

Abstract
A phosphoramidite reagent of N6-(2-deoxy-d-erythro-pentofuranosyl)-2,6-diamino-1,4-dihydro-4-oxo-5-N-methylformamidopyrimidine (MeFapy-dGuo) lesions was synthesized in four steps from 2′-deoxyguanosine. Fapy nucleosides can rearrange to the pyranose form when the 5′-hydroxyl group is unprotected. The phosphoramidite was incorporated into oligonucleotides using solid-phase synthesis by adjusting the deprotection time for removal of the 5′-dimethoxytrityl group of the MeFapy-dGuo nucleotide, thereby minimizing its rearrangement to the ribopyranose. The furanose and pyranose forms were differentiated by a series of 2D NMR experiments.
Introduction
Formamidopyrimidines1 (Fapy) are DNA lesions that result from ring opening of the imidazole portions of purines. Although formally a hydrolysis product, the unsubsituted Fapy-dGuo lesion arises from initial oxidation of dGuo. A second class of Fapy lesions are derived from initial N7-alkylation followed by addition of hydroxide to the C8-position of the N7-cationic species and subsequent imidazolium ring-opening; this later process results in an N5-alkylated Fapy-dGuo adduct such as MeFapy-dGuo (1, 2). A competing pathway is depurination of the N7-cationic intermediate resulting in the well-studied abasic site (3, 4).
A number of electrophiles react predominantly with DNA at the N7-dGuo position; however, only a small number of N5-substituted Fapy-dGuo lesions derived from initial N7-alkylation have been characterized to date (2). This may be due to the slow rate of imidazolium ring-opening at neutral pH; the efficiency of the ring-opening increases at more basic pH values. The best studied of the N5-alkylated Fapy’s are those derived from initial N7-reaction with methylating agents (MeFapy-dGuo) and aflatoxin B1 epoxide (AFB-Fapy-dGuo).
Methylation of DNA results from exposure to exogenous agents such as dimethyl sulfate, N-methylnitrosoamines and the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) (5, 6). Methylation can also occur from reaction with S-adenosyl-L-methionine, which is present in cell nuclei for the enzymatic methylation of cytidine and adenine bases in DNA; methylation of dGuo occurs by a non-enzymatic pathway (7,8). Most methylating agents afford N7-methyl-dGuo adduct as the major product with minor products resulting from reaction with exocyclic oxygen and nitrogen atoms. A compilation of rate data for the competing imidazolium ring-opening and depurination pathways at the nucleoside level suggest that the partitioning favors depurination at neutral pH (2). One study noted 7-methyl-dGuo in a poly-(dGuo-dCyd) oligonucleotide was remarkably resistant to imidazolium ring-opening at pH 8.9 and 37° C (9). However, analysis of the DNA from the bladders and livers of rats that had been exposed to N-methylnitrosourea, N,N-dimethylnitrosamine or 1,2-dimethylhydrazine indicated that the MeFapy-dGuo adduct was the major persistent lesion over time (10, 11). These observations suggest that N5-substituted Fapy-dGuo adducts may contribute to the long-term genotoxicity of alkylating agents.
We report here a versatile approach for the site-specific synthesis of oligonucleotides containing the MeFapy-dGuo adduct and their characterization. A phosphoramidite from the MeFapy-dGuo mono-nucleoside was prepared in four steps and subsequently incorporated into oligonucleotides by solid-phase synthesis. Pyranose formation was minimized by modification of the reaction conditions for removal of the 5′-dimethoxytrityl group
MATERIAL AND METHODS
N2-[(Dimethylamino)methylene]-N6-[5′-O-(bis(4-methoxyphenyl)phenylmethyl)-2-deoxy-β-d-erythro-pentofuranosyl]-2,6-diamino-3,4-dihydro-4-oxo-5-N-methylformamidopyrimidine (3)
Methyl iodide (1.5 mL, 24 mmol) was added dropwise to a stirred solution of 1 (12) (1 g, 1.60 mmol) in dry, degassed dimethylsulfoxide (8 mL) at room temperature. After 45 min, the excess methyl iodide was removed in vacuo (rotary evaporator), and the remaining DMSO was diluted with 10 mL of distilled water. A 1 M solution of NaOH (4 mL) was added dropwise over 1 min. The resulting clear solution was immediately neutralized by the dropwise addition of 0.1 M HCl. The pH was monitored by pH paper. The precipitate was filtered off to give pure 3 (0.79 g, 75 %). 1H NMR (DMSO-d6) mixture of isomers: δ 11.10 (broad s, 1 H, NHCO), 8.37 (s, 0.5H, N=CH), 8.29 (s, 0.5H, N=CH), 7.82 (s, 0.5H, CHO), 7.74 (s, 0.5H, CHO), 7.38-7.23 (m, 9H, aromatic), 6.87-6.84 (m, 5H, NH + aromatic), 6.32-6.29 (m, 1H, H’-1), 5.39 (d, 0.5H, OH-3′, J = 10 Hz) 5.32 (d, 0.5H, OH-3′, J = 10 Hz), 4.31-4.22 (m, 1H, H’-3), 3.95-3.85 (m, 1H, H’-4), 3.71 (s, 6H, CH3O), 3.03-2.95 (m, 1H, H-5′), 2.80-2.75 (m, 1H, H-5′), 2.89 (s, 1.5H, CH3N-CHO), 2.86 (s, 1.5H, CH3-N-CH=), 2.84 (s, 1.5H, CH3N-CHO), 2.79 (s, 1.5H, CH3-N-CH=), 2.33-2.15 (m, 1H, H-2′), 2.08-1.87 (m, 1H, H-2′). HRMS (FAB+) m/z calcd for C35H40N6O7 [M + H]+ 656.2985, found 656.2990.
N2-[(Dimethylamino)methylene]-N6-[5-O-(bis(4-methoxyphenyl)phenylmethyl)-3-O-[(N,N-diisopropylamino)(2-cyanoethoxy)phosphino]-2-deoxy-β-d-erythro-pentofuranosyl]-2,6-diamino-3,4-dihydro-4-oxo-5-N-methylformamidopyrimidine (4)
Compound 3 (100 mg, 0.15 mmol) was co-evaporated with anhydrous pyridine and dried overnight under high vacuum. The gummy residue was dissolved in dry CH2Cl2 (10 mL) and a solution of anhydrous 1H-tetrazole (12.60 mg, 0.18 mmol) was added, followed by 2-cyanoethyl-N,N,N’,N’-tetraisopropyl-phosphorodiamidite (63.29 mg, 0.21 mmol). The solution was stirred at room temperature for 2 h. The solvent was removed in vacuo (rotary evaporator) and the crude product was purified by flash chromatography on silica gel (97:2:1 CH2Cl2:MeOH:pyridine) to give 4 as a mixture of stereoisomers (100 mg, 78%). 1H NMR (CH2Cl2-d2) mixture of isomers: δ 8.52 (s, 0.5H, N=CH), 8.44 (s, 0.5H, N=CH), 7.95 (s, 0.5H, CHO), 7.90 (s, 0.5H, CHO), 7.38-7.15 (m, 9H, aromatic), 6.88-6.84 (m, 4H, aromatic), 6.51-6.29 (m, 1H, H-1′), 6.01 (d, 0.5H, NH, J = 12 Hz), 5.92 (d, 0.5H, NH, J = 12 Hz), 4.74-4.51 (m, 1H, H-3′), 4.31-4.15 (m, 1H, H-4′), 3.78-3.63 (m, 8H, CH3O + POCH2), 3.63-3.43 (m, 2H, isopropyl CH), 3.20-3.03 (m, 2H, H-5′), 3.04 (s, 1.5 H, CH3-N-CH= ), 3.02 (s, 1.5 H, CH3-N-CH= ), 3.00 (s, 1.5 H, CH3N-CHO), 2.83 (s, 1.5 H, CH3N-CHO), 2.65 (t, 2H, CH2-CN), 2.17-2.02 (m, 1H, H-2′), 2.02-1.89 (m, 1H, H-2′), 1.22-1.01 (m, 12H, isopropyl CH3). 31P NMR (CH2Cl2-d2 121 MHz) δ 150.47, 150.22, 149.74, 149.62, 149.59, 148.80. HRMS (FAB+) m/z calcd for C44H58N8O8P [M + H]+ 857.4115, found 857.4143.
