

Abstract
Objective
Genetic loss of TSC1/TSC2 function in tuberous sclerosis complex (TSC) results in overactivation of the mammalian target of rapamycin complex 1 pathway, leading to cellular dysplasia. We hypothesized that the dysplastic cells in TSC tubers are heterogeneous, including separable classes on a neuronal-glial spectrum, and that these dysplastic cells express glutamate receptor (GluR) patterns consistent with increased cortical network excitability.
Methods
Surgically resected human cortical tubers and nondysplastic epileptic cortical samples were analyzed by double-label immunocytochemistry for coexpression of neuronal and glial markers, the TSC1/TSC2 pathway downstream molecule phospho-S6 (pS6) and GluR subunits, and compared with control cortical tissue. Western blotting was used to quantify changes in GluR subunit expression in tubers versus controls.
Results
We demonstrate that cortical tubers contain a broad spectrum of cell types including disoriented pyramidal cells, dysplastic neurons, giant neuroglial cells, dysplastic astroglia, and reactive astrocytes. Dysplastic neurons, giant cells, and dysplastic astroglia express high levels of pS6 and demonstrate altered GluR subunit composition, resembling those of normal immature neurons and glia. In contrast, nondysplastic neurons in TSC and non-TSC epileptic lesions express lower pS6 levels and display changes in GluR subunit expression that are distinct from the patterns seen in tuber dysplastic cells.
Interpretation
This work significantly expands the spectrum of abnormal cells recognized in tubers beyond the classic tuber giant cell and demonstrates cell-specific abnormalities in GluR expression that may contribute to seizure pathogenesis in TSC. Furthermore, these results suggest that subunit-specific antagonists may be of potential use in the treatment of epilepsy in TSC.
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder caused by inactivating mutations in either the TSC1 or TSC2 gene.1,2 TSC brain lesions are malformations of cortical development that include cortical tubers, white matter abnormalities, subependymal nodules, and subependymal giant cell tumors.3 Cortical tuber pathology is characterized by disrupted cortical and subcortical architecture and the presence of cytomegalic cells.4 Seizures occur in 75 to 90% of TSC patients and often begin in the first year of life as infantile spasms.5 Tubers are recognized as the source of interictal discharges, although often epileptogenic areas involve the adjacent cortex.6 Pharmacoresistent epilepsy is often seen in TSC patients, requiring epilepsy surgery.7
TSC1 and TSC2 encode the proteins hamartin and tuberin, respectively, which form a complex and function as tumor suppressors by reducing Rheb-GTP levels and inhibiting the mammalian target of rapamycin complex 1 (mTORC1) (for review see Kwiatkowski and Manning8). The mTORC1 kinase phosphorylates downstream targets including ribosomal S6 kinases (S6K1 and S6K2) and eukaryotic initiation factor 4E–binding protein, enhancing protein synthesis and cell growth. In addition, hamartin and tuberin have a role in the regulation of the actin cytoskeleton through effects on Rho-family GTP-ases.9 To date, there is no evidence for complete genetic loss of TSC1/TSC2 in tubers, although like cells in other TSC lesions,10 tuber cells show markers of mTORC1 hyperactivation.11–13
The pathogenesis of the TSC-related neurological symptoms including epilepsy is unclear. A critical factor regulating neuronal excitability is expression and function of synaptic receptors for the main excitatory neurotransmitter glutamate. Ionotropic glutamate receptors (iGluRs) include the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) and the N-methyl-d-aspartate receptors (NMDARs), and are composed of multiple protein subunits (glutamate receptors 1–4[GluR1–4] for AMPARs; NR1, NR2A-D, and NR3A-B for NMDARs), producing isoforms with different functional properties.14 iGluR expression is developmentally regulated, and during early cortical development, certain receptor subtypes are overexpressed resulting in physiological hyperexcitability during the critical periods of enhanced synaptic plasticity.15
