

Abstract
Phospholipid oxidation products accumulate in the necrotic core of atherosclerotic lesions, in apoptotic cells, and circulate in oxidized LDL. Phospholipid oxidation generates toxic products, but little is known about which specific products are cytotoxic, their receptors, or the mechanism(s) that induces cell death. We find the most common phospholipid oxidation product of oxidized LDL, phosphatidylcholine with esterified sn-2 azelaic acid, induced apoptosis at low micromolar concentrations. The synthetic ether phospholipid hexadecyl azelaoyl phosphatidylcholine (HAzPC) was rapidly internalized, and over-expression of PLA2g7 (PAF acetylhydrolase) that specifically hydrolyzes such oxidized phospholipids suppressed apoptosis. Internalized HAzPC associated with mitochondria, and cytochrome C and apoptosis-inducing factor escaped from mitochondria to the cytoplasm and nucleus, respectively, in cells exposed to HAzPC. Isolated mitochondria exposed to HAzPC rapidly swelled, and released cytochrome C and apoptosis-inducing factor. Other phospholipid oxidation products induced swelling, but HAzPC was the most effective and was twice as effective as its diacyl homolog. Cytoplasmic cytochrome C completes the apoptosome, and activated caspase 9 and 3 were present in cells exposed to HAzPC. Irreversible inhibition of caspase 9 blocked downstream caspase 3 activation, and prevented apoptosis. Mitochondrial damage initiated this apoptotic cascade because over-expression of Bcl-XL, an anti-apoptotic protein localized to mitochondria, blocked cytochrome C escape, and apoptosis. Thus, exogenous phospholipid oxidation products target intracellular mitochondria to activate the intrinsic apoptotic cascade.
Vascular cells are exposed to exogenous phospholipid oxidation products in the circulation, but particularly so in the concentrated environment of atherosclerotic lesions (1). Advanced lesions contain extracellular deposits of oxidized lipids (2-4) formed by oxidation of lipoprotein particles trapped in the vascular matrix (5). These lesions contain apoptotic cells (6), and the apoptotic process plays a critical role in atherogenesis (7-9).
A significant portion of the cytotoxic material generated during lipoprotein oxidation is oxidized phospholipid because phospholipase A2 treatment decreases the toxicity of oxidized LDL (10). Conversely, inhibition of LDL-associated PAF acetylhydrolase with an irreversible inhibitor increases the toxicity of oxidized LDL (11). Plasma PAF acetylhydrolase hydrolyzes oxidized phospholipids with sn-2 fragments up to 9-carbon atoms long, including the common azelaoyl fragment, yet unlike most phospholipases A2 cannot hydrolyze intact, long chain fatty acyl residues (12). This implies that oxidatively modified phospholipids are abundant cytotoxic agents of oxidized LDL, and that neither unmodified phospholipids, the fragmented fatty acids themselves, nor the lysolipid backbone constitute the toxic lipids of oxidized lipoprotein particles.
The majority of the fatty acyl fragments that remain esterified to the phosphatidylcholine backbone after oxidative fragmentation of LDL phospholipids are 9-carbon long azelaoyl fragments (13). Azelaic acid, a 9-carbon di-acid, is formed by oxidative fragmentation of the 9,10 double bond (14) of the most abundant unsaturated (oleoyl) and polyunsaturated (linoleoyl and linolenoyl) fatty acyl residues of LDL. Accordingly, these azelaoyl phosphatidylcholines (AzPC) account for almost two-thirds of the oxidized phospholipid in oxidized LDL (13). Indeed, octadecyl azPC has long been identified as the cytotoxic oxidation product of oxidant-stressed erythrocytes (15), but whether this phospholipid is toxic to nucleated cells is unknown.
The identity of specific cytotoxic lipids is not well defined, but their cellular targets are even less apparent. Some phospholipid oxidation products activate the PAF receptor displayed on plasma membranes of inflammatory cells (16) to promote inflammation, while others interact with the scavenger receptor CD36 (17). A third class of phospholipid oxidation products interact with a phosphatidylinositol-linked membrane protein (18) that may be distinct from CD14 (19).
The mechanism of cell death following exposure to oxidized phospholipids is also incompletely characterized. Cells may die by necrosis or by a regulated pathway of apoptosis. Mitochondria play a critical role in the intrinsic pathway to apoptotic death where release of cytochrome C from the mitochondrial inter-membrane space to the cytoplasm allows it to associate with the structural protein apaf-1 and pro-caspase 9 to form the apoptosome (20). This apoptotic machine proteolytically activates caspase 9, which then cleaves and activates the executioner caspase 3 (20). Other proteins released subsequent to the loss of mitochondrial compartmentalization include apoptosis-inducing factor (AIF), which is then free to move to the nucleus to participate in the DNA fragmentation characteristic of apoptotic cell death (21). Oxidant-induced cell death is suppressed by the mitochondria-targeted Bcl-2 family member Bcl-XL (22) that prevents mitochondrial permeabilization and release of pro-apoptotic proteins (23). Mitochondrial dysfunction subsequent to oxidative stress plays a primary role in atherogenesis (24).
Here we used synthetic hexadecyl azelaoyl phosphatidylcholine (HAzPC), to understand how this abundant extracellular oxidized phospholipid initiates cell death. We find this lipid is toxic in a receptor independent way: it is rapidly internalized, damages mitochondria, and initiates the intrinsic apoptotic caspase cascade.
Experimental Procedures
Reagents
Annexin V-Alexa488 (Invitrogen; Carlsbad, CA); zVAD-fmk, zLEHD-fmk and other reagents (Sigma; St. Louis, MO); fluorogenic FAM-zLEHD-fmk (Biocarta; San Diego, CA); Ac-DEVD-AMC (Alexis Biochemicals; Lausen, Switzerland). Protease inhibitor (Roche Diagnostics GmbH; Indianapolis, IN). HAzPC (1-O-hexadecyl-2-azelaoyl-sn-glycero-3-phosphocholine) and 1-palmitoyl-2-azelaoyl-sn-glycero-3-phosphocholine (Cayman Chemical; Ann Arbor, MI). LysoPAF, carbamoyl-PAF (cPAF), PAF, C4 PAF (BioMol; Plymouth Meeting, PA), other phospholipids (Avanti Polar Lipids; Alabaster, AL). MitoTracker Red (Invitrogen, Carlsbad, CA); anti-cleaved caspase 3 (Cell Signaling Technology; Beverly, MA); anti-apoptosis-inducing factor (AIF), anti-calnexin, and anti-cytochrome C (Santa Cruz); anti-adenine nucleotide translocase, MitoSciences (Eugene, OR); Secondary goat anti-mouse (Biosource International; Camarillo, CA); VECTASHIELD+DAPI (Vector Laboratories; Burlingame, CA).