N6-[(3S,4R)-3,4,5-trihydroxypentyl]-2,6-diamino-3,4-dihydro-4-oxo-5-N-methyl-formamidopyrimidine (9)
MeFapy-dGuo (2 mg, 0.0066 mmol) was dissolved in water (1 mL) and NaB(CN)H3 (1.3 mg, .02 mmol) was added in daily for 30 days. The reaction was monitored with Phenomenex Gemini-C18 column and purified using gradient 3 to give 9 (0.5 mg, 25 %). 1H NMR (DMSO-d6) δ 7.70 (s, 1H, CHO), 6.50 (bs, 3H, NH2, NH), 4.51-4.49 (m, 1H, H-4′), 4.46-4.44 (m, 1H, H-3′), 4.32 (m, 1H, OH-3′), 3.49-3.47 (m, 2H , H-5′), 3.25-3.24 (m, 2H , H-1′), 2.74 (m, 3H, CH3), 1.79-1.76 (m, 1H, H-2′), 1.42-1.38 (m, 1H,H-2′). HRMS (FAB+) m/z calcd for C11H19N5O5 [M + H]+ 301.1464, found 301.1458.
Oligonucleotide Synthesis
The oligodeoxynucleotides were synthesized on a Perseptive Biosystems Model 8909 DNA synthesizer on a 1-μmol scale using their Expedite reagents with the standard synthetic protocol for the coupling of the unmodified bases. The coupling of the MeFapy-dGuo phosphoroamidite (4) was performed off-line for 30 min as previously described (13). The DMTr group of the MeFapy-dGuo was removed on the DNA synthesizier with protocol 10/20 (160 μL of Cl3CCO2H for 20 s, short deprotection) for MeFapy-dGuo furanose and with protocol 20/50 (320 μL of Cl3CCO2H for 50 s, long deprotection) plus an additional 3 min in standby mode for MeFapy-dGuo pyranose. The remainder of the synthesis was performed on-line using standard protocols. Unless otherwise noted, the modified oligodeoxynucleotides were cleaved from the solid support and the exocyclic amino groups were deprotected in a single step using 0.1 M NaOH at room temperature overnight.
HPLC purification
A YMC ODS-AQ column (250 × 4.6 mm, flow rate 1.5 ml/min, 250 × 4.6 mm, flow rate 10 ml/min) or Phenomenex Gemini-C18 column (250 × 4.6 mm, flow rate 1.5 ml/min, 250 × 4.6 mm, flow rate 5 ml/min) was used to monitor reactions and for oligonucleotide purifications. Oligonucleotides were detected by their UV absorbance at 254 nm. The mobile phase consisted of acetonitrile and 100 mM ammonium formate buffer.
Gradient 1
initial conditions were 1% acetonitrile; a linear gradient to 10% acetonitrile over 15 min; a linear gradient to 20% acetonitrile over 5 min; isocratic at 20% acetonitrile for 5 min; a linear gradient to 80% acetonitrile over 2.5 min; isocratic at 80% acetonitrile for 3 min; then a linear gradient to the initial conditions over 3 min.
Gradient 2
initial conditions were 1% acetonitrile; a linear gradient to 5% acetonitrile over 5 min; a linear gradient to 12% acetonitrile over 15 min; a linear gradient to 80% acetonitrile over 2 min; isocratic at 80% acetonitrile for 2 min; then a linear gradient back to the initial conditions over 3 min.
Gradient 3
initial conditions were 1% acetonitrile; a linear gradient to 5% acetonitrile over 10 min; a linear gradient to 10% acetonitrile over 5 min; a linear gradient to 80% acetonitrile over 2 min; isocratic at 80% acetonitrile for 2 min; then a linear gradient back to the initial conditions over 3 min.
Enzymatic digestion and analysis of oligonucleotides
The enzymatic digestion of oligonucleotides was carried out in a single step as follows: the oligonucleotide (0.5 A260 units) was dissolved in 70 μL of buffer (pH 7, 0.01 M Tris-HCl, 0.01 M MgCl2). DNase I (5 units), alkaline phosphatase (1.7 units), and snake venom phosphodiesterase I, type II (0.02 units) were added and the solution was incubated at 37 °C for 1.5 h. HPLC analysis was performed using solvent gradient 1.
MALDI-TOF Sequencing of Oligonucleotides
The modified oligonucleotides (0.3 A260 units) were treated with 2 milliunits of phosphodiesterase I (PI) in ammonium hydrogen citrate buffer (pH 9.4, 24 μL, 20 mM MgSO4) at 37 °C. An aliquot of 4 μL was taken before enzyme addition and at 1, 8, 18, 28 and 38 min time points after enzyme addition. The aliquots were added to the same vial, which was kept frozen on dry ice. A complementary experiment was performed in which the modified oligonucleotides (0.3 A260 units) were incubated with 2 milliunits of phosphodiesterase II (PII) in ammonium acetate (20 mM, pH 6.6). The aliquots were taken in at the same manner. The two digested mixtures were desalted using Millipore C18 Ziptips and eluted onto a MALDI plate on a matrix of 3-hydroxypicolinic acid and ammonium hydrogen citrate.
Thermal Melting (Tm) analysis
A 1:1 mixture of MeFapy-dGuo oligonucleotide 8a and its complementary strand (0.25 A260 units) was placed in melting buffer (0.5 mL, 10 mM Na2HPO4/NaH2PO4, 1.0 M NaCl, 50 μM Na2EDTA, pH 7.2). UV measurements monitoring the absorbance at 260 nm, were taken at 1 min intervals with a 1° C/min temperature gradient. The temperature was cycled between 10 and 85° C. The first derivative of the melting curve was used to establish Tm values.
5′-TT-(MeFapy-dGuo)-TTC-3′ (5)
Purified by reversed-phase HPLC using gradient 1. MALDI-TOF MS (HPA) m/z calcd for (M-H), 1803.8; found MeFapy-dGuo furanose 1803.1; found MeFapy-dGuo pyranose 1803.8.