Increasing experimental data suggest a direct effect of the Tsc1 or Tsc2 mutations on brain development and excitability. Loss of Tsc1 or Tsc2 in mature post-mitotic hippocampal neurons in vitro causes enlarged somas, abnormal dendritic spines, and enhancement of glutamatergic neurotransmission.16 In vivo, both astrocyte-specific17 and neuron-specific18 Tsc1 conditional knock-out mice exhibit abnormal brain organization and spontaneous seizure activity. In addition, mice lacking Tsc1 expression in neurons demonstrate increased neocortical excitability.19
Although previous reports have recognized the occurrence of various dysplastic cell types within tubers beyond the classical giant cell,4,20,21 we set out to classify the cellular pathology of these lesions in detail, using combined morphological and immunocytochemical criteria, including the use of neuronal and glial markers and phospho-S6 (pS6) to assess the mammalian target of rapamycin (mTOR) pathway dysfunction. In addition, given that TSC is a malformation of development, we explored the hypothesis that the abnormal cell types present in tubers express immature iGluRs patterns known to promote cortical network hyperexcitability, thereby possibly contributing to the epileptogenicity of these lesions. White and coworkers22 previously reported that dysplastic neurons and giant cells express abnormal patterns of glutamate and GABA receptor subunit messenger RNA. To further determine whether neurotransmitter function might be altered on tuber cells, we quantified differences in membrane expression of AMPAR and NMDAR subunits in cortical tubers and controls, and evaluated their expression patterns within different classes of dysplastic cells. In parallel, we performed a subset of double-labeling experiments for specific receptor subunits in nondysplastic epilepsy tissue, in an attempt to differentiate between the effects of seizure activity versus TSC1/TSC2 dysfunction on iGluR expression in TSC.
Subjects and Methods
Human Neocortical Tissue
The relevant clinical and neuropathological data of all human brain specimens used in this study are summarized in Table 1 and Table 2. All procedures and experiments were conducted under guidelines approved by the Committees for Clinical Research at Children’s Hospital, and Brigham and Women’s Hospital Boston. Cortical tubers (n = 10; 7 male and 3 female patients) were obtained from patients clinically diagnosed with tuberous sclerosis,23 who underwent surgical treatment for focal epilepsy at Children’s Hospital Boston (n = 9) and New York University Medical Center (n = 1). The diagnosis of TSC was always confirmed by subsequent neuropathological examination. Nondysplastic epileptic cortical samples from cases diagnosed with medically intractable epilepsy not associated with TSC (n = 6; 4 male and 2 female patients) and treated in the Epilepsy Surgery Service at Children’s Hospital Boston included samples from Rasmussen’s encephalitis cases (n = 2), perilesional tissue from diagnosed focal cortical dysplasia type Ia24 (n = 1), and temporal lobe epilepsy with or without hippocampal sclerosis (n = 3). All nondysplastic epileptic samples exhibited normal cortical architecture, with no signs of cellular dysplasia by standard neuropathological examination. Control neocortical tissue from autopsy cases with normal neurological history (n = 7; 3 male and 4 female patients) was obtained from the Neuropathology Department at Children’s Hospital and Brigham and Women’s Hospital Boston (n = 2) as part of the standard postmortem examination and from the University of Maryland Brain Tissue Bank for Developmental Disorders (n = 5). A detailed description of human subjects is available online (see Supplementary Methods).
Table 1.
