK)-mediated cell death (Xia et al., 1995). Interestingly, JNK is stimulated by seizure activity in the hippocampus (Jeon et al., 2000), and thus repressed ERK activity may increase the vulnerability of hilar interneurons to excitotoxic cell death via JNK- dependent signaling. In this context, it is interesting to note that somatostatinergic hilar interneurons are highly vulnerable to a number of excitatory insults, including ischemia and trauma, as well as SE (Lowenstein et al., 1992; Matsuyama et al., 1993). Given the profound neuroprotective effects of the MAPK pathway, these findings suggest that STEP-mediated repression of ERK activity may be a common element in the inherent vulnerability of these cells to excitotoxic stimuli. Future experiments will test this idea.
Footnotes
This work was supported by National Institutes of Health Grants MH62335 and NS47176 (K.O.), MH52711 and MH01527 (P.J.L.), and NS42826 (J.R.N.) and by Ohio State Neuroscience Center Core Grant 5P30NS045758. Y.-S.C. is a recipient of an Epilepsy Foundation fellowship, and B.L. is a recipient of an American Heart Association fellowship.
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Acsády L, Kamondi A, Sik A, Freund TF, Buzsáki G. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. 1998;18:3386–3403. doi: 10.1523/JNEUROSCI.18-09-03386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol. 2003;182:21–34. doi: 10.1016/s0014-4886(03)00086-4. [DOI] [PubMed] [Google Scholar]
- Boulanger LM, Lombroso PJ, Raghunathan A, During MJ, Wahle P, Naegele JR. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J Neurosci. 1995;15:1532–1544. doi: 10.1523/JNEUROSCI.15-02-01532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate-treated rats. J Comp Neurol. 1997;385:385–404. [PubMed] [Google Scholar]
- Buckmaster PS, Jongen-Rélo AL. Highly specific neuron loss preserves lateral inhibitory circuits in the dentate gyrus of kainate-induced epileptic rats. J Neurosci. 1999;19:9519–9529. doi: 10.1523/JNEUROSCI.19-21-09519.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Schwartzkroin PA. Physiological and morphological heterogeneity of dentate gyrus-hilus interneurons in the gerbil hippocampus in vivo. Eur J Neurosci. 1995;7:1393–1402. [PubMed] [Google Scholar]
- Buckmaster PS, Otero-Corchon V, Rubinstein M, Low MJ. Heightened seizure severity in somatostatin knockout mice. Epilepsy Res. 2002;48:43–56. doi: 10.1016/s0920-1211(01)00318-7. [DOI] [PubMed] [Google Scholar]
- Butcher SP, Henshall DC, Teramura Y, Iwasaki K, Sharkey J. Neuroprotective actions of FK506 in experimental stroke: in vivo evidence against an antiexcitotoxic mechanism. J Neurosci. 1997;17:6939–6946. doi: 10.1523/JNEUROSCI.17-18-06939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Brautigan DL. Protein phosphatase 2A suppresses MAP kinase signalling and ectopic protein expression. Cell Signal. 1999;11:575–580. doi: 10.1016/s0898-6568(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- Coffer P, de Jonge M, Mettouchi A, Binetruy B, Ghysdael J, Kruijer W. junB promoter regulation: Ras mediated transactivation by c-Ets-1 and c-Ets-2. Oncogene. 1994;9:911–921. [PubMed] [Google Scholar]
- Cossart R, Bernard C, Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108–115. doi: 10.1016/j.tins.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Criswell T, Beman M, Araki S, Leskov K, Cataldo E, Mayo LD, Boothman DA. Delayed activation of insulin-like growth factor-1 receptor/Src/MAPK/Egr-1 signaling regulates clusterin expression, a pro-survival factor. J Biol Chem. 2005;280:14212–14221. doi: 10.1074/jbc.M412569200. [DOI] [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- El Bahh B, Lespinet V, Lurton D, Coussemacq M, Le Gal La Salle G, Rougier A. Correlations between granule cell dispersion, mossy fiber sprouting, and hippocampal cell loss in temporal lobe epilepsy. Epilepsia. 1999;40:1393–1401. doi: 10.1111/j.1528-1157.1999.tb02011.x. [DOI] [PubMed] [Google Scholar]