5′-A-(MeFapy-dGuo)-C-3′ (6)
This oligonucleotide was synthesized with a 5′-dimethoxytrityl (DMTr) group still in place. The oligonucleotide was cleaved from the solid support and the protecting groups removed with conc. ammonia at room temperature overnight. After filtration through a 0.2 μM filter (Millex™, Millipore Corporation, Bedford, MA, USA) and drying under vacuum, the oligonucleotide was passed through a Poly-Pak cartridge (Glen Research Co., Inc.) with elution by 2% trifluoroacetic acid (TFA) to remove the 5′-DMTr group. The oligonucleotide was eluted from the cartridge with 20% acetonitrile in water. Purification was accomplished by reversed-phase chromatography with gradient 1. ESI/MS calcd for (M+H) 900.2; found MeFapy-dGuo furanose 900.1; found MeFapy-dGuo pyranose 900.1
5′-CCTCTTC-(MeFapy-dGuo)-CTCTC-3′ (7)
Purified by by reverse-phase HPLC gradient 2. MALDI-TOF MS (HPA) m/z calcd for (M-H), 3841.6; found MeFapy-dGuo furanose 3842.9; found MeFapy-dGuo pyranose 3843.0
5′-GCTAGC-(MeFapy-dGuo)-AGTCC-3′ (8)
Purified by reverse-phase HPLC gradient 1. MALDI-TOF MS (HPA) m/z calcd for (M-H), 3677.9; found MeFapy-dGuo furanose 3676.7,
NMR Spectroscopy
Trinucleotide samples were analyzed in 20 mM sodium phosphate buffer (pH 7.0) conatining 100 mM NaCl, 10 mM NaN3, and 50 μM Na2EDTA. Two-dimensional techniques: 1H-1H double quantum filtered correlated spectroscopy (DQF-COSY) (14), 1H-1H nuclear Overhauser effect spectroscopy (NOESY) (15, 16), 1H-1H total correlated spectroscopy (TOCSY) (17), and multiplicity edited 1H-13C heteronuclear single quantum correlation spectroscopy (HSQC) (18) experiments were performed at a 1H frequency of 500 MHz on a Bruker Avance spectrometer with a 5mm CP-TCI-Z cryoprobe. Heteronuclear 1H-31P COSY (19) experiments were performed at a 1H frequency of 600 MHz on Bruker Avance spectrometer with a QXI-XYZ probe. Experiments were conducted at 15 ± 0.5 °C. 1H and 31P spectra were referenced to internal 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid, sodium salt (3-TMSP) and external 85% H3PO4 (capillary in H2O), respectively. Typical acquisition parameters for homonuclear experiments were as follows: 2K complex data points with 512 increments, 32 scans per FID, sweep width of 5250 Hz in both dimensions, relaxation delay of 2.0 seconds, States-TPPI mode. Presaturation was sufficient for water suppression. NOESY and TOCSY spectra were recorded with mixing times of 300 and 80 ms, respectively. Acquisition parameters for heteronuclear 13C-1H HSQC experiments were as follows: 1K complex data points (sweep width = 5250 Hz), 360 increments (sweep width = 90000 Hz), 32 scans per FID, garp decoupling, relaxation delay of 1.8 seconds, Echo-Antiecho acquisition mode, pulses were optimized for 1JCH coupling constants (140 Hz). Acquisition parameters for heteronuclear 31P-1H COSY experiments were as follows: 1K complex data points (sweep width = 6000 Hz) with 256 increments (sweep width = 3000 Hz), 128 scans per FID, relaxation delay of 1.8 seconds, STATES mode, pulses were optimized for 3JPH coupling constants (12 Hz). Carrier frequencies were set at 4.7 ppm for 1H, 80 ppm for 13C, and 0 ppm for 31P. XWINNMR (v 3.5 patch level 6, Bruker Inc., Karlsruhe, Germany) was used for data processing. Apodization was attained using a skewed sinebell-squared function with a 90° shift in both dimensions (180° for COSY experiments); zero-filling and linear prediction was applied in the indirect dimension. Resonance assignments and peak integration was performed using the program SPARKY (20).
Results
Site-specific Synthesis of Oligonucleotides Containing MeFapy-dGuo
The MeFapy-dGuo phosphoramidite (4) was prepared in four steps and 24% overall yield from dGuo (Scheme 1). Conversion of dGuo to 1 was accomplished as previously described (12). Protected dGuo derivative 1 was treated with an excess of methyl iodide in DMSO for 45 min at room temperature to produce the cationic N7-methyl-dGuo 2, which was not isolated. After removal of the excess methyl iodide, 2 was briefly exposed to 1 M NaOH to affect ring-opening of the imidazolium ion, then neutralized with 0.1 M HCl to afford 3 as a white precipitate. The protected MeFapy-dGuo nucleoside 3 was isolated by filtration and converted to the desired MeFapy-dGuo phosphoramidite 4 without further purification.
Scheme 1.

Phosphoramidite 4 was used for the preparation of oligonucleotides containing the MeFapy-dGuo lesion at defined sites (Table 1). Incorporation of the modified nucleotide was performed off-line, using a manual coupling protocol (13). The critical step of the oligonucleotide synthesis was the deprotection of the 5′-hydroxyl group of the MeFapy-dGuo nucleotide, since the Fapy with a free 5′-hydroxyl group can undergo a rearrangement to the pyranose form (21, 22). The results of the synthesis 5′-TT-(MeFapy-dGuo)-TTC-3′ oligonucleotide at two deprotection times are shown in Figure 2. When the 5′-DMTr group was removed using a “short” deprotection cycle that consisted of 160 μL of Deblock reagent (3% Cl3CCO2H in methylene chloride) for 20 s, followed by incorporation of the subsequent nucleotide under standard conditions, five products were observed by HPLC analysis (Figure 2A). Components 1-5 were separated and re-analyzed by HPLC. Component 1 (earliest eluting) did not change composition upon re-analysis (Figure 2C). However, when components 2-5 were individually re-analyzed, they had all equilibrated to the same mixture with the other three (Figure 2D). Components 1 and 2-5 were analyzed by MALDI-TOF mass spectrometry and found to possess the masses consistent with the desired modified oligonucleotide (Table 1).
Table 1.
Mass spectrometric characterization of the oligonucleotides containing the MeFapy-dGuo lesion (furanose and pyranose forms)
| Oligonucleotide | m/z | |||
|---|---|---|---|---|
| observed | calcd | |||
| a: furanose | b: pyranose | |||
| 5 | 5′-TT-(MeFapy-dGuo)-TTC-3′ | 1803.1 | 1803.8 | 1803.8 |
| 6 | 5′-A-(MeFapy-dGuo)-C-3′ | 900.2 | 900.0 | 900.2 |
| 7 | 5′-CCTCTTC-(MeFapy-dGuo)-CTCTC-3′ | 3842.9 | 3843.0 | 3841.6 |
| 8 | 5′-GCTAGC-(MeFapy-dGuo)-AGTCC-3′ | 3677.9 | 3676.7 | |
Figure 2.

HPLC analyses of the 5′-TT-(MeFapy-dGuo)-TTC-3′ oligonucleotide. A. Analysis of the oligonucleotide synthesis with a “short” deprotection cycle. B. Analysis of the oligonucleotide synthesis with a “long” deprotection cycle. C. Analysis of peak 1, identified as the pyranose form of the MeFapy-dGuo lesion, after initial purification. D. Analysis of interconverting peaks 2-4, identified as the furanose form of the MeFapy-dGuo lesion, after each peak was individually purified.
We tentatively assigned component 1 as containing the pyranose form of the MeFapy-dGuo and components 2-5 as those containing the furanose forms from this limited information. In addition to the furanose and pyranose forms of the deoxyribose unit, α- and β-anomers and isomers due to restricted rotation about the C5-N5 and N5-formyl bonds are also possible, giving a total of eight possible MeFapy-dGuo species (22, 23). Previous NMR studies on the β-MeFapy ribonucleoside triacetate showed four species in a 1:1:8:8 ratio that were assigned as atropisomers (major) resulting from slow rotation of the C5-N5 bond and geometric isomers (minor) of the formyl group (22). We also observe only four of the possible eight isomers of the presumptive furanosyl MeFapy-dGuo in the oligonucleotide. The deglycosylated MeFapy-Gua base is observed as two peaks on HPLC, which must be due to geometric isomers of the formamide since the atopisomers of the base would be enantiomer and are not resolvable on an achiral stationary phase. We therefore assume that we are observing the formamide geometric isomers of α- and β-anomers of the furnaose form of the MeFapy-dGuo nucleotide and that the interconversion of the atropisomers is faster than the HPLC time scale. We reasoned that the MeFapy-Gua base would exist largely in the equatorial anomer of the pyranose form, leaving a total of two resolvable atropisomers of a single anomer. We subsequently observed that peak 1 was actually composed of two components, consistent with this prediction. It is possible that two small peaks eluting between peak 1 and 2 are the other two stereoisomers of the pyranose form but they were not examined. We further predicted that a longer deprotection cycle for the MeFapy-dGuo would favor isomerization to the pyranose form. When a longer deprotection cycle (320 μL of Cl3CCO2H for 50 s, plus an additional 3 min in standby mode) for removing the 5′-DMTr group of the MeFapy-dGuo nucleotide was utilized, a larger percentage of the pyranose form (peak 1) was observed (Figure 2B). It should be noted that the small peaks between peaks 1 and 2 also increased in intensity.NMR studies ultimately confirmed the assignment of the furanose and pyranose forms.