Clinical and Neuropathological Characteristics of TSC and Non-TSC Epilepsy Cases
| Case No. | Sex | Pathology | Age at Surgery (yr) | Age at Seizure Onset (yr) | Antiepileptic Medication | Brain Region |
|---|---|---|---|---|---|---|
| 1 | M | TSC | 1.6 | 0.5 | Lamotrigine, lorazepam, phenobarbital, vigabatrin | Right parietal lobe |
| 2 | M | TSC | 2.5 | 0.2 | Phenytoin, lamotrigine, vigabatrin | Right temporal lobe |
| 3 | F | TSC | 2.6 | 0.4 | Carbamazepine, topiramate | Left temporal lobe |
| 4 | M | TSC | 3.3 | 0.4 | Phenytoin | Left frontal lobe |
| 5 | M | TSC | 3.7 | 0.2 | Carbamazepine, clonazepam, topiramate | Left occipital lobe |
| 6 | F | TSC | 3.8 | 0.7 | Ketogenic diet, vigabatrin | Left occipital lobe |
| 7 | M | TSC | 3.8 | NA | NA | Right frontal lobe |
| 8 | M | TSC | 4.8 | 0.6 | Carbamazepine, clonazepam, lamotrigine | Right frontal lobe |
| 9 | F | TSC | 5.3 | NA | NA | Left temporal lobe |
| 10 | M | TSC | 6.8 | 0.1 | Carbamazepine, phenytoin | Left temporal lobe |
| 11 | M | Rasmussen | 3.5 | 3.2 | Phenobarbital, oxcarbazepine, levetiracetam | Right occipital lobe |
| 12 | M | Rasmussen | 14.4 | 13.9 | Diazepam, oxcarbazepine, levetiracetam | Right frontal lobe |
| 13 | M | FCD Iaa | 3.2 | 0.3 | Vigabatrin, levetiracetam | Right occipital lobe |
| 14 | F | TLE | 13 | NA | NA | Right temporal lobe |
| 15 | M | TLE | 14.2 | 1.5 | Lamotrigine, carbamazepine, levetiracetam | Right temporal lobe |
| 16 | F | TLE | 10.1 | 1.1 | Lamotrigine, oxcarbazepine | Left temporal lobe |
Only perilesional tissue was used for analysis from this case.
TSC = tuberous sclerosis complex; NA = information not available; FCD Ia = focal cortical dysplasia type Ia; TLE = temporal lobe epilepsy.
Table 2.
Clinical and Neuropathological Characteristics of Control Subjects
| Subject No. | Sex | Pathology | Age at Autopsy (yr) | PMI (hr) | Cause of Death | Brain Region |
|---|---|---|---|---|---|---|
| 1 | M | Control | 1 | 15 | Drowning | Parietooccipital lobe |
| 2 | F | Control | 2.5 | 20 | Traumatic multiple injuries | Parietooccipital lobe |
| 3 | F | Control | 3.3 | 11 | Drowning | Parietooccipital lobe |
| 4 | M | Control | 6 | 20 | Traumatic multiple injuries | Parietooccipital lobe |
| 5 | M | Control | 21 | 18 | Drowning | Parietooccipital lobe |
| 6 | F | Control | 41 | 24 | Nonneurological | Parietooccipital lobe |
| 7 | F | Control | 51 | 8 | Nonneurological | Parietooccipital lobe |
PMI = postmortem interval
Immunocytochemistry
Immunofluorescence double labeling was performed on a total of 18 cases: 6 TSC cortical tubers (Cases 2, 4, 5, 6, 8, and 9), 6 non-TSC surgical specimens (Cases 11–16), and 6 control autopsy specimens (Cases 1–6). According to our published protocols,25 fresh tissue blocks were fixed in 4% paraformaldehyde for 48 to 72 hours and then cryoprotected in 30% sucrose for 24 hours. Additional immunocytochemistry experiments were performed on snap-frozen material, as described previously.25 Tissue samples processed in a similar way were run in batches together. Between uses, tissue blocks were kept frozen at −80°C to avoid tissue degradation and to optimize staining quality (see Supplementary Methods for details). The distribution pattern and relative staining intensity for each antibody was qualitatively analyzed in three to six cases/group (at least two sections/case). Quantitation of pS6+ cells was performed on six low-power (×100) microscopic fields in two serial sections/case. Positive cells were counted when the nuclear DAPI stain (Vector Laboratories, Burlingame, CA) was distinct (n = 552 cells). pS6+/SMI 311+ dysplastic neurons and giant cells were distinguished from each other based on morphological criteria (shape, process characteristics, and presence of multiple nuclei), whereas the elongated bipolar pS6+/SMI 311− cells were considered to be dysplastic astroglia.