Cells
HL60 cells (2×106; ATCC, Manassas, VA) were transfected with 2 μg Flag-tagged Bcl-XL (a kind gift of Clark W. Distelhorst, Case Western Reserve), Flag-PLA2g7 or empty vector by Amaxa (Gaithersburg, MD) nucleofection (T-19 program). Complete RPMI with 500 μg/ml G418 was substituted 24 h later and aliquots transferred to microtiter dishes to isolate Flag positive clones.
Human umbilical vein endothelial cells (HUVEC, ATCC) were cultured in 8 well-chamber slides in F12K medium (ATCC) with 2 mM glutamine, 1.5 g/L sodium bicarbonate, 0.1 mg/ml heparin, 0.03 mg/ml endothelial cell growth supplement (Sigma) and 10% FBS. HepG2 cells (ATCC) were cultured as described by the supplier.
Mitochondria
Minced liver from an adult Sprague-Dawley rat in a protocol approved by Cleveland Clinic IACUC was homogenized (0.1 g/ml) in EB buffer (200 mM D-mannitol, 70 mM sucrose, 20 mM Hepes pH 7.4, 0.5 mg/ml defatted BSA, and 1 mM EGTA.) Homogenates were cleared twice (1,000 × g, 5min) before recovery of the mitochondria (9,500 × g, 10 min). This pellet was washed once with an equal volume of BSA-free media and then resuspended in medium without BSA (35 mg/ml). HL60 cells (3 × 108) stably transfected with Bcl-XL or its empty vector were washed 2× in PBS and once in EB media before the cells were suspended in 10 ml EB medium containing BSA (2 mg/ml), and then mechanically homogenized and mitochondria purified as above.
Caspase 3 activation
HL-60 cells were pre-incubated (1 h) with zLEHD-fmk (20 μM), zVAD-fmk (50 μM), or DMSO before incubation (4 h) with 5 μM HAzPC or buffer. The cells were suspended in 20 mM Hepes, pH 7.5, 10 % glycerol, 2 mM DTT, proteinase inhibitor mix, and then lysed by freeze/thaw. Caspase 3 activity in cleared supernatants (50 μg) was determined (emission 460 nm; excitation 380 nm) using 200 μM Acetyl-DEVD-AMC.
Flow cytometry
HL-60 cells in serum-free RPMI 1640 were incubated with buffer or HAzPC for the stated times, or irradiated (1 mW/cm2; IL1700 radiometer, SED240 UVB detector; International Light, Newburyport, MA) for 5 min. Apoptosis: Cells were treated for 6 h, washed, then stained with Annexin V-Alex488 and 1 μg/ml propidium iodide in 140 mM NaCl, 10 mM Hepes pH 7.4 and 2.5 mM CaCl2 for 15 min. Caspase 9 activation: Cells were treated (4h) with HAzPC or buffer and then FAM-LEHD-fmk for 1 h before washing and single color flow cytometry.
Apoptosis-inducing factor, cytochrome C and MitoTracker Red immunocytochemistry - Cytochrome C release
HL60 cells were incubated with 5 μM HAzPC or buffer, washed and resuspended in 20 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 1mM EDTA, 1 mM EGTA, 1 mM DTT and 250 mM sucrose. The cells were homogenized (20×) by expression through a 1cc/28G1/2 syringe, supernatants were cleared (1,000 × g) and organelles separated from cytoplasm by centrifugation (20,000 × g).
Apoptosis-inducing factor
HUVEC or HepG2 were treated with HAzPC, washed, fixed and then permeabilized with 0.1% Triton X-100 before staining with anti-AIF and Alexa488-anti-mouse and DAPI. MitoTracker Red. HUVEC medium was changed to a protein-free medium, and the cells treated or not with 5 μM HAzPC for 30 min at 37° before 25 nM MitoTracker Red was added to the medium. After 30 min, the cells were washed thrice and then fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences, San Jose, CA) for 30 min at 4°. The cells were then washed with PBS thrice and mounted with medium containing DAPI.
Transmission electron microscopy
Mitochondria were recovered after treatment (9,500 × g) and fixed in 100 mM sodium cacodylate buffer pH 7.4, 2.5% glutaraldehyde and 4% paraformaldehyde (4°, 16 h). Samples were washed in this buffer (3×), post-fixed with aqueous osmium tetroxide (1 h, 4°), washed in sodium cacodylate buffer and then in maleate buffer (pH 5.16), and then dehydrated with step-wise ethanol solutions from 30% to 100% followed by propylene oxide. Samples were embedded in LX-112 medium, polymerized (68°, 48 h), sectioned (70-90 nm), and stained with uranyl acetate and lead citrate.
HAzPC internalization
HL60 cells were treated in suspension with 5 μM HAzPC for the stated times and recovered by centrifugation. The cells were washed once with RPMI containing 0.5% human serum albumin and once with RPMI. [2H]PAF was added as an internal standard, and the lipids extracted (25) and analyzed by LC/ESI/MS/MS (Quattro Ultima; Micromass, Wythenshawe, UK). Lipids in methanol were separated with a Prodigy ODS C18 HPLC column (150 × 2 mm, 5μm, Phenomenex, Torrance, CA). The solvent gradient started at 85% methanol (0.2 ml/min), then linear gradient to 100% methanol over 5 min was applied and held for 20 min. The mobile phase was linearly changed back to 85% MeOH over 0.5 min and held for 4.5 min. Mobile phase solvents contained 0.2% formic acid. Source temperature was 120°, desolvation temperature was 250° and N2 flow was 735 L/h, and the cone N2 flow was 71 L/h. Argon was used for collision-induced dissociation. The multiplier was set at an absolute value of 500 V, total ion current was obtained over m/z 200-1000 using a cone energy of 50 V in the positive ion mode, and 5.0 kV was applied to the electrospray capillary. Multiple reaction monitoring used 22 eV collision energy, and the transition used to identify HAzPC were the molecular cation [M+H+] of m/z 652 and the daughter ion of m/z 184.
Results
An exogenous oxidized phospholipid caused apoptotic cell death
We treated the promyelocytic cell line HL60 with increasing concentrations of synthetic hexadecyl azelaoyl phosphatidylcholine (HAzPC) for 6 h and then tested for an early marker of apoptotic cell death, exposure of phosphatidylserine on the exterior leaflet of the plasma membrane. HAzPC caused a concentration-dependent increase in annexin V staining of exposed phosphatidylserine where 2.5 μM HAzPC increased by 5-fold the number of cells stained by annexin V (Fig. 1A). Doubling the amount of exogenous HAzPC to 5 μM produced a further increase in the number of cells displaying phosphatidylserine on their surface. HAzPC also increased the number of cells unable to exclude the nuclear dye propidium iodide, but at this early time few cells had progressed to become doubly positive (not shown). The liver cell line HepG2 (Fig. 1B) and human umbilical endothelial cells (not shown) also lost plasma membrane phosphatidylserine asymmetry when exposed to increasing concentrations of HAzPC, so the effect of HAzPC was not specific to HL60 cells.