- Esclapez M, Houser CR. Somatostatin neurons are a subpopulation of GABA neurons in the rat dentate gyrus: evidence from colocalization of pre-prosomatostatin and glutamate decarboxylase messenger RNAs. Neuroscience. 1995;64:339–355. doi: 10.1016/0306-4522(94)00406-u. [DOI] [PubMed] [Google Scholar]
- Ford SR, Leach FR. Improvements in the application of firefly luciferase assays. Methods Mol Biol. 1998;102:3–20. doi: 10.1385/0-89603-520-4:3. [DOI] [PubMed] [Google Scholar]
- Furuichi Y, Katsuta K, Maeda M, Ueyama N, Moriguchi A, Matsuoka N, Goto T, Yanagihara T. Neuroprotective action of tacrolimus (FK506) in focal and global cerebral ischemia in rodents: dose dependency, therapeutic time window and long-term efficacy. Brain Res. 2003;965:137–145. doi: 10.1016/s0006-8993(02)04151-3. [DOI] [PubMed] [Google Scholar]
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- Haas KZ, Sperber EF, Moshé SL, Stanton PK. Kainic acid-induced seizures enhance dentate gyrus inhibition by downregulation of GABAB receptors. J Neurosci. 1996;16:4250–4260. doi: 10.1523/JNEUROSCI.16-13-04250.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund L, Swanson LW, Köhler C. The projection of the supramammillary nucleus to the hippocampal formation: an immunohistochemical and anterograde transport study with the lectin PHA-L in the rat. J Comp Neurol. 1984;229:171–185. doi: 10.1002/cne.902290204. [DOI] [PubMed] [Google Scholar]
- Han ZS, Buhl EH, Lorinczi Z, Somogyi P. A high degree of spatial selectivity in the axonal and dendritic domains of physiologically identified local-circuit neurons in the dentate gyrus of the rat hippocampus. Eur J Neurosci. 1993;5:395–410. doi: 10.1111/j.1460-9568.1993.tb00507.x. [DOI] [PubMed] [Google Scholar]
- Hetman M, Xia Z. Signaling pathways mediating anti-apoptotic action of neurotrophins. Acta Neurobiol Exp (Wars) 2000;60:531–545. doi: 10.55782/ane-2000-1374. [DOI] [PubMed] [Google Scholar]
- Hetman M, Kanning K, Cavanaugh JE, Xia Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- Houser CR, Esclapez M. Vulnerability and plasticity of the GABA system in the pilocarpine model of spontaneous recurrent seizures. Epilepsy Res. 1996;26:207–218. doi: 10.1016/s0920-1211(96)00054-x. [DOI] [PubMed] [Google Scholar]
- Howard EM, Gao TM, Pulsinelli WA, Xu ZC. Electrophysiological changes of CA3 neurons and dentate granule cells following transient forebrain ischemia. Brain Res. 1998;798:109–118. doi: 10.1016/s0006-8993(98)00403-x. [DOI] [PubMed] [Google Scholar]
- Inoue D, Kido S, Matsumoto T. Transcriptional induction of FosB/DeltaFosB gene by mechanical stress in osteoblasts. J Biol Chem. 2004;279:49795–49803. doi: 10.1074/jbc.M404096200. [DOI] [PubMed] [Google Scholar]
- Jeon SH, Kim YS, Bae CD, Park JB. Activation of JNK and p38 in rat hippocampus after kainic acid induced seizure. Exp Mol Med. 2000;32:227–230. doi: 10.1038/emm.2000.37. [DOI] [PubMed] [Google Scholar]
- Jiang W, Van Cleemput J, Sheerin AH, Ji SP, Zhang Y, Saucier DM, Corcoran ME, Zhang X. Involvement of extracellular regulated kinase and p38 kinase in hippocampal seizure tolerance. J Neurosci Res. 2005;81:581–588. doi: 10.1002/jnr.20566. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 2003;23:2440–2452. doi: 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler C, Swanson LW, Haglund L, Wu JY. The cytoarchitecture, histochemistry and projections of the tuberomammillary nucleus in the rat. Neuroscience. 1985;16:85–110. doi: 10.1016/0306-4522(85)90049-1. [DOI] [PubMed] [Google Scholar]