Immunoblotting
Western blot quantification was performed on a total of 11 cases: 6 TSC surgical specimens (Cases 1, 3, 6, 7, 9, 10) and 5 control samples (Cases 2, 3, 5–7). Snap-frozen cortical tubers and control samples were processed for membrane protein extraction, as previously described25 (see Supplementary Methods). Individual values for each receptor subunit were expressed as percentage of the mean level of expression of control samples run on the same blot (100%). Means (three to five cases/group) were compared by t tests and differences between groups were considered significant at p ≤ 0.05.
Results
Neuropathological Features of Human Cortical Tubers and Cell-Type Characterization
Cortical tubers and control specimens were analyzed by single and double label immunocytochemistry using a broad spectrum of ontogenetic and functional markers, including developmentally regulated neuronal and glial specific proteins and pS6, as indicator of mTOR pathway activation.11–13 Single labeling with neuron-specific nuclear protein (NeuN), non-phosphorylated neurofilament SMI 311, vimentin and glial fibrillary acidic protein (GFAP) demonstrated cortical architectural defects associated with cytomegaly and variable degrees of gliosis in all cortical tuber specimens analyzed (see Supplemental Fig 1a).
Differential coexpression of neuronal and glial cell markers in individual tuber cells highlighted the cellular heterogeneity of these lesions (see Fig 1). Dysplastic neurons were immunopositive for neuronal marker SMI 311 but immunonegative for vimentin, a marker typically expressed in neuroglial progenitor cells, including radial glia26 (see Figs 1a, A–C). In contrast, giant cells demonstrated both SMI 311 and vimentin immunoreactivity (see Fig 1a, D–F). Dysplastic neurons demonstrated consistent immunopositivity for NeuN, expressed only in postmitotic neurons,27 whereas giant cells were NeuN-negative (data not shown). Neither of these cell types coexpressed the mature astrocytic marker GFAP. Another separable population was represented by dysplastic astroglial cells, which were immunopositive for vimentin but negative for SMI 311 (see Fig 1a, D–F, arrowhead), as well as GFAP (see Fig 1b, A–C). In contrast, numerous cells with morphological characteristics of reactive astrocytes coexpressed both vimentin and GFAP (see Fig 1b, D–F). Interspersed normal-sized disoriented and normal-appearing neurons were immunopositive for only neuronal markers NeuN and SMI 311, whereas resting astrocytes stained only with GFAP (data not shown).
Fig 1. Differential expression of cell-specific markers and phospho-S6 (pS6) in human cortical tubers.

(a) Double-label immunofluorescence for neuronal marker nonphosphorylated neurofilament SMI 311 (green) and immature glial marker vimentin (red) identify three different abnormal cell types. Dysplastic neurons express SMI 311 but not vimentin (A–C), undifferentiated giant cells express both SMI 311 and vimentin (D–F), and dysplastic astroglia express vimentin, but not SMI 311 (D–F, arrowhead). Scale bar =40 µm. (b) Double-label immunofluorescence for vimentin (red) and mature astrocytic marker glial fibrillary acidic protein (GFAP; green) shows that dysplastic astroglial cells express vimentin, but low or no GFAP (A–C), whereas reactive astrocytes demonstrate strong expression of both glial markers (D–F). Scale bar = 40 µm. (c) Confocal imaging shows undifferentiated giant cells expressing both SMI 311 (A) and vimentin (B), together with pS6, as well as multiple nuclei (B, small arrows). Scale bars = 20 µm. (d) Neuron-specific nuclear protein (NeuN)–positive dysplastic neurons (A–C, arrowheads) are intensely pS6-immunopositive, whereas normal-sized disoriented neurons (A–C) demonstrate low or no pS6 expression. Vimentin-positive dysplastic astroglia (D–F) demonstrate increased pS6 levels, in contrast with GFAP-positive reactive astrocytes (G–I), which demonstrate no pS6 labeling. Scale bar = 40 µm.