The presence of MeFapy-dGuo was confirmed by HPLC analysis of the enzymatic digestion of the oligonucleotides (Figure 3). Interestingly, the MeFapy-dGuo was not among the nucleosides arising from the digestion reaction; rather, the deglycosylated base, MeFapy-Gua, was observed. The modified oligonucleotides were sequenced by MALDI-TOF analyses of the controlled exonuclease digestions with phosphodiesterases I and II (24). The oligonucleotides were separately digested with phosphodiesterase I (snake venom phosphodiesterase) and II (bovine spleen phosphodiesterase) along with alkaline phosphatase. Aliquots of the digestion at various time points were stored at -70 °C, then subsequently combined for MALDI-TOF-MS analysis, which provided a mass ladder corresponding to the sequential loss of nucleotides from either the 3′- (phosphodiesterase I) or 5′-end (phosphodiesterase II). Figure 4 shows the MALDI-TOF-MS analysis from controlled digestion of oligonucleotide 8a (m/z 3677 Da) with phosphodiesterase I (top) and phosphodiesterase II (bottom). In this example, a mass was observed in the phosphodiesterase II digestion corresponding to loss of the MeFapy-dGuo nucleotide (m/z 1823.1 → 1463.6). In the complementary digestion, the nuclease was unable to digest through the MeFapy-dGuo lesion; however, a mass difference of 164.9 Da (m/z 2153.4 → 1988.5) corresponds to the loss of the MeFapy-Gua base from the oligonucleotide with the lesion at the 3′-terminus. In other examples, the nucleases were unable to digest through the MeFapy-dGuo nucleotide within the time scale of the digestion reaction. However, the complete sequence of the oligonucleotide was established by the observation of mass ladders from both the 5′- and 3′-sides up to the modified nucleotide.
Figure 3.

HPLC analysis of the enzymatic digestion of oligonucleotide 8a with phosphodiesterase I and alkaline phosphatase.
Figure 4.

Sequencing of oligonucleotide 8a by controlled enzymatic digestion with phosphodiesterase I (top) and II (bottom) followed by MALDI-TOF-MS.
NMR analysis of trinucleotide 5′-A-(MeFapy-dGuo)-C-3′
The NMR spectrum of the furanose and pyranose forms of the 5′-TT-(MeFapy-dGuo)-TTC-3′ oligonucleotide were complex due to the presence of multiple interconverting species, precluding a full analysis. A trinucleotide 5′-A-(MeFapy-dGuo)-C-3′ containing the putative furanose and pyranose forms of the MeFapy-dGuo was therefore synthesized using the “short” and “long” deprotection cycles described above and purified for NMR studies to establish the furanose and pyranose forms.
The high conformational mobility of the single strand trinucleotides 5′-A-(MeFapy-dGuo)-C-3′ containing either the furanose or pyranose form of the MeFapy-dGuo nucleotide prevented the evolution of inter-residue NOEs for use in sequence specific resonance assignments. The spin systems of the individual 2-deoxyribose units were identified from TOCSY spectra; the assignments of 2-deoxyribose proton resonances were based on connectivity and chemical shift. The carbon resonances of the 2-deoxyribose units were correlated from the proton assignments by multiplicity-edited HSQC spectra. Inter-residue connectivity was established by 3JPH3′ - 3JPH5” scalar couplings (Figure 5). The pyranose and furanose forms could be distinguished from the available data. A three-bond 31P-1H coupling is observed from the phosphate that bridges the 5′-dAdo and the MeFapy-dGuo residue (correlation a in Figure 5). Likewise, three-bond 31P-1H coupling established connectivity from the phosphate to the MeFapy-dGuo sugar (correlation b). A one-bond 1H-13C HSQC identified the MeFapy-dGuo carbon resonance involved in the phosphate bridge (correlation c) of the furanose form (C5′) as a methylene (negative phase peak, red) while this group was a methine (C4′) for the pyranose form (positive phase peak, black).
Figure 5.

NMR analysis of the furanose (I) and pyranose (II) forms of the MeFapy-dGuo lesion in 5′-A-(MeFapy-dGuo)-C-3′ sequence. Tile plots of 31P-1H COSY (top), 1H-1H TOCSY (middle), and 1H-13C HSQC (bottom) spectra. The connectivity was traced from H3′ of dAdo to the phosphate (a) to the H5” of the MeFapy-dGuo (b) then to C5′ of the MeFapy-dGuo for the furanose form (I), and from the phosphate to the H4′ of the MeFapy-dGuo (b) then to C4′ of the MeFapy-dGuo for the pyranose form (II). The multiplicity edited HSQC confirmed that the furanose C5′ is a methylene carbon and a methine carbon for the pyranose form (negative phase peaks = red, positive phase peaks = black).
The carbon resonances of the 2-deoxyribose unit of the MeFapy-dGuo were also compared to model 2-deoxyribosides (Figure 6). We observed a 13 ppm difference in chemical shift for the 4′-carbon resonance of the MeFapy-dGuo residue depending on the ring size. The 4′-carbon chemical shifts for the MeFapy-dGuo residue were 83.2 ppm for the furanose form and 69.6 ppm in the pyranose form in the 5′-A-(MeFapy-dGuo)-C-3′ trinucleotide. The furanose value is compared to that of the 3′,5′-dibutyrate of the AFB-Fapy-dGuo nucleoside, and methyl 2-deoxyribofurnanoside 3,5-dimethyl ether, which have C4 carbon chemical shifts of 83.7 and 82.4 ppm, respectively (25, 26). The C4′ carbon chemical shifts of the MeFapy-dGuo pyranose form compares favorably with the pyranose form of the AFB-Fapy-dGuo nucleoside and methyl 2-deoxyribopyranose 3,5-diacetate (66.6 and 69.0, respectively) (27).
Figure 6.

Comparison of the 13C chemical shifts for furanose and pyranose forms of the 2-deoxyribose units.
Stability of the MeFapy-dGuo Nucleotide in Single-Stranded DNA
The cationic N7-alkyl-dGuo adducts can undergo deglycosylation resulting in an abasic site. The corresponding N5-alkyl-Fapy-dGuo adducts have been reported to be stable in duplex DNA (2, 28). The mechanism for anomerization involves a ring-opened deoxyribose unit, which could be intercepted by the addition of water across the imine. An abasic site can result from this carbinolamine intermediate through loss of the N5-alkyl-Fapy-Gua base. We found that the 13mer containing the MeFapy-dGuo adduct (7a) was stable at neutral buffer conditions. However, deglycosylation was observed over a 48 h period at pH 6.5. This process was followed by HPLC and LC-ESI-MS (Figure 7). The MeFapy-Gua base was observed by HPLC. The intrinsic lyase activity of endonuclease V was then used to cleave the degycosylated product into two shorter oligonucleotides, which possessed the expected masses providing further evidence for the deglycosylation of MeFapy-Gua base (Figure 7, panel D).
Figure 7.

HPLC analysis of the deglycosylation of MeFapy-Gua base in oligonucleotide 5′-CCTCTTC-(MeFapy-dGuo)-CTCTC-3′ at pH 6.5. A. Starting 13mer oligonucleotide containing the MeFapy-dGuo nucleotide; B) partial deglycosylation after 24 h at pH 6.5; C) after 48 h; D) after addition of endonuclease V.