- Kondratyev A, Gale K. Temporal and spatial patterns of DNA fragmentation following focally or systemically-evoked status epilepticus in rats. Neurosci Lett. 2001;310:13–16. doi: 10.1016/s0304-3940(01)02055-9. [DOI] [PubMed] [Google Scholar]
- Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci. 2005;25:1137–1148. doi: 10.1523/JNEUROSCI.4288-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Callaway EM, Albright TD, Gage FH. Defining a molecular atlas of the hippocampus using DNA microarrays and high-throughput in situ hybridization. J Neurosci. 2005;24:3879–3889. doi: 10.1523/JNEUROSCI.4710-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leranth C, Malcolm AJ, Frotscher M. Afferent and efferent synaptic connections of somatostatin-immunoreactive neurons in the rat fascia dentata. J Comp Neurol. 1990;295:111–122. doi: 10.1002/cne.902950110. [DOI] [PubMed] [Google Scholar]
- Lombroso PJ, Murdoch G, Lerner M. Molecular characterization of a protein-tyrosine-phosphatase enriched in striatum. Proc Natl Acad Sci USA. 1991;88:7242–7246. doi: 10.1073/pnas.88.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama T, Tsuchiyama M, Nakamura H, Matsumoto M, Sugita M. Hilar somatostatin neurons are more vulnerable to an ischemic insult than CA1 pyramidal neurons. J Cereb Blood Flow Metab. 1993;13:229–234. doi: 10.1038/jcbfm.1993.28. [DOI] [PubMed] [Google Scholar]
- McDonald DR, Bamberger ME, Combs CK, Landreth GE. Beta-amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J Neurosci. 1998;18:4451–4460. doi: 10.1523/JNEUROSCI.18-12-04451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki A, Lu YF, Tomizawa K, Matsui H. An immunosuppressant, FK506, protects against neuronal dysfunction and death but has no effect on electrographic and behavioral activities induced by systemic kainate. Neuroscience. 1998;86:855–865. doi: 10.1016/s0306-4522(98)00071-2. [DOI] [PubMed] [Google Scholar]
- Noto T, Ishiye M, Furuichi Y, Keida Y, Katsuta K, Moriguchi A, Matsuoka N, Aramori I, Goto T, Yanagihara T. Neuroprotective effect of tacrolimus (FK506) on ischemic brain damage following permanent focal cerebral ischemia in the rat. Brain Res Mol Brain Res. 2004;128:30–38. doi: 10.1016/j.molbrainres.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Obenaus A, Esclapez M, Houser CR. Loss of glutamate decarboxylase mRNA containing neurons in the rat dentate gyrus following pilocarpine-induced seizures. J Neurosci. 1993;13:4470–4485. doi: 10.1523/JNEUROSCI.13-10-04470.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiishi T, Saitoh Y, Yukawa A, Saji M, Ren Y, Shirao T, Miyamoto H, Nakata H, Sekino Y. High level of adenosine A1 receptor-like immunoreactivity in the CA2/CA3a region of the adult rat hippocampus. Neuroscience. 1999;93:955–967. doi: 10.1016/s0306-4522(99)00179-7. [DOI] [PubMed] [Google Scholar]
- Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- Paul S, Olausson P, Venkitaramani DV, Ruchkina I, Moran TD, Tronson N, Mills E, Hakim S, Salter MW, Taylor JR, Lombroso PJ. The striatal-enriched protein tyrosine phosphatase gates long-term potentiation and fear memory in the lateral amygdala. Biol Psychiatry. 2007 doi: 10.1016/j.biopsych.2006.08.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Askalan R, Paul S, Kalia LV, Nguyen TH, Pitcher GM, Salter MW, Lombroso PJ. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34:127–138. doi: 10.1016/s0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- Prendergast MA, Rogers DT, Mulholland PJ, Littleton JM, Wilkins LH, Jr, Self RL, Nath A. Neurotoxic effects of the human immunodeficiency virus type-1 transcription factor Tat require function of a polyamine sensitive-site on the N-methyl-D-aspartate receptor. Brain Res. 2002;954:300–307. doi: 10.1016/s0006-8993(02)03360-7. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Muller RU, Kubie JL, Ranck JB., Jr The positional firing properties of medial entorhinal neurons: description and comparison with hippocampal place cells. J Neurosci. 1992;12:1945–1963. doi: 10.1523/JNEUROSCI.12-05-01945.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Dashtipour K. Neuroplasticity in the damaged dentate gyrus of the epileptic brain. Prog Brain Res. 2005;136:319–328. doi: 10.1016/s0079-6123(02)36027-8. [DOI] [PubMed] [Google Scholar]