The cellular specificity of mTOR activation and the regional distribution of dysplastic cell types in human cortical tubers were further analyzed by colabeling for pS6 (Ser 235/236) and different cellular markers. Consistent with previous reports,11–13 we found increased pS6 expression in giant cells (see Fig 1c, A–B) and dysplastic neurons (see Fig 1d, A–C, arrowheads), but not in normal-sized disoriented and normal-appearing neurons (see Fig 1d, A–C). Increased pS6 expression also was observed in dysplastic astroglia (see Fig 1d, D–F), in contrast with minimal pS6 levels in the reactive astrocytes (see Fig 1d, G–I). The analysis of regional distribution of dysplastic pS6+ cells (data not shown) demonstrated that overall, each cell type represented approximately a third of the total pS6+ cells (31% dysplastic neurons, 37% giant cells, and 32% dysplastic astroglia). However, in the cortical portion of the tubers, most pS6+ cells were dysplastic neurons (39%) and giant cells (48%), while dysplastic astroglia represented only a small fraction (13%). In the white matter portion of the tubers, the majority of dysplastic cells were astroglia (62%) followed by giant cells (20%) and dysplastic neurons (18%). The cell-specific pS6 expression, together with all other structural and functional markers used in this study, is summarized in Figure 5.
Fig 5.
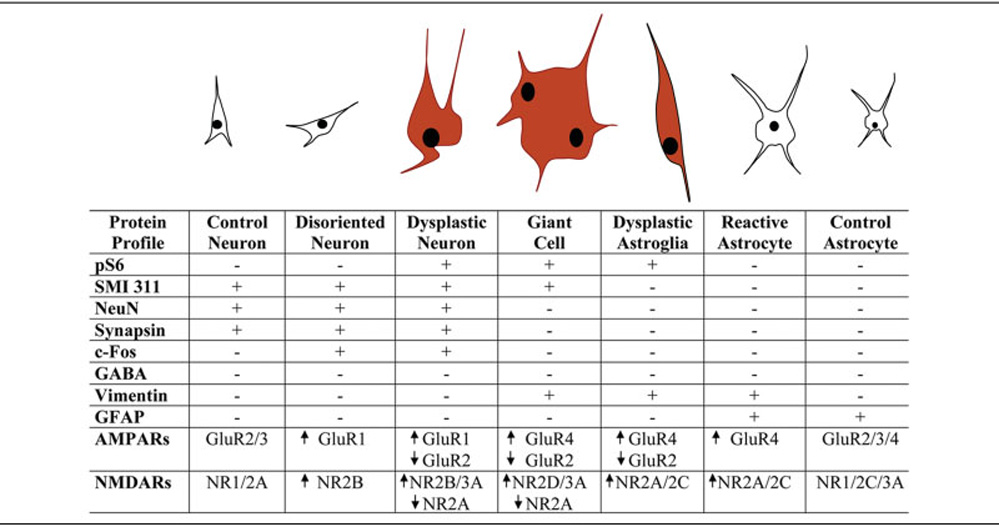
The spectrum of cellular elements in human cortical tubers and their specific immunocytological profile. High pS6 levels are exclusively present in dysplastic neurons, giant cells, and dysplastic astroglial cells (red). Dysplastic neurons express only neuronal markers, demonstrate capacity to form synaptic contacts, and appear to be excitatory neurons. They express α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) and N-methyl-d-aspartate receptor (NMDAR) profiles suggestive for increased Ca2+ permeability (high glutamate receptor 1 [GluR1]/GluR2 and high NR2B/NR2A ratios) and low Mg2+sensitivity (high NR3A), similar to immature postmitotic cortical neurons. Giant cells express both neuronal and glial markers, no synaptic markers, and AMPAR and NMDAR subunits consistent with an undifferentiated neuroprogenitor phenotype (high GluR4/GluR2 ratio and high NR2D). Dysplastic astroglial cells are immunopositive for neuroglial progenitor marker vimentin and express AMPAR and NMDAR subunits suggestive for an immature radial glial phenotype (high GluR4/GluR2 ratio). GFAP = glial fibrillary acidic protein; NeuN = neuron-specific nuclear protein.