Figure 1. The oxidized phospholipid HAzPC induces apoptosis.
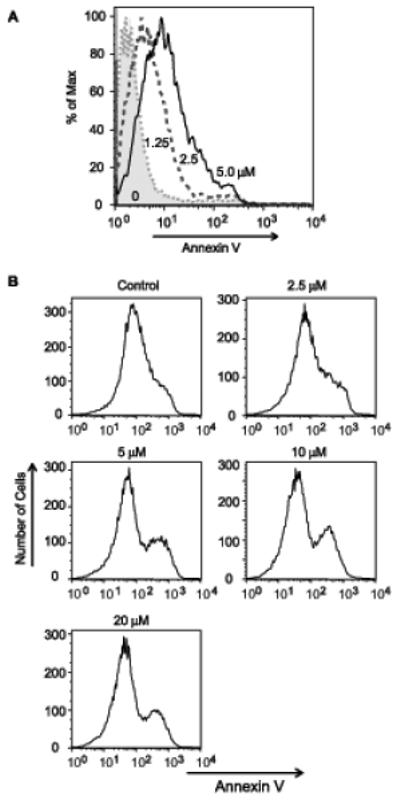
A. HL60 cells were incubated with the stated concentrations of synthetic HAzPC for 6 h, or exposed to 1 mW/cm2 UVB (not shown) for 5 min as a positive control for apoptosis. The cells were stained for surface phosphatidylserine with fluorescent Alexa Fluor488-annexin V as described in “Methods.” Flow cytometry for the intensity of annexin V fluorescence on the x-axis vs cell number was normalized to the maximal number of cells in a channel in order to overlay the plots. This experiment is representative of three independent experiments. B. HepG2 cells were exposed to HAzPC at the stated concentrations before the level of Annexin V staining determined as in panel A.
We determined whether HAzPC caused a lethal increase in intracellular free Ca++ using the fluorescent dye FURA2. We found that while the cells retained the dye, and therefore remained intact, there was only a small transient increase in Ca++ (not shown), suggesting the cells did not die by lysis or necrosis.
Exogenous HAzPC is internalized and associates with mitochondria as an intact phospholipid
We added HAzPC to the medium of HL60 cells, recovered the cells at varied times by centrifugation, and washed them once with albumin. HL60 cells rapidly internalized the added HAzPC, with maximal accumulation by about 30 min (Fig. 2). Apoptosis can be initiated through mitochondrial damage, so we determined whether mitochondria were exposed to the internalized oxidized phospholipid. We treated HL60 cells with HAzPC, washed them once with albumin and once with buffer, and then homogenized the cells and fractionated the homogenate by differential centrifugation. We found that the mitochondrial fraction was enriched with HAzPC compared to other cellular membranes, organelles and cytosol (Fig. 2B). At least a portion of the internalized HAzPC trafficked to mitochondria as the intact molecule because the mass spectrometer monitored the molecular ion.
Figure 2. HAzPC internalized by HL60 cells is cytotoxic.

A. Time-dependent internalization of HAzPC. HAzPC (5 μM) was added to suspended HL60 cells and the cells recovered by centrifugation at the stated times. The cells were washed once with 0.5% human serum albumin, an [2H]PAF internal standard was added and the lipids were extracted and purified by reversed phase HPLC. HAzPC was determined by tandem mass spectrometry as the molecular cation [M+H+] of m/z 652 producing a phosphocholine daughter ion of m/z 184. The average of duplicate values is shown, and this experiment is representative of three experiments. B. Exogenous HAzPC accumulates in mitochondria. HL60 cells (7 × 108) were incubated with or without 5 μM HAzPC for 20 min at room temperature. The cells were washed once in RPMI containing 0.5% human serum albumin and once in RPMI before the cells were suspended in 10 ml EB medium (200 mM D-mannitol, 70 mM sucrose, 20 mM Hepes (pH 7.4), 1 mM EGTA and 100 μM Pefabloc.) and then mechanically homogenized. Homogenates were centrifuged (2000 × g, 5 min) to remove unbroken cells. The resulting lysates were centrifuged (9500 × g, 10 min) and the mitochondrial pellets were resuspended in the same volume of buffer. Lipids were extracted from these duplicate samples and the content of HAzPC determined by HPLC-MS/MS as described in “Methods”. A separate experiment showed the mitochondrial fraction contained no endoplasmic reticulum associated calnexin by western blotting, and that the mitochondrial recovery, visualized by blotting adenine nucleotide translocase, was incomplete. Therefore, the relative amount of HAzPC recovered in the mitochondrial fraction is an underestimate. C. HL60 cells expressing PAF acetylhydrolase are protected from HAzPC toxicity. HL60 cells stably expressing the oxidized phospholipid phospholipase PLA2g7 (plasma PAF acetylhydrolase) or its vector control were treated with 5 μM HAzPC for 6 h and stained with Alexa Fluor488-annexin V as in Fig. 1.
We next determined whether internalized HAzPC was responsible for the cytotoxicity of the oxidized phospholipid. To do this, we stably expressed PAF acetylhydrolase (PLA2g7) in these cells because this enzyme hydrolyzes PAF and oxidatively damaged phospholipids, including HAzPC, without attacking unmodified membrane phospholipids. Cells expressing PLA2g7 became almost fully resistant to the toxic effect of HAzPC (Fig. 2C).
Intracellular mitochondria are damaged by exogenous HAzPC
We considered that the unusual structure of HAzPC potentially distorts membrane bilayer structure, and also that one form of apoptosis results from altered mitochondrial membrane barrier function that allows proapoptotic proteins to escape. To define mitochondrial structure after exposure to HAzPC, we stained adherent HUVEC cells with MitoTracker RED. We found extensive accumulation of the cationic dye by mitochondria that produced a bright, localized staining pattern (Fig. 3). In contrast, MitoTracker RED was diffusely present in the cytoplasm of cells exposed to HAzPC, suggesting the mitochondrial transmembrane potential that drives the cationic dye into the matrix may have been dissipated by HAzPC exposure.
Figure 3. Mitochondria are compromised in cells exposed to HAzPC.

Mitochondria were visualized with MitoTracker RED, a cationic dye accumulated in energized mitochondria, by culturing HUVEC on chamber slides, treating the cells with buffer (A) or with 5 μM HAzPC (B) for 30 min, and then adding 25 nM MitoTracker Red for an additional 30 min before the cells were fixed and visualized by fluorescence microscopy.