The reaction of NaB(CN)H3 with MeFapy-dGuo nucleoside, as a mixture of furanose and pyranose forms, in water was found to be slow. After ∼1 month, the reduced product 9 was isolated in 25% yield (Figure 8) and characterized by NMR and MS. The mass spectral fragmentation of 9 resulted in a product ion at m/z 274.1, which was assigned as loss of the formyl group; a second product ion was observed at m/z 256.2, which was due to subsequent dehydration. Fragmentation of the MeFapy-dGuo nucleoside resulted in product ions at m/z 184.2 and 156.2 corresponding to loss of 2′-deoxyribose and the formyl groups, respectively.
Figure 8.

Reduction products of the putative C1′-N6 imine of MeFapy-dGuo.
Oligonucleotide 7a (m/z 3842.97) containing the MeFapy-dGuo adduct was also treated with NaB(CN)H3 in water for 48 h. LC-ESI-MS analysis could not definitively resolve the reduced oligonucleotide from that of the starting material. After reduction, the pH of the solution was adjusted to 6.5; no deglycosylation of the MeFapy-Gua base was observed suggesting that reduction of the imine had occurred. The reduced oligonucleotide was treated with phosphodiesterases I and II, nuclease PI and alkaline phosphatase. Analysis of the digestion mixture revealed a new species, which was not the expected reduced nucleoside 9. The new species possessed an m/z of 591.2, which suggested it was dinucleotide 10 composed of the reduced nucleoside and a neighboring dCyd. It is not known if the neighboring dCyd is 5′ or 3′ to the reduced MeFapy-dGuo. We attribute the difference in reactivity between the MeFapy-dGuo nucleoside and oligonucleotide 7a toward reduction to the initial isomerization of the nucleoside to the more favored pyranose form, which is not available to the lesion when in oligonucleotide. Ring opening of the more favored pyranose form back to the intermediate imine is likely to be slower than for the furanose form.
Thermal melting temperature (Tm) of MeFapy-dGuo containing duplex 8a
MeFapy-dGuo containing oligonucleotide 8a was annealed to its complementary strand and the UV absorbance of the duplex was monitored as a function of temperature. The Tm of the unmodified duplex of 8a was previously determined to be 59° C (29, 30). Two transitions were observed for the MeFapy-dGuo containing duplex at 39° C and 51° C (Figure 9). We attribute the biphasic melting of the duplex to the presence of the α- (Tm= 39° C) and β-anomers (Tm= 51° C). The ratio β:α anomers is estimated to be ∼60:40 based on the relative absorption change for each phase of the melting curve.
Figure 9.

Thermal melting analysis of duplex of 8a. The Tm for the unmodified duplex was 59° C.
Discussion
A unique feature of Fapy lesions is that they exist in solution as a mixture of interconverting isomers, which include the β- and α-anomers, atropisomers that result from restricted rotation about the C5-N5-bond, and geometric isomers from rotamers due to slow rotation about the formyl N-CO bond (11, 22, 23, 25). The anomerization reaction presumably occurs through the ring-opened form of the deoxyribose unit. At the nucleoside level, the re-closure of the deoxyribose can also involve the 5′-hydroxyl group leading to the pyranose form of the nucleoside (21, 22). The last feature provides a significant challenge to the chemical synthesis of oligonucleotides containing Fapy lesions.
The MeFapy-dGuo lesion has been incorporated into DNA previously. The first method involved treating single stranded DNA containing a single dGuo with dimethyl sulfate followed by base (23, 31, 32). A shortcoming of the method is that methylation also occurs on O6 of guanine, N3 of adenine and other sites. Direct alkylation has used successfully for preparation of prepare oligonucleotides containing the AFB-Fapy-dGuo adduct (33, 34). The regiospecificity of reactions of aflatoxin epoxide is much higher than that of dimethyl sulfate. Ide and co-workers reported an enzymatic strategy in which 7-methyl-dGTP was incorporated opposite a dCyd residue of a template DNA by a DNA polymerase (35). The resulting oligonucleotide was then treated with hydroxide to convert the cationic 7-methyl-dGuo to a MeFapy-dGuo (35). It had been previously shown that 7-methyl-dGTP can substitute for GTP in a DNA polymerase reaction although the insertion rate of 7-methyl-dGTP is slower than the natural substrate (9). Since the polymerase is not selective for insertion of 7-methyl-dGTP over dGTP opposite dCyd, the method is limited to a template strand containing a single dCyd. A third approach, used by Ezaz-Nikpay and Verdine for the preparation of a duplex containing 7-methyl-dGuo, utilized two DNA strands with overlapping complementarity so that they self-assembled into a double-stranded oligomer with single-base gaps opposite dCyd. 7-Methyl-dGTP was ligated into the gaps with Sequenase and the nicks sealed by T4 DNA ligase (36, 37). The sequence design included a restriction site that allowed the ligation product to be digested into a 12-mer duplex. Although the authors did not carry out the transformation, base induced ring-opening of the cationic 7-methyl-dG would presumably provide oligonucleotides containing the MeFapy-dGuo. Greenberg and co-workers have developed the only solid-phase synthetic approach for the incorporation of the parent Fapy-dGuo and Fapy-dAdo lesions (38-40). Phosphoramidite of a dinucleotides in which the 3′-nucleotide was Fapy-dGuo or Fapy-dAdo were utilized to prevent isomerization of the ribofuranose to the pyranose form. A drawback to this approach is that four different phosphoramidite reagents are required in order to have a full range of potential sequence contexts. Greenberg and Carell have independently synthesized configurationally stable Fapy-dGuo analogues in which the N6 and deoxyribose ring oxygen atoms were replaced by a methylene group (41-44).
Burgdorf and Carell separated the α- and β-anomers of a protected Fapy-dGuo nucleoside analogue 11 (Figure 10) and the individual anomers were found to be stable toward anomerization in DMSO at 60° C for up to 24 h (45). The anomerization was slow in a 1:1 acetonitrile/water mixture at 50° C. Similarly, Greenberg and coworkers determined that the anomers of Fapy-dAdo 5′-phosphate 12 equilibrated over ∼ 6 hrs (28). Anomerization of AFB-Fapy-dGuo (13) in DNA was found to be acid catalyzed (25). Since the anomerization and the furanose to pyranose isomerization reactions most likely involve a common ring-opened deoxyribose intermediate, these results suggested to us that the ring-opening reaction could be minimized by controlling the conditions of the DMTr-deprotection step during solid-phase synthesis. To test this, phosphoramidite 4 was synthesized and incorporated into oligonucleotides (Scheme 2). The deprotection time for removal of the 5′-DMTr group of the MeFapy-dGuo nucleotide was varied before the next coupling reaction. The oligonucleotides were analyzed by HPLC, which indicated the presence of interconverting species. The furanose form of the MeFapy-dGuo nucleotide was favored when a “short” deprotection cycle was employed for removal of its 5′-DMTr group, while the pyranose form predominated when longer deprotection cycles were utilized. The structures of the furanose and pyranose forms of the MeFapy-dGuo nucleotide were confirmed by a series of 2D NMR studies (Figures 5 and 6). This provides a convenient and flexible approach to the synthesis of oligonucleotides containing the MeFapy-dGuo lesion.
Figure 10.