The capability of dysplastic cells to form synaptic contacts was analyzed using the presynaptic marker synapsin. Synapsin was highly expressed around dysplastic neuron cell bodies and processes (see Supplemental Fig 2a, A–C), whereas there was no synapsin immunoreactivity surrounding the giant cells (see Supplemental Fig 2a, D–F). This expression pattern correlated with increased c-Fos expression, an immediate early gene product widely used as marker for transsynaptic neuronal excitation.28 c-Fos was highly expressed in dysplastic neurons (see Supplemental Fig 2b, A–C), being mostly concentrated in the nuclei, in contrast with giant cells that showed relatively weak c-Fos expression confined to the cytoplasm (see Supplemental Fig 2b, D–F). Intense synapsin and c-Fos immunoreactivity also was observed in normal-sized disoriented and normal-appearing neurons within tuber tissue (data not shown).
Next, to assess inhibitory versus excitatory phenotype of dysplastic tuber cells, we used the inhibitory neuronal marker GABA and Ca2+ binding proteins parvalbumin and calbindin. Neither dysplastic neurons (see Supplemental Fig 2c, A–C) nor giant cells (see Supplemental Fig 2c, D–F) were GABA-immunopositive, although a small fraction of dysplastic neurons intensely labeled with parvalbumin (see Supplemental Fig 2d, A–C) and calbindin (see Supplemental Fig 2d, D–F).
Differential Expression of AMPA and NMDA Receptor Subunits in Human Cortical Tubers
Enhanced glutamatergic neurotransmission may critically contribute to seizure initiation and development of epilepsy in TSC.16,19,22 We thus further examined human TSC brain tissue for alterations in expression levels and cell-specific patterns of AMPAR and NMDAR subunits, relative to control tissue.
Western blot analysis of cortical tissue homogenates demonstrated that GluR1 and GluR4 AMPAR subunit expression (Figs 2a, b) was significantly greater (p < 0.05) compared with control tissue (312% of control for GluR1, and 468% of control for GluR4). Immunocytochemical staining of tuber sections demonstrated that GluR1 was highly expressed in the majority of normal-sized (see Fig 2c, A–C) and dysplastic neurons (see Fig 2c, D–F, J–L, small arrow), but was low in most giant cells (see Fig 2c, G–I), dysplastic astroglia (see Fig 2c, J–L, arrowhead) and reactive astrocytes (not shown). In contrast, GluR4 expression was low in the majority of normal-sized (see Fig 2d, A–C) and dysplastic neurons (see Fig 2d, D–F, arrowhead), and consistently was increased in giant cells (see Fig 2d, G–I), dysplastic astroglia (see Fig 2d, D–F, small arrow J–L), and reactive astrocytes (not shown). Unlike GluR1 and GluR4, GluR2 and GluR3 levels (Figs 3a, b) were significantly lower in cortical tubers (p < 0.05), relative to controls (30% of control for GluR2 and 20% of control for GluR3). Cell-specific staining of tuber tissue showed that GluR2, although highly expressed in normal-sized neurons (see Fig 3c, A–C), was low in most dysplastic neurons (see Fig 3c, D–F, arrowhead), giant cells (see Fig 3c, G–I, arrowhead), dysplastic astroglia (see Fig 3c, J–L), and reactive astrocytes (data not shown). A similar cell-specific expression pattern to that of GluR2 was observed for GluR3 (data not shown). In control tissue, pyramidal neurons expressed high levels of GluR2 and GluR3, and low levels of GluR1 and GluR4 subunits, whereas normal cortical astrocytes expressed low GluR2, GluR3, and GluR4 levels, as we have previously reported25 (data not shown).The expression pattern of AMPAR subunits in cortical tubers suggests a relative GluR2 deficiency in the pS6+ dysplastic neurons, giant cells, and dysplastic astroglia.
Fig 2. Increased expression of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) subunits glutamate receptor 1 (GluR1) and GluR4 in human cortical tubers.