Apoptosis-inducing factor (AIF) is an integral mitochondrial inner membrane protein, but proteolysis of the membrane anchor, coupled with outer membrane damage, allows the protein to escape this confine. A portion of this released AIF moves to the nucleus to participate in the DNA fragmentation characteristic of apoptotic cells (26). We used adherent HepG2 (Fig. 4A) and HUVEC (Fig. 4B) to image AIF to find that AIF was localized in punctate, peri-nuclear structures in control cells, but that it had a diffuse distribution in cells exposed to HAzPC. Staining of the nuclei with DAPI showed that a portion of the AIF was now associated with the nucleus of HAzPC treated cells. Structurally compromised mitochondria also allow cytochrome C to escape its association with the inner membrane, and we observed that immunoreactive cytochrome C had leaked from the mitochondrial compartment into the cytoplasm in HepG2 (Fig. 4C) and HL60 cells (Fig. 4D) exposed to HAzPC.
Figure 4. Mitochondrial proteins escape the mitochondrial compartment in cells exposed to HAzPC.

AIF immunocytochemistry. HepG2 (A) or HUVEC (B) cells adhering to a glass surface were treated with 10 μM HAzPC in complete DMEM for 16 h. The cells were then permeabilized and stained with mouse anti-apoptosis inducing factor (AIF), Alexa Fluor488 labeled anti-mouse and DAPI. The punctate mitochondrial green fluorescence of control cells becomes dispersed in the cytoplasm after HAzPC exposure, with many cells showing co-localization of AIF and the blue nuclear DAPI stain. Cytochrome C immunoblot. HL60 cells (C) or HepG2 cells (D) were treated with 5 μM HAzPC for 4 h, the cells recovered by centrifugation and mechanically lysed before cytoplasmic components were separated from mitochondria by centrifugation. Proteins in the cytoplasmic fraction were denatured, resolved by SDS-PAGE, transferred to a solid support, and then sequentially probed with anti-cytochrome C and anti-β-actin monoclonal antibodies. Each panel is representative of three independent experiments.
Isolated mitochondria swell when exposed to HAzPC
The inner mitochondrial membrane supports a chemiosmotic gradient and mitochondria swell when this permeability barrier is breached. Rat liver mitochondria maximally increased their volume when treated with a high concentration of Ca++ and the pore-forming peptide alamethicin (Fig. 5A). Isolated mitochondria swelled to 70% of this maximal change when treated with HAzPC. Depolarization of mitochondria alone was insufficient to cause swelling because the protonophore CCCP had little effect on mitochondrial volume. The homologous phospholipid palmitoyl glutaroyl (a 5-carbon di-acid) phosphatidylcholine (PGPC), formed from oxidative fragmentation of palmitoyl arachidonoyl phosphatidylcholine, caused mitochondria to increase their volume to 50 % of that of the positive control. Other short chain phospholipids, and lysophosphatidic acid, were less effective in this regard and were about as effective as the underivitized lysophosphatidylcholine in changing mitochondrial volume. We found (Fig. 5B) that the acyl form of HAzPC, palmitoyl azelaoyl phosphatidylcholine, induced swelling, but was about half as effective as the ether phospholipid. A minor portion of this swelling was blocked by cyclosporin A, a small molecule that binds to cyclophilin D of the mitochondrial permeability transition pore and keeps in it closed (Fig. 5C).
Figure 5. Phospholipid oxidation products cause mitochondrial swelling.

A. Isolated mitochondria were incubated with the stated lipids at a concentration of 2.5 μM for 10 min and the change in light scattering determined as stated in “Methods.” The maximal swelling was defined as that induced by calcium in the presence of alamethicin. The abbreviations are: LPA, lysophosphatidic acid; PGPC, palmitoyl glutaroyl phosphatidylcholine; POVPC, palmitoyl oxovaleroyl phosphatidylcholine; c-PAF, cabamoyl-PAF; PAF, platelet-activating factor (hexadecyl acetyl phosphatidylcholine); C4PAF, hexadecyl butyroyl phosphatidylcholine; CCCP, carbonyl cyanide 3-chlorophenylhydrazone. B. Effect of the sn-1 bond on mitochondrial swelling. Isolated mitochondria were treated with the ether lipid HAzPC or the diacyl homolog palmitoyl azelaoyl phosphatidylcholine (PAzPC) and swelling determined as in panel A. C. Cyclosporin A minimally affects HAzPC-induced swelling. Isolated mitochondria were pre-treated with 680 nM cyclosporin A at room temperature to interfere with flow through the mitochondrial permeability transition pore before swelling in response to 2.5 μM HAzPC was determined as before. D. Transmission electron microscopy. Electron micrographs of rat liver mitochondria treated with 5 μM lysophosphatidylcholine or HAzPC, or exposed to 5 mM CaCl2 and 7 μg/ml alamethicin for 10 min. The bar is 1 μmeter in length.
We examined the effect of HAzPC on the physical structure of mitochondria by electron microscopy. The photomicrographs we obtained (Fig. 5D) show this normally compact and electron dense organelle was affected by lysophosphatidylcholine, and to a greater extent by HAzPC. Mitochondria treated with lysophosphatidylcholine were enlarged and less opaque, while those treated with HAzPC were greatly enlarged with transparent domains. The positive control Ca++ and alamethicin showed the complete disruption of the organelle.
Cytochrome C and AIF escape from isolated mitochondria exposed to HAzPC
The two pro-apoptotic proteins cytochrome C and AIF lost their exclusive mitochondrial localization in cells exposed to HAzPC, and a similar loss of mitochondrial retention occurred when isolated mitochondria were exposed to HAzPC (Fig. 6). We found by immunoblotting that HAzPC induced a concentration dependent loss of cytochrome C from isolated mitochondrial to their supernatant and that cyclosporin A partially reduced the loss of cytochrome C from the mitochondrial pellet at lower HAzPC concentrations. Mitochondria exposed to HAzPC also lost AIF to their supernatant and again cyclosporin A partially suppressed this release.
Figure 6. Pro-apoptotic proteins are released from mitochondria exposed to HAzPC.
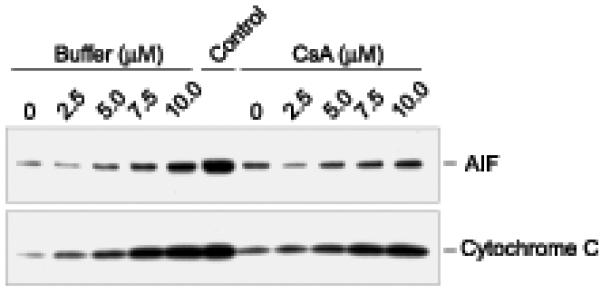
Isolated mitochondria were treated or not with cyclosporin A for 5 min and then with the stated concentration of HAzPC for 10 min. Mitochondria were then separated from soluble material by centrifugation and the amount of AIF and cytochrome C in the supernatant was determined by western blotting as defined under “Methods.” The center lane, labeled “control”, was the amount of the two target proteins in detergent solubilized mitochondria at the start of the experiment.