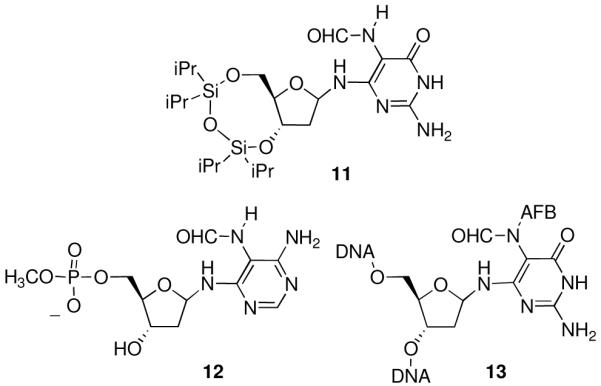
Anomerization of Fapy derivatives is slow at neutral pH
Greenberg showed that the deglycosylation of Fapy-dGuo in a single-strand oligonucleotide was slow with a t1/2 of 514 ± 30 h at pH 7.4 and 55 °C (28); deglycosylation of Fapy-dAdo was about 25 times faster. We observed that the MeFapy-G base was completely lost from oligonucleotide 8 at pH 6.5 over ∼48 h. The mechanism of deglycosylation by BER protein is usually envisioned as a direct nucleophilic displacement of the modified base by water (46). The lability of the MeFapy-Gua base under modestly acidic conditions suggests a possible acid-catalyzed mechanism for its deglycosylation. If general, this observation may be important for the detection of Fapy-dGuo lesions from biological samples. Incubation at pH 6.5 could selectively hydrolyze Fapy-dGuo lesions from DNA samples with minimal deglycosylation of unmodified bases. The Fapy-Gua base could then be separated from oligomeric DNA by solid-phase extraction or a size exclusion column providing a simple enrichment protocol for enhanced detection.
The thermal melting temperature of a duplex of 12mer oligonucleotide 8a was measured at a single DNA concentration. Interestingly, we observed two transitions (Tm = 39 and 51° C) for the dissociation of MeFapy-dGuo containing oligonucleotide 8a and its complement, which were 20 and 8° C lower than the unmodified duplex. We attribute the lower and higher melting transitions to the presence of the α- and β-anomers of the MeFapy-dGuo lesion. A similar biphasic melting profile was observed for a duplex containing the AFB-Fapy-dGuo in a 5′-CTAT-(AFB-Fapy-dGuo)-GATTCA-3′ sequence (Tm = 28 and 56° C) (47). If this duplex was allowed to equilibrate over several days, a single transitions at 56° C was observed, which was 15° C higher than the unmodified duplex. The structure of the more stable adduct was determined by NMR, which showed the AFB-Fapy-dGuo to be the β anomer with the aflatoxin moiety intercalated into the DNA helix. The structure of the lower melting duplex has not yet been determined. The Tm of the MeFapy-dGuo containing duplex 8a did not change over time. It is possible that the lower melting AFB-Fapy-dGuo duplex possesses the α-anomer and that isomerization to the β-anomer is a result of favorable stacking interactions of the intercalated aflatoxin moiety. Such a driving force is not present in the MeFapy-dGuo duplex.
Thermal melting analyses for duplexes containing Fapy-dGuo and configurationally stable Fapy-dGuo models have also been reported. The Tm of Fapy-dGuo in a 5′-TGCAGT-(Fapy-dGuo)-ACAGC-3′ sequence opposite dCyd was determined to be only 3° C lower than the unmodified duplex (48). Interestingly, the configurationally stable β-Fapy-dGuo models were more destabilizing than the natural lesion. The Tm of the Fapy-dGuo model where N6 is replaced with methylene group was 5° C lower than the duplex with the natural Fapy-dGuo (41). The Fapy-dGuo analogue in which the deoxyribose ring oxygen is replaced by a methylene group (cFapy-dGuo) lowered the Tm nearly 15° C versus the corresponding unmodified 11mer duplex (49); interestingly, the β-cFapy-dGuo containing duplex showed biphasic melting behavior when opposite mismatched dAdo and dGuo. The α-cFapy-dGuo showed larger destablization regardless of the opposing base (44). Oligonucleotides containing the α-anomer of dAdo have also been prepared and one study reported that the Tm of a duplex containing α-dAdo opposite dThd was 6° lower than the corresponding control duplex (50).
MeFapy-dGuo has been reported to be a strong block to replication (32, 51-54). Ide and coworkers examined the in vitro bypass of an oligonucleotide containing a site-specific MeFapy-dGuo lesion by Klenow fragment of E. coli DNA polymerase I (KF) (54); they concluded that the extension step was the major kinetic barrier to the lesion bypass of MeFapy-dGuo by Kf. Replication studies of Fapy-dGuo and its configurationally stable analogues have also been reported (43, 44). The mutagenicity of the Fapy-dGuo lesion was low in bacteria (55, 56); the mutagenic frequency of Fapy-dGuo was higher in mammalian cell lines and also found to be sequence dependent (55). The major mutation was Gua→Thy transversions in both systems. In vitro studies found that the α-cFapy-dGuo model was a strong block to replication (44), consistent with previous reports with oligonucleotides containing α-dAdo (57). Replication studies of the MeFapy-dGuo with human bypass polymerases would be of interest and such studies are ongoing in our laboratory.
Conclusion
Oligonucleotides containing MeFapy-dGuo lesions were synthesized using a protected phosphoramidite of the modified mono-nucleoside. It was discovered that either the furanose or pyranose form of the MeFapy-dGuo lesion could be favored by adjusting the deprotection cycle during solid-phase synthesis. Selective, phase-sensitive 31P-1H COSY, 1H-1H TOCSY, and 13C-1H HSQC NMR spectra were used to confirm the structure of the MeFapy-dGuo lesion (i.e. furanose vs. pyranose). The synthesis of the MeFapy-dGuo described here is potentially extendable to Fapy-dGuo lesions derived from other N7-alkylating agent (2).
Supplementary Material
Figure 1.

Formamidopyrimidine lesions from initial oxidation and N7-methylation of dGuo
Acknowledgements
This work was support by NIH Grants PO1 ES05355 and center grant P30 ES00267. Vanderbilt University, the Vanderbilt Center in Molecular Toxicology (P30 ES00267) and NIH Grant RR05805 provided funding for the NMR instrumentation.