- Robbins RJ, Brines ML, Kim JH, Adrian T, de Lanerolle N, Welsh S, Spencer DD. A selective loss of somatostatin in the hippocampus of patients with temporal lobe epilepsy. Ann Neurol. 1991;29:325–332. doi: 10.1002/ana.410290316. [DOI] [PubMed] [Google Scholar]
- Roux PP, Colicos MA, Barker PA, Kennedy TE. p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. J Neurosci. 1999;19:6887–6896. doi: 10.1523/JNEUROSCI.19-16-06887.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE. Epileptogenesis in the parahippocampal region. Parallels with the dentate gyrus. Ann NY Acad Sci USA. 2000;911:305–327. doi: 10.1111/j.1749-6632.2000.tb06734.x. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA. Origins of the epileptic state. Epilepsia. 1997;38:853–858. doi: 10.1111/j.1528-1157.1997.tb01250.x. [DOI] [PubMed] [Google Scholar]
- Singleton RH, Stone JR, Okonkwo DO, Pellicane AJ, Povlishock JT. The immunophilin ligand FK506 attenuates axonal injury in an impact-acceleration model of traumatic brain injury. J Neurotrauma. 2001;18:607–614. doi: 10.1089/089771501750291846. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: the “dormant basket cell” hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus. 1991;1:41–66. doi: 10.1002/hipo.450010106. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Dean E, Sollas AL, Goodman JH. Apoptosis and necrosis induced in different hippocampal neuron populations by repetitive perforant path stimulation in the rat. J Comp Neurol. 1996;366:516–533. doi: 10.1002/(SICI)1096-9861(19960311)366:3<516::AID-CNE10>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS. MSK2 and MSK1 mediate the mitogen- and stress induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- Tucker MS, Khan I, Fuchs-Young R, Price S, Steininger TL, Greene G, Wainer BH, Rosner MR. Localization of immunoreactive epidermal growth factor receptor in neonatal and adult rat hippocampus. Brain Res. 1993;631:65–71. doi: 10.1016/0006-8993(93)91187-w. [DOI] [PubMed] [Google Scholar]
- Tuff LP, Racine RJ, Adamec R. The effects of kindling on GABA-mediated inhibition in the dentate gyrus of the rat. I. Paired-pulse depression. Brain Res. 1983;277:79–90. doi: 10.1016/0006-8993(83)90909-5. [DOI] [PubMed] [Google Scholar]
- Uchino H, Minamikawa-Tachino R, Kristian T, Perkins G, Narazaki M, Siesjo BK, Shibasaki F. Differential neuroprotection by cyclosporin A and FK506 following ischemia corresponds with differing abilities to inhibit calcineurin and the mitochondrial permeability transition. Neurobiol Dis. 2002;10:219–233. doi: 10.1006/nbdi.2002.0514. [DOI] [PubMed] [Google Scholar]
- Vogt KE, Regehr WG. Cholinergic modulation of excitatory synaptic transmission in the CA3 area of the hippocampus. J Neurosci. 2001;21:75–83. doi: 10.1523/JNEUROSCI.21-01-00075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A, Patrylo PR, Spencer DD. Decrease in inhibition in dentate granule cells from patients with medial temporal lobe epilepsy. Ann Neurol. 1999;45:92–99. doi: 10.1002/1531-8249(199901)45:1<92::aid-art15>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gelowitz DL, Lai CT, Boulton AA, Yu PH. Gradation of kainic acid induced rat limbic seizures and expression of hippocampal heat shock protein-70. Eur J Neurosci. 1997;9:760–769. doi: 10.1111/j.1460-9568.1997.tb01424.x. [DOI] [PubMed] [Google Scholar]