HL60 cells exposed to HAzPC contain activated caspase 9 and caspase 3
Cytoplasmic cytochrome C is free to associate with cytoplasmic apaf-1 and pro-caspase-9 to form an active pro-caspase-9 cleavage complex, the apoptosome. We tested whether the cytochrome C released to the cytoplasm of HL60 cells exposed to HAzPC promoted the formation of active caspase 9, an upstream initiator caspase of the intrinsic apoptotic pathway. We found by flow cytometry (Figs. 7A,B) that few control cells contained active caspase 9, but that a significant portion of HAzPC-treated cells contained this active caspase and hydrolyzed its fluorogenic zLEHD-fmk substrate to a greater extent than our positive control, UVB irradiation.
Figure 7. HAzPC activates caspases of the intrinsic apoptotic cascade.
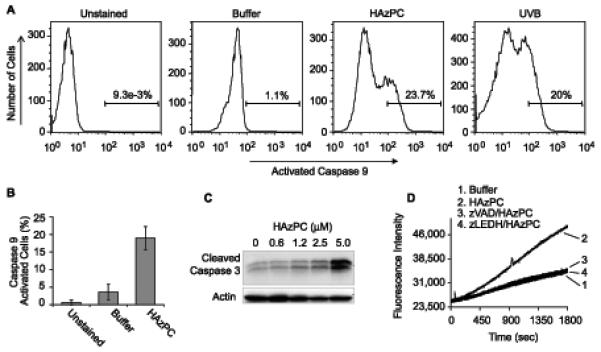
A. Intracellular activated caspase 9. HL60 cells were exposed to 5 μM HAzPC for 4 h as before and then incubated with the fluorescent substrate and pro-irreversible caspase 9 inhibitor FAM-LEHD-fmk. The cells were cultured at 37° for an additional hour, washed and the amount of caspase 9-bound dye determined by single channel flow cytometry. B. The bar graph presents the average and standard error of three independent experiments. C. Active caspase 3 fragments accumulate in cells exposed to HAzPC. HL60 cells were exposed to the stated concentration of HAzPC for 4 h, lysed before the presence of 19 and 17 kDa caspase 3 fragments was assessed by western blotting. D. HAzPC induces caspase 3 enzymatic activity in a caspase 9-dependent fashion. HL 60 cells were treated for 1 h with buffer or the pan caspase inhibitor zVAD-fmk or zLEHD-fmk to inhibit caspase 9 and then incubated with buffer or 5 μM HAzPC for 4 h before cell lysates were prepared and assayed for caspase-3 activity. Each panel, including panel B, is representative of three independent experiments.
Caspase 3 cleavage is catalyzed by activated caspase 9, which then allows caspase 3 to function as a downstream executioner caspase. We probed HL60 cells, treated or not with a range of HAzPC concentrations, for proteolytically activated caspase 3 by immunoblotting, and found a concentration-dependent increase in 17 kDa and 19 kDa caspase 3 fragments (Fig. 7C). The specific caspase 9 inhibitor zLEHD-fmk blocked formation of the 17 and 19 kDa caspase 3 fragments (not shown), and it prevented the formation of active caspase 3 in HAzPC-treated HL60 cells (Fig. 7D). Caspase 3 activation in response to HAzPC therefore depends on active caspase 9.
Caspase inhibition suppresses HAzPC induced cell death
The oxidized phospholipid HAzPC activated components of the intrinsic apoptotic cascade, so we next determined whether these activated proteases were required for cell death. We first inhibited caspase function with the general caspase inhibitor zVAD-fmk, and then with the specific caspase 9 inhibitor zLEHD-fmk. We found that the 46% of apoptotic cells with surface exposed phosphatidylserine after HAzPC treatment was reduced to ∼16% by each caspase inhibitor (Fig. 8). We conclude that HAzPC is cytotoxic because it activates pro-apoptotic caspases.
Figure 8. Caspases of the intrinsic apoptotic cascade are required for HAzPC-induced apoptosis.
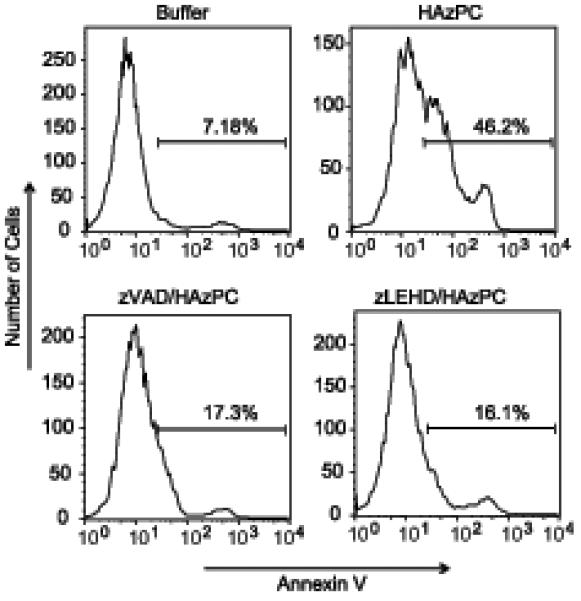
HL60 cells were treated with the caspase inhibitor zVAD-fmk or the caspase 9 specific inhibitor zLEHD-fmk, and with 5 μM HAzPC or maintained in buffer for 6 h. The cells were then stained with annexin V and the resulting cellular fluorescence analyzed by flow cytometry as in Figure 1. This experiment is representative of two other independent experiments.
Mitochondria are primary targets of HAzPC-induced apoptosis
HAzPC damages mitochondrial integrity in intact cells and initiates the intrinsic caspase cascade leading to apoptosis, but is this mitochondrial damage responsible for cell death? To investigate this relationship we used Bcl-XL , an anti-apoptotic Bcl-2 homolog that localizes to mitochondria where it interferes with pro-apoptotic family members and suppresses mitochondrial permeabilization (27). We stably over-expressed Flag-tagged Bcl-XL in HL60 cells (Fig. 9A) and isolated mitochondria from these cells. Mitochondria from Bcl-XL—expressing cells were significantly less sensitive to HAzPC than control cells, and allowed little cytochrome C to escape after HAzPC exposure (Fig. 9B). Bcl-XL over-expression was similarly efficacious in vivo where it blocked annexin V staining of HL-60 cells exposed to HAzPC (Fig. 9C). HAzPC therefore interacts with mitochondria to initiate an apoptotic cascade.