Footnotes
- Fapy
- formamidopyrimidine
- Fapy-dGuo
- N6-(2-Deoxy-d-erythro-pentofuranosyl)-2,6-diamino-3,4-dihydro-4-oxo-5-formamidopyrimidine
- MeFapy-dGuo
- N6-(2-Deoxy-d-erythro-pentofuranosyl)-2,6-diamino-3,4-dihydro-4-oxo-5-N-methylformamidopyrimidine
- AFB
- aflatoxin B1
- Tm
- thermal melting temperature
- COSY
- correlated spectroscopy
- NOESY
- nuclear Overhauser effect spectroscopy
- TOCSY
- total correlated spectroscopy
- HSQC
- heteronuclear single quantum correlation spectroscopy
REFERENCES
- (1).Greenberg MM. In vitro and in vivo effects of oxidative damage to deoxyguanosine. Biochem. Soc. Trans. 2004;32:46–50. doi: 10.1042/bst0320046. [DOI] [PubMed] [Google Scholar]
- (2).Gates KS, Nooner T, Dutta S. Biologically relevant chemical reactions of N7-alkylguanine residues in DNA. Chem. Res. Toxicol. 2004;17:839–856. doi: 10.1021/tx049965c. [DOI] [PubMed] [Google Scholar]
- (3).Boiteux S, Guillet M. Abasic sites in DNA: Repair and biological consequences in Saccharomyces cerevisiae. DNA Repair. 2004;3:1–12. doi: 10.1016/j.dnarep.2003.10.002. [DOI] [PubMed] [Google Scholar]
- (4).Loeb LA, Preston DB. Mutagenesis by apurinic/apyrimidinic sites. Annu. Rev. Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- (5).Singer B, Kuśmierek JT. Chemical mutagenesis. Annu. Rev. Biochem. 1982;51:655–693. doi: 10.1146/annurev.bi.51.070182.003255. [DOI] [PubMed] [Google Scholar]
- (6).Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- (7).Rydberg B, Lindahl T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982;1:211–216. doi: 10.1002/j.1460-2075.1982.tb01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Barrows LR, Magee PN. Nonenzymatic methylation of DNA by S-adenosylmethionine in vitro. Carcinogenesis. 1982;3:349–351. doi: 10.1093/carcin/3.3.349. [DOI] [PubMed] [Google Scholar]
- (9).Hendler S, Furer E, Srinivasan PR. Synthesis and chemical properties of monomers and polymers containing 7-methylguanine and an investigation of their substrate or template properties for bacterial deoxyribonucleic acid or ribonucleic acid polymerases. Biochemistry. 1970;9:4141–4153. doi: 10.1021/bi00823a017. [DOI] [PubMed] [Google Scholar]
- (10).Beranek DT, Weis CC, Evans FE, Chetsanga CJ, Kadlubar FF. Identification of N5-methyl-N5-formyl-2,5,6-triamino-4-hydroxypyrimidine as a major adduct in rat liver DNA after treatment with the carcinogens, N,N-dimethylnitrosamine or 1,2-dimethylhydrazine. Biochem. Biophy. Res. Commun. 1983;110:625–631. doi: 10.1016/0006-291x(83)91195-6. [DOI] [PubMed] [Google Scholar]
- (11).Kadlubar FF, Beranek DT, Weis CC, Evans FE, Cox R, Irving CC. Characterization of the purine ring-opened 7-methylguanine and its persistence in rat bladder epithelial DNA after treatment with the carcinogen N-methylnitrosourea. Carcinogenesis. 1984;5:587–592. doi: 10.1093/carcin/5.5.587. [DOI] [PubMed] [Google Scholar]
- (12).McBride LJ, Kierzek R, Beaucage SL, Caruthers MH. Nucleotide chemistry. 16. Amidine protecting groups for oligonucleotide synthesis. J. Am. Chem. Soc. 1986;108:2040–2048. [Google Scholar]
- (13).Elmquist CE, Stover JS, Wang Z, Rizzo CJ. Site-specific synthesis and properties of oligonucleotides containing C8-deoxyguanosine adducts of the dietary mutagen IQ. J. Am. Chem. Soc. 2004;126:11189–11201. doi: 10.1021/ja0487022. [DOI] [PubMed] [Google Scholar]
- (14).Derome AE, Williamson MP. Rapid-pulsing artifacts in double-quantum-filtered COSY. J. Magn. Reson. 1990;88:177–185. [Google Scholar]
- (15).Jeener J, Meier BH, Bachmann P, Ernst RR. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979;71:4546–4553. [Google Scholar]
- (16).Wagner R, Berger S. Gradient-selected NOESY—A fourfold reduction of the measurement time for the NOESY experiment. J. Magn. Reson. 1996;123:119–121. doi: 10.1006/jmra.1996.0222. [DOI] [PubMed] [Google Scholar]
- (17).Bax A, Davis DG. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Mag. Reson. 1985;65:355–360. [Google Scholar]
- (18).Willker W, Leibfritz D, Kerssebaum R, Bermel W. Gradient selection in inverse heteronuclear correlation spectroscopy. Mag. Reson. Chem. 1993;31:287–292. [Google Scholar]
- (19).Wang H, Zuiderweg ERP, Glick GD. Solution structure of a disulfide cross-linked DNA hairpin. J. Am. Chem. Soc. 1995;117:2981–2991. [Google Scholar]
- (20).Goddard TD, Kneller DG. University of California: San Francisco. SPARKY 3.11. University of California; San Francisco: [Google Scholar]
- (21).Berger M, Cadet J. Isolation and characterization of the radiation-induced degradation products of 2′-deoxyguanosine in oxygen-free aqueous solutions. Z. Naturforsch. 1985;40B:1519–1531. [Google Scholar]
- (22).Tomasz M, Lipman R, Lee MS, Verdine GL, Nakanishi K. Reaction of acid-activated mitomycin C with calf thymus DNA and model guanines: Elucidation of the base-catalyzed degradation of N7-alkylguanine nucleosides. Biochemistry. 1987;26:2010–2027. doi: 10.1021/bi00381a034. [DOI] [PubMed] [Google Scholar]
- (23).Boiteux S, Belleney J, Roques BP, Laval J. Two rotameric forms of open ring 7-methylguanine are present in alkylated polynucleotides. Nucleic Acids Res. 1984;12:5429–5439. doi: 10.1093/nar/12.13.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tretyakova N, Matter B, Ogdie A, Wishnok JS, Tannenbaum SR. Locating nucleobase lesions within DNA sequences by MALDI-TOF mass spectral analysis of exonuclease ladders. Chem. Res. Toxicol. 2001;14:1058–1070. doi: 10.1021/tx010062i. [DOI] [PubMed] [Google Scholar]
- (25).Brown KL, Deng JZ, Iyer RS, Iyer LG, Voehler MW, Stone MP, Harris CM, Harris TM. Unraveling the aflatoxin-FAPY conundrum: Structural basis for differential replicative processing of isomeric forms of the formamidopyrimidine-type DNA adduct of aflatoxin B1. J. Am. Chem. Soc. 2006;128:15188–15199. doi: 10.1021/ja063781y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Birk C, Voss J, Wirsching J. Preparation, structural elucidation and reactions of benzyl 2-deoxy-3,5-di-O-methyl-1,4-dithio-L-threo-pentofuranoside and synthesis of the corresponding 2′-deoxy-4′-thionucleosides. Carbohydr. Res. 1997;304:239–247. [Google Scholar]
- (27).Mastihubová M, Biely P. Deoxy and deoxyfluoro analogues of acetylated methyl β-D-xylopyranoside-substrates for acetylxylan esterases. Carbohydr. Res. 2004;339:2101–2110. doi: 10.1016/j.carres.2004.06.001. [DOI] [PubMed] [Google Scholar]
- (28).Greenberg MM, Hantosi Z, Wiederholt CJ, Rithner CD. Studies on N4-(2-deoxy-D-pentofuranosyl)-4,6-diamino-5-formamidopyrimidine (Fapy·dA) and N6-(2-deoxy-D-pentofuranosyl)-6-diamino-5-formamido-4-hydroxypyrimidine (Fapy·dG) Biochemistry. 2001;40:15856–15861. doi: 10.1021/bi011490q. [DOI] [PubMed] [Google Scholar]