(a, b) Western blot quantification of GluR1 (a) and GluR4 (b) expression demonstrates a significant increase in the tubers in comparison with control cortex (312% of control for GluR1; 468% for GluR4). * = p ≤ 0.05. Insets are representative Western blots for individual subunits. (c) Both normal-sized (A–C) and dysplastic neurons (D–F, J–L, small arrow) express high GluR1 levels, whereas giant cells (G–I) and dysplastic astroglia (J–L, arrowhead) demonstrate relatively low GluR1 expression. (d) In contrast with GluR1, GluR4 expression is low in normal-sized (A–C) and dysplastic neurons (D–F, arrowhead), but highly expressed in giant cells (G–I) and dysplastic astroglia (D–F, small arrow; J–L). Scale bars = 40µm. NeuN = neuron-specific nuclear protein; TSC = tuberous sclerosis complex.
Fig 3. Decreased expression of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) subunits glutamate receptor 2 (GluR2) and GluR3 in the tuberal tissue.
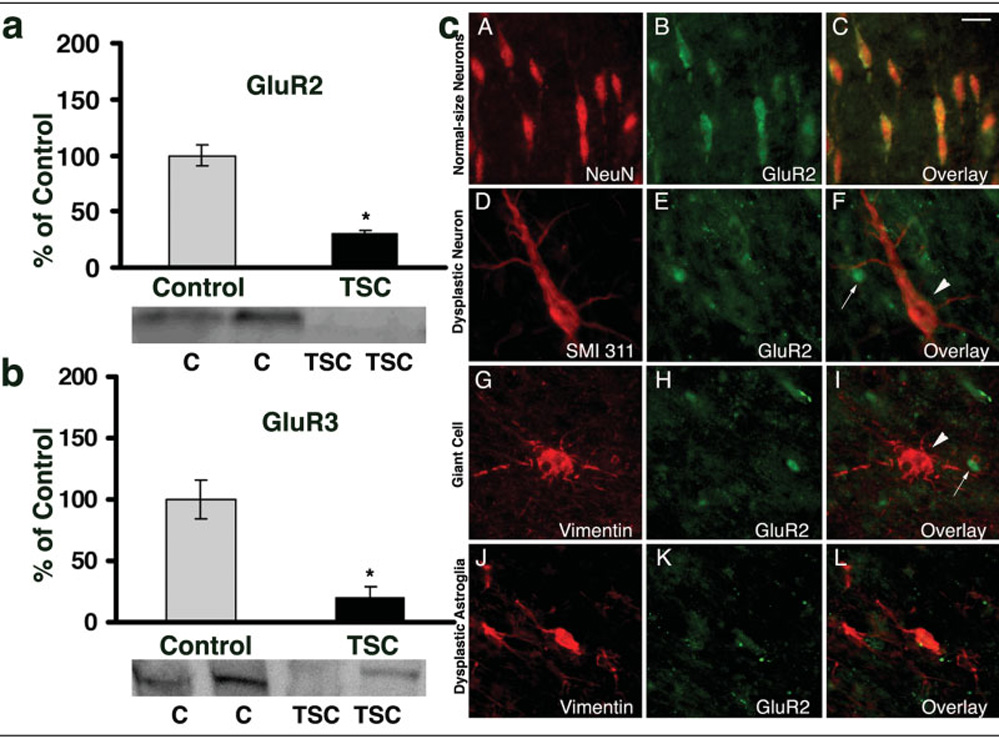
(a, b) Western blot analysis of GluR2 (a) and GluR3 (b) subunits demonstrate that, in cortical tubers, there is significant decrease in expression of both subunits, relative to control (30% of control for GluR2; 20% of control for GluR3). * = p ≤ 0.05. Insets show representative Western blots for each subunit. (c) Normal-sized neurons (A–C) express high GluR2 levels, whereas dysplastic neurons (D–F, arrowhead), giant cells (G–I, arrowhead), and dysplastic astroglia (J–L) demonstrate lower GluR2 expression, compared with surrounding cells (small arrows in F, I). Scale bar = 20 µm. NeuN = neuron-specific nuclear protein; TSC = tuberous sclerosis complex.