Figure 9. Mitochondrial protection by Bcl-XL blocks HAzPC-induced apoptosis.
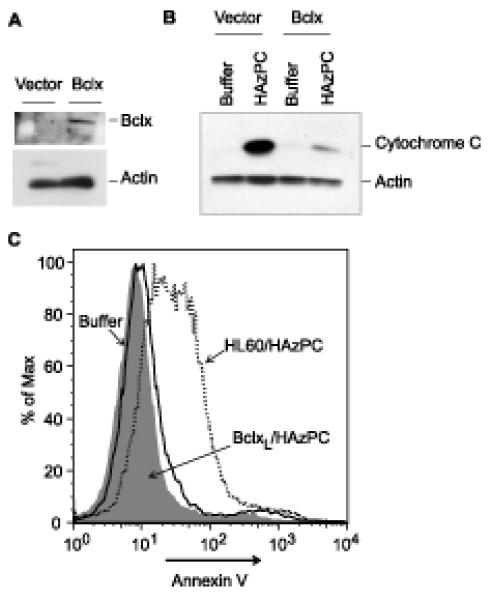
A. Bcl-XL expression. HL60 cells stably over-expressing human Bcl-XL were immunoblotted for total cellular Blc-XL and actin. B. Cytochrome C loss from isolated mitochondria. Mitochondria were isolated from Bcl-XL over-expressing cells, treated or not with HAzPC, and loss of cytochrome C to the supernatant was determined as in Figure 6. C. Bcl-XL expression suppresses HAzPC-induced apoptosis. Surface display of phosphatidylserine was quantitated by flow cytometry using Alexa488-conjugated annexin V as in Figure 1. Each panel is representative of three independent experiments. This is an arm of the experiment in Fig. 2B, so the buffer and HAzPC histograms are the same.
Discussion
We show that an exogenous oxidized phospholipid is cytotoxic because it is readily internalized, migrates to mitochondria and then damages mitochondria in a way that activates the intrinsic caspase cascade. The sequence of events subsequent to internalization of the oxidation product included association with mitochondria, loss of mitochondrial sequestration of pro-apoptotic proteins, formation of the apoptosome with caspase-9 activation, and then caspase-3 activation. Activation of these caspases was required for HAzPC-induced cell death because irreversibly inhibiting caspase-9 suppressed cell death. Cellular mitochondria were the targets of the internalized HAzPC because over-expression of Bcl-XL suppressed cell death. The phospholipid itself was the pro-apoptotic agent because hydrolysis by PAF acetylhydrolase suppressed apoptosis.
We tested several cell lines, and circulating human cells (not shown) and found that each was susceptible to HAzPC-induced cell death, although the concentration of HAzPC required to induce death of these cells varied several fold, perhaps reflecting varied rates of internalization or hydrolysis. However, for each cell type HAzPC at low micromolar concentrations induced phosphatidylserine exposure, cytoplasmic accumulation of cytochrome C, and redistribution of AIF to the cytoplasm and nucleus.
AIF is released from its association with the inner mitochondrial membrane by apoptotic stimuli (26) allowing it to migrate to the nucleus to cause nuclear condensation and cell death (28). Cytosolic cytochrome C is an essential component of the apoptosome (29) that cleaves pro-caspase 9 to its active form. The peptide zLEHD-fmk is a selective caspase-9 substrate and irreversible inhibitor that prevents cleavage of down stream targets such as caspase 3. Cells treated with HAzPC contained activated caspase 9, identified by fluorescent adduct derived from a fluorogenic irreversible inhibitor, and the cells contained active caspase 3. Activation of the executioner caspase 3 depended on caspase 9 cleavage because proteolytically activated caspase 3 fragments did not appear in cells containing irreversibly inactivated caspase 9. This inhibition of caspase activity also suppressed HAzPC-induced cell death, so HAzPC initiates death only via the apoptotic intrinsic caspase cascade.
The C18 homolog of C16 HAzPC, octadecyl azelaoyl phosphatidylcholine, previously has been identified as a lytic agent present in oxidized egg yolk phospholipid when erythrocytes were the target cells (30). However, we find in nucleated cells that contain mitochondria that the primary effect of exogenous HAzPC was not lysis. The cells remained morphologically unremarkable and their plasma membrane presented a functional permeability barrier that retained cytoplasmic dyes. We did not observe (not shown) a prolonged influx of extracellular Ca++, as found in human umbilical vein endothelial cells exposed to lysophosphatidylcholine (31), or non-selective disruption of the plasma membrane as suggested for other short chain phospholipids (32). Instead, we identified a marked change in cellular mitochondrial function in the absence of frank plasma membrane damage.
We conclude that the oxidized phospholipid HAzPC is pro-apoptotic because of its effect on intracellular mitochondria for several reasons. First, there is a temporal correlation between early mitochondrial dysfunction (3 to 4 h) and subsequent phosphatidylserine exposure (staring at 6h). Second, internalized HAzPC was concentrated as an intact phospholipid by mitochondria. Third, Bcl-XL, which physically interacts with the mitochondrial outer membrane to promote cell survival (33), suppressed HAzPC-induced cell death. Mitochondria isolated from cells over-expressing Bcl-XL released little cytochrome C, which, in turn, resulted in fewer cells expressing phosphatidylserine on their surface after being exposed to HAzPC. Since specifically protecting mitochondria with ectopic Bcl-XL blocked HAzPC cytotoxicity, the primary target of internalized HAzPC for this event are mitochondria. We currently do not know whether mitochondria are particularly sensitive to HAzPC, whether compromising their integrity is more apparent than with other organelles, or whether the high concentration of HAzPC encountered by mitochondria account for the mitotoxicity. It is apparent, however, that this lipid does not act as an indiscriminant detergent.
As anticipated from experiments with intact cells, HAzPC had a direct effect on isolated mitochondria and rapidly induced swelling in a concentration-dependent fashion. HAzPC, with an sn-1 ether bond and a 9 carbon di-acidic fragment derived from linoleoyl/linolenoyl oxidation, was more effective in this than its palmitoyl homolog, or the 5-carbon glutaroyl analog derived from arachidonate oxidation. It also was more effective than the shorter arachidonoyl fragmentation products containing 4-carbon butyroyl residue, or PAF that contains the two-carbon acetyl residue. The phospholipase A2 hydrolytic product of HAzPC, 1-hexadecyl-sn-glycero-3-phosphocholine (lysoPAF), was only mildly effective in altering mitochondrial volume. This means that catabolism of this particular oxidized phospholipid by phospholipase A2 activity reduces its mitotoxicity. This relationship for the relative effectiveness of precursor and hydrolytic product does not hold for the other less potent oxidized phospholipids. A recent report (34) shows that a calcium independent phospholipase A2 localizes to mitochondria and protects cells from oxidative insult and apoptotic death, suggesting phospholipid products such as HAzPC generated in vivo are more toxic than their hydrolyzed components.