- (29).Wang H, Kozekov ID, Kozekova A, Tamura P, Marnett LJ, Harris TM, Rizzo CJ. Site-specific synthesis of oligonucleotides containing malondialdehyde adducts of deoxyguanosine and deoxyadenosine via a post-synthetic modification strategy. Chem. Res. Toxicol. 2006;19:1467–1474. doi: 10.1021/tx060137o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Goodenough AK, Kozekov ID, Zang H, Choi J-Y, Guengerich FP, Harris TM, Rizzo CJ. Site-specific synthesis and polymerase bypass of oligonucleotides containing a 6-hydroxy-3,5,6,7-tetrahydro-9H-imidazo[1,2-α]purin-9-one base, an intermediate in the formation of 1,N2-etheno-2′-deoxyguanosine. Chem. Res. Toxicol. 2005;18:1701–1714. doi: 10.1021/tx050141k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chetsanga CJ, Lindahl T. Release of 7-methylguanine residues whose imidazole rings have been opened from damaged DNA by a DNA glycosylase from Escherichia coli. Nucl. Acids Res. 1979;6:3673–3684. doi: 10.1093/nar/6.11.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Boiteux S, Laval J. Imidazole open ring 7-methylguanine: An inhibitor of DNA synthesis. Biochem. Biophy. Res. Commun. 1983;110:552–558. doi: 10.1016/0006-291x(83)91185-3. [DOI] [PubMed] [Google Scholar]
- (33).Gopalakrishnan S, Harris TM, Stone MP. Intercalation of aflatoxin B1 in two oligodeoxynucleotide adducts: Comparative 1H NMR analysis of d(ATCAFBGAT)·d(ATCGAT) and d(ATAFBGCAT)2. Biochemistry. 1990;29:10438–10448. doi: 10.1021/bi00498a002. [DOI] [PubMed] [Google Scholar]
- (34).Jones WR, Johnston DS, Stone MP. Site-specific synthesis of aflatoxin B1 adducts within an oligodeoxyribonucleotide containing the human p53 codon 249 sequence. Chem. Res. Toxicol. 1999;12:707–714. doi: 10.1021/tx990048u. [DOI] [PubMed] [Google Scholar]
- (35).Asagoshi K, Yamada T, Terato H, Ohyama Y, Monden Y, Arai T, Nishimura S, Aburatani H, Lindahl T, Ide H. Distinct repair activities of human 7,8-dihydro-8-oxoguanine DNA glycosylase and formamidopyrimidine DNA glycosylase for formamidopyrimidine and 7,8-dihydro-8-oxoguanine. J. Biol. Chem. 2000;275:4956–4964. doi: 10.1074/jbc.275.7.4956. [DOI] [PubMed] [Google Scholar]
- (36).Ezaz-Nikpay K, Verdine GL. Aberrantly methylated DNA - site-specific introduction of N7-methyl-2′-deoxyguanosine into the Dickerson/Drew dodecamer. J. Am. Chem. Soc. 1992;114:6562–6563. [Google Scholar]
- (37).Ezaz-Nikpay K, Verdine GL. The effects of N7-methylguanine on duplex DNA structure. Chem. Biol. 1994;1:235–240. doi: 10.1016/1074-5521(94)90016-7. [DOI] [PubMed] [Google Scholar]
- (38).Haraguchi K, Greenberg MM. Synthesis of oligonucleotides containing Fapy·dG (N6-(2-deoxy-α,β-D-erythro-pentofuranosyl)-2,6-diamino-4-hydroxy-5-formamidopyrimidine) J. Am. Chem. Soc. 2001;123:8636–8637. doi: 10.1021/ja0160952. [DOI] [PubMed] [Google Scholar]
- (39).Haraguchi K, Delaney MO, Wiederholt CJ, Sambandam A, Hantosi Z, Greenberg MM. Synthesis and characterization of oligonucleotides containing formamidopyrimidine lesions (Fapy·dA, Fapy·dG) at defined sites. Nucleic Acids Res. Supplement. 2001:129–130. [PubMed] [Google Scholar]
- (40).Jiang YL, Wiederholt CJ, Patro JN, Haraguchi K, Greenberg MM. Synthesis of oligonucleotides containing Fapy·dG (N6-(2-deoxy-α,β-D-erythropentofuranosyl)-2,6-diamino-4-hydroxy-5-formamidopyrimidine) using a 5′-dimethoxytrityl dinucleotide phosphoramidite. J. Org. Chem. 2005;70:141–149. doi: 10.1021/jo048253o. [DOI] [PubMed] [Google Scholar]
- (41).Delaney MO, Greenberg MM. Synthesis of oligonucleotides and thermal stability of duplexes containing the β-C-nucleoside analogue of Fapy-dG. Chem. Res. Toxicol. 2002;15:1460–1465. doi: 10.1021/tx025588x. [DOI] [PubMed] [Google Scholar]
- (42).Wiederholt CJ, Delaney MO, Pope MA, David SS, Greenberg MM. Repair of DNA containing Fapy·dG and its β-C-nucleoside analogue by formamidopyrimidine DNA glycosylase and MutY. Biochemistry. 2003;42:975597–975560. doi: 10.1021/bi034844h. [DOI] [PubMed] [Google Scholar]
- (43).Ober M, Muller H, Pieck C, Gierlich J, Carell T. Base pairing and replicative processing of the formamidopyrimidine-dG DNA lesion. J. Am. Chem. Soc. 2005;127:18143–18149. doi: 10.1021/ja0549188. [DOI] [PubMed] [Google Scholar]
- (44).Busch F, Pieck JC, Ober M, Gierlich J, Hsu GW, Beese LS, Carell T. Dissecting the differences between the alpha and beta anomers of the oxidative DNA lesion FaPydG. Chem. Eur. J. 2008;14:2125–2132. doi: 10.1002/chem.200701373. [DOI] [PubMed] [Google Scholar]
- (45).Burgdorf LT, Carell T. Synthesis, stability, and conformation of the formamidopyrimidine G DNA lesion. Chem. Eur. J. 2002;8:293–301. doi: 10.1002/1521-3765(20020104)8:1<293::aid-chem293>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- (46).David SS, Williams SD. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev. 1998;98:1221–1262. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- (47).Mao H, Deng Z, Wang F, Harris TM, Stone MP. An intercalated and thermally stable FAPY adduct of aflatoxin B1 in a DNA duplex: structural refinement from 1H NMR. Biochemistry. 1998;37:4374–4387. doi: 10.1021/bi9718292. [DOI] [PubMed] [Google Scholar]
- (48).Wiederholt CJ, Greenberg MM. Fapy.dG instructs Klenow exo- to misincorporate deoxyadenosine. J. Am. Chem. Soc. 2002;124:7278–7279. doi: 10.1021/ja026522r. [DOI] [PubMed] [Google Scholar]
- (49).Ober M, Linne U, Gierlich J, Carell T. The two main DNA lesions 8-oxo-7,8-dihydroguanine and 2,6-diamino-5-formamido-4-hydroxypyrimidine exhibit strongly different pairing properties. Angew. Chem. Int. Ed. Engl. 2003;42:4947–4951. doi: 10.1002/anie.200351287. [DOI] [PubMed] [Google Scholar]
- (50).Aramini JM, Cleaver SH, Pon RT, Cunningham RP, Germann MW. Solution structure of a DNA duplex containing an α-anomeric adenosine: insights into substrate recognition by endonuclease IV. J. Mol. Biol. 2004;338:77–91. doi: 10.1016/j.jmb.2004.02.035. [DOI] [PubMed] [Google Scholar]
- (51).O’Connor TR, Boiteux S, Laval J. Ring-opened 7-methylguanine residues in DNA are a block to in vitro DNA synthesis. Nucleic Acids Res. 1988;16:5879–5894. doi: 10.1093/nar/16.13.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Tudek B, Boiteux S, Laval J. Biological properties of imidazole ring-opened N7-methylguanine in M13mp18 phage DNA. Nucleic Acids Res. 1992;20:3079–3084. doi: 10.1093/nar/20.12.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Tudek B, Graziewicz M, Kazanova O, Zastawny TH, Obtulowicz T, Laval J. Mutagenic specificity of imidazole ring-opened 7-methylpurines in M13mp18 phage DNA. Acta Biochim. Pol. 1999;46:785–799. [PubMed] [Google Scholar]
- (54).Asagoshi K, Terato H, Ohyama Y, Ide H. Effects of a guanine-derived formamidopyrimidine lesion on DNA replication: Translesion DNA synthesis, nucleotide insertion, and extension kinetics. J. Biol. Chem. 2002;277:14589–14597. doi: 10.1074/jbc.M200316200. [DOI] [PubMed] [Google Scholar]
- (55).Kalam MA, Haraguchi K, Chandani S, Loechler EL, Moriya M, Greenberg MM, Basu AK. Genetic effects of oxidative DNA damages: comparative mutagenesis of the imidazole ring-opened formamidopyrimidines (Fapy lesions) and 8-oxo-purines in simian kidney cells. Nucleic Acids Res. 2006;34:2305–2315. doi: 10.1093/nar/gkl099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Weledji YN, Wiederholt CJ, Delaney MO, Greenberg MM. DNA polymerase bypass in vitro and in E. coli of a C-nucleotide analogue of Fapy·dG. Bioorg. Med. Chem. 2008;16:4029–4034. doi: 10.1016/j.bmc.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ide H, Yamaoka T, Kimura Y. Replication of DNA templates containing the α-anomer of deoxyadenosine, a major adenine lesion produced by hydroxyl radicals. Biochemistry. 1994;33:7127–7133. doi: 10.1021/bi00189a016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.