Components of oxidized LDL have previously been found to promote mitochondrial dysfunction (35) and caspase 3 activation (36,37), although the relevant mechanism(s) remain incompletely defined. The oxidized LDL components palmitoyl glutaroyl phosphatidylcholine and palmitoyl oxovaleroyl phosphatidylcholine (oxidative fragments of palmitoyl arachidonoyl phosphatidylcholine (4,12) that are 4 carbons shorter than HAzPC) also enhance caspase 3 activity, although it requires 50 μM of these shorter chained oxidized phospholipids to achieve a modest increase in caspase 3 activity (38), or a 50% decrease in viability (39). This is a larger concentration difference than their effects on mitochondrial swelling, but internalization and intracellular distribution may also differ for individual oxidized phospholipids (40). Reactive non-phospholipid oxidation products, such as acrolein (41,42), 4-hydroxhexenal (43) or 15-deoxy-Δ12,14-prostaglandin J2 (44), depolarize isolated mitochondria and induce apoptosis. However, these chemically reactive lipids are unlikely to accumulate to a level able to affect mitochondrial function nor are they the cytotoxic agents destroyed by phospholipase A2 (10) or PAF acetylhydrolase (45-49) digestion.
Oxidized LDL levels in carotid plaque are some 70-fold greater than circulating levels (50), and accumulation of apoptotic macrophages and smooth muscle cells parallels the progression of atherosclerosis (6). An important component of the toxic soup of atherosclerotic plaque are oxidized phospholipids because phospholipid hydrolysis reduces oxidized LDL cytotoxicty, at least to endothelial cells (10). Linoleoyl (C18:2) and linolenoyl (C18:3) are the most abundant esterified polyunsaturated fatty acids, and both contain a 9,10 double bond. Preferential bond scission at this site (14) creates phospholipids with a 9-carbon sn-2 fragment that contain an aldehyde, which also is pro-apoptotic (51), or the more oxidized azelaoyl residue. The azelaoyl residue is not a product of fatty acid β-oxidation, and has long served as a marker of chemical oxidation. Here we show phospholipids with this shortened fatty acyl residue not only are markers of oxidative processes, they directly participate in cell death by targeting mitochondria to initiate the intrinsic apoptotic caspase cascade.
Acknowledgements
This work was supported by HL44513. The work was made possible through the services offered by the mass spectrometry core II, the electron microscopy imaging core, as well as the flow cytometry core, the imaging core, and the media preparation core facilities of the Cleveland Clinic. We gratefully acknowledge Dr. Clark W. Distelhorst for supplying the Bcl-XL plasmid. We greatly appreciate thoughtful discussions with Gopal Marathe and the aid of Renliang Zhang (mass spectrometry core II) and Mei Yin (EM imaging core).
Abbreviations
- AIF
apoptosis-including factor
- CsA
cyclosporin A
- FAM-zLEHD-fmk
carboxyfluorescein-N-benzyloxycabonyl-Leu-Glu-His-Asp-fluoromethylketone
- Ac-DEVD-AMC
N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin amide
- HAzPC
1-hexadecyl-2-azelaoyl-sn-glycero-3-phosphocholine
- zVAD-fmk
N-benzyloxycabonyl-Val-Ala-Asp-fluoromethylketone
- zLEHD-fmk
N-benzyloxycabonyl-Leu-Glu-His-Asp-fluoromethylketone
References
- 1.Nakajima K, Nakano T, Tanaka A. Clin Chim Acta. 2006;367:36–47. doi: 10.1016/j.cca.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 2.Waddington EI, Croft KD, Sienuarine K, Latham B, Puddey IB. Atherosclerosis. 2003;167:111–120. doi: 10.1016/s0021-9150(02)00391-x. [DOI] [PubMed] [Google Scholar]
- 3.Ravandi A, Babaei S, Leung R, Monge JC, Hoppe G, Hoff H, Kamido H, Kuksis A. Lipids. 2004;39:97–109. doi: 10.1007/s11745-004-1207-5. [DOI] [PubMed] [Google Scholar]
- 4.Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA. J Biol Chem. 1997;272:13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- 5.Salvayre R, Auge N, Benoist H, Negre-Salvayre A. Biochim Biophys Acta. 2002;1585:213–221. doi: 10.1016/s1388-1981(02)00343-8. [DOI] [PubMed] [Google Scholar]
- 6.Kockx MM, De Meyer GR, Muhring J, Jacob W, Bult H, Herman AG. Circulation. 1998;97:2307–2315. doi: 10.1161/01.cir.97.23.2307. [DOI] [PubMed] [Google Scholar]
- 7.Walsh K, Smith RC, Kim HS. Circ Res. 2000;87:184–188. doi: 10.1161/01.res.87.3.184. [DOI] [PubMed] [Google Scholar]
- 8.Littlewood TD, Bennett MR. Curr Opin Lipidol. 2003;14:469–475. doi: 10.1097/00041433-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Tabas I. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 10.Schmitt A, Negre-Salvayre A, Troly M, Valdiguie P, Salvayre R. Biochim Biophys Acta. 1995;1256:284–292. doi: 10.1016/0005-2760(95)00032-8. [DOI] [PubMed] [Google Scholar]
- 11.Carpenter KL, Challis IR, Arends MJ. FEBS Lett. 2003;553:145–150. doi: 10.1016/s0014-5793(03)01007-x. [DOI] [PubMed] [Google Scholar]
- 12.Stremler KE, Stafforini DM, Prescott SM, McIntyre TM. J. Biol. Chem. 1991;266:11095–11103. [PubMed] [Google Scholar]
- 13.Tokumura A, Toujima M, Yoshioka Y, Fukuzawa K. Lipids. 1996;31:1251–1258. doi: 10.1007/BF02587909. [DOI] [PubMed] [Google Scholar]
- 14.Frankel EN. Prog Lipid Res. 1984;23:197–221. doi: 10.1016/0163-7827(84)90011-0. [DOI] [PubMed] [Google Scholar]
- 15.Itabe H, Kushi Y, Handa S, Inoue K. Biochim Biophys Acta. 1988;962:8–15. [PubMed] [Google Scholar]
- 16.Smiley PL, Stremler KE, Prescott SM, Zimmerman GA, McIntyre TM. J. Biol. Chem. 1991;266:11104–11110. [PubMed] [Google Scholar]
- 17.Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. J Biol Chem. 2002;277:38517–38523. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- 18.Walton KA, Hsieh X, Gharavi N, Wang S, Wang G, Yeh M, Cole AL, Berliner JA. J Biol Chem. 2003;278:29661–29666. doi: 10.1074/jbc.M300738200. [DOI] [PubMed] [Google Scholar]
- 19.Knapp S, Matt U, Leitinger N, van der Poll T. J Immunol. 2007;178:993–1001. doi: 10.4049/jimmunol.178.2.993. [DOI] [PubMed] [Google Scholar]
- 20.Kim HE, Du F, Fang M, Wang X. Proc Natl Acad Sci U S A. 2005;102:17545–17550. doi: 10.1073/pnas.0507900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cregan SP, Dawson VL, Slack RS. Oncogene. 2004;23:2785–2796. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 22.Yang E, Korsmeyer SJ. Blood. 1996;88:386–401. [PubMed] [Google Scholar]
- 23.Green DR, Kroemer G. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 24.Madamanchi NR, Runge MS. Circ Res. 2007;100:460–473. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- 25.Bligh EG, Dyer WJ. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 26.Otera H, Ohsakaya S, Nagaura Z, Ishihara N, Mihara K. Embo J. 2005;24:1375–1386. doi: 10.1038/sj.emboj.7600614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danial NN, Korsmeyer SJ. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 28.Loeffler M, Daugas E, Susin SA, Zamzami N, Metivier D, Nieminen AL, Brothers G, Penninger JM, Kroemer G. Faseb J. 2001;15:758–767. doi: 10.1096/fj.00-0388com. [DOI] [PubMed] [Google Scholar]
- 29.Schafer ZT, Kornbluth S. Dev Cell. 2006;10:549–561. doi: 10.1016/j.devcel.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Itabe H, Kushi Y, Handa S, Inoue K. Biochim Biophys Acta. 1988;962:8–15. [PubMed] [Google Scholar]
- 31.Watanabe N, Zmijewski JW, Takabe W, Umezu-Goto M, Goffe CL, Sekine A, Landar A, Watanabe A, Aoki J, Arai H, Kodama T, Murphy MP, Kalyanaraman R, Darley-Usmar VM, Noguchi N. Am J Pathol. 2006;168:1737–1748. doi: 10.2353/ajpath.2006.050648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kogure K, Nakashima S, Tsuchie A, Tokumura A, Fukuzawa K. Chem Phys Lipids. 2003;126:29–38. doi: 10.1016/s0009-3084(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 33.Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C. J Cell Biol. 2003;160:53–64. doi: 10.1083/jcb.200210084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. J Biol Chem. 2006;281:22275–22288. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asmis R, Begley JG. Circ Res. 2003;92:e20–29. doi: 10.1161/01.res.0000051886.43510.90. [DOI] [PubMed] [Google Scholar]
- 36.Wintergerst ES, Jelk J, Rahner C, Asmis R. Eur J Biochem. 2000;267:6050–6059. doi: 10.1046/j.1432-1327.2000.01682.x. [DOI] [PubMed] [Google Scholar]
- 37.Vindis C, Elbaz M, Escargueil-Blanc I, Auge N, Heniquez A, Thiers JC, Negre-Salvayre A, Salvayre R. Arterioscler Thromb Vasc Biol. 2005;25:639–645. doi: 10.1161/01.ATV.0000154359.60886.33. [DOI] [PubMed] [Google Scholar]
- 38.Loidl A, Sevcsik E, Riesenhuber G, Deigner HP, Hermetter A. J Biol Chem. 2003;278:32921–32928. doi: 10.1074/jbc.M306088200. [DOI] [PubMed] [Google Scholar]
- 39.Fruhwirth GO, Moumtzi A, Loidl A, Ingolic E, Hermetter A. Biochim Biophys Acta. 2006;1761:1060–1069. doi: 10.1016/j.bbalip.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 40.Moumtzi A, Trenker M, Flicker K, Zenzmaier E, Saf R, Hermetter A. J Lipid Res. 2007;48:565–582. doi: 10.1194/jlr.M600394-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Finkelstein EI, Ruben J, Koot CW, Hristova M, van der Vliet A. Am J Physiol Lung Cell Mol Physiol. 2005;289:L1019–1028. doi: 10.1152/ajplung.00227.2005. [DOI] [PubMed] [Google Scholar]
- 42.Sun L, Luo C, Long J, Wei D, Liu J. Mitochondrion. 2006;6:136–142. doi: 10.1016/j.mito.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 43.Kristal BS, Park BK, Yu BP. J Biol Chem. 1996;271:6033–6038. doi: 10.1074/jbc.271.11.6033. [DOI] [PubMed] [Google Scholar]
- 44.Landar A, Shiva S, Levonen AL, Oh JY, Zaragoza C, Johnson MS, Darley-Usmar VM. Biochem J. 2006;394:185–195. doi: 10.1042/BJ20051259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hirashima Y, Ueno H, Karasawa K, Yokoyama K, Setaka M, Takaku A. Brain Res. 2000;885:128–132. doi: 10.1016/s0006-8993(00)02852-3. [DOI] [PubMed] [Google Scholar]
- 46.Ogden F, DeCoster MA, Bazan NG. J Neurosci Res. 1998;53:677–684. doi: 10.1002/(SICI)1097-4547(19980915)53:6<677::AID-JNR6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 47.Bonin F, Ryan SD, Migahed L, Mo F, Lallier J, Franks DJ, Arai H, Bennett SA. J Biol Chem. 2004;279:52425–52436. doi: 10.1074/jbc.M410967200. [DOI] [PubMed] [Google Scholar]
- 48.Matsuzawa A, Hattori K, Aoki J, Arai H, Inoue K. J Biol Chem. 1997;272:32315–32320. doi: 10.1074/jbc.272.51.32315. [DOI] [PubMed] [Google Scholar]
- 49.Marques M, Pei Y, Southall MD, Johnston JM, Arai H, Aoki J, Inoue T, Seltmann H, Zouboulis CC, Travers JB. J Invest Dermatol. 2002;119:913–919. doi: 10.1046/j.1523-1747.2002.01859.x. [DOI] [PubMed] [Google Scholar]
- 50.Nishi K, Itabe H, Uno M, Kitazato KT, Horiguchi H, Shinno K, Nagahiro S. Arterioscler Thromb Vasc Biol. 2002;22:1649–1654. doi: 10.1161/01.atv.0000033829.14012.18. [DOI] [PubMed] [Google Scholar]
- 51.Uhlson C, Harrison K, Allen CB, Ahmad S, White CW, Murphy RC. Chem Res Toxicol. 2002;15:896–906. doi: 10.1021/tx010183i. [DOI] [PubMed] [Google Scholar]