The expression of the obligatory NMDAR subunit NR1 was not significantly different in tubers relative to controls (71% of control; p > 0.05), and cell-specific NR1 expression pattern in neuronal and glial subtypes in the tubers was comparable with that in neurons and astrocytes in the control group (data not shown). Total NR2A expression was not significantly different in TSC samples compared with controls (133% of control; p > 0.05) (see Supplemental Fig 3a), although there were marked differences in NR2A cell–specific expression. In cortical tubers, NR2A subunit was highly expressed in most normal-sized neurons (see Supplemental Fig 3b, A–C) but was low or absent in most dysplastic neurons (see Supplemental Fig 3b, D–F) and giant cells (see Supplemental Fig 3b, G–I). The glial components including dysplastic astroglia (see Supplemental Fig 3b, J–L) and reactive astrocytes (see Supplemental Fig 3b, M–O) consistently expressed high NR2A levels. Total expression of both NR2B (see Fig 4a) and NR3A subunits (see Fig 4b) were significantly greater in cortical tubers relative to controls (412% of control for NR2B, p < 0.05; 304% of control for NR3A, p < 0.05). In tuber tissue, NR2B was high in most normal-sized and dysplastic neurons (see Fig 4c, A–C), but relatively low in giant cells (see Fig 4c, D–F), dysplastic astroglia (see Fig 4c, G–I), and reactive astrocytes (see Fig 4c, J–L). NR2C was low in normal-sized neurons, dysplastic neurons, and giant cells, but was high in the majority of dysplastic astroglial cells and reactive astrocytes, whereas NR2D was high in most giant cells, but low or undetectable in other cell types (data not shown). In contrast, NR3A was at high levels in all tuber cells types (see Fig 4d, A–L). In control cortex, pyramidal neurons expressed high NR1 and NR2A and low NR2B, NR2C, NR2D, and NR3A levels, whereas cortical astrocytes demonstrated relatively high levels of NR1, NR2A, and NR2C subunits, associated with low or no NR2B, NR2D, and NR3A expression (data not shown). In summary, although overall NR1 subunit expression was similar in the TSC and control cases, there were marked differences in the expression levels and/or cellular distribution of the regulatory subunits NR2 and NR3.
Fig 4. Upregulation of N-methyl-d-aspartate receptors (NMDAR) subunits NR2B and NR3A in human cortical tubers.

(a, b) Western blot analysis of the membrane-expressed NR2B (a) and NR3A (b) subunits demonstrate that, in cortical tubers, the expression levels of both subunits are significantly greater compared with controls (412% for NR2B; 304% for NR3A). * = p ≤ 0.05. Insets are representative Western blots for individual subunits. (c) NR2B subunit is highly expressed in normal-sized and dysplastic neurons (A–C), but not in giant cells (D–F), dysplastic astroglia (G–I), or reactive astrocytes (J–L). Scale bar = 40µm. (d) High NR3A levels are expressed in dysplastic neurons (A–C), giant cells (D–F), dysplastic astroglia (G–I), and reactive astrocytes (J–L). Scale bar = 40 µm. GFAP = glial fibrillary acidic protein; NeuN = neuron-specific nuclear protein; TSC = tuberous sclerosis complex.
AMPA and NMDA Receptor Subunit Expression in Human Nondysplastic Epileptic Tissue
Numerous studies in animal models show that seizures can alter iGluR expression and function,29 suggesting that altered iGluR subunit expression in cortical tubers might be, at least in part, seizure induced. Therefore, we examined the cell-specific expression patterns of select iGluR subunits in human spiking nondysplastic neocortical tissue for comparison.
Immunocytochemical analysis of nondysplastic epileptic tissue demonstrated preserved cortical lamination with no signs of cellular abnormalities or dysplasia, except for moderate neuronal loss and reactive gliosis in some cases (see Supplemental Fig 1b). Notably, pS6 (Ser 235/236) expression in nondysplastic epileptic cortex was generally low and comparable with controls, although moderate increases in pS6 levels, at levels well below those observed in TSC tissue, were seen occasionally in neurons located in the upper cortical layers II-IV (data not shown).
Similar to tuber samples, there was marked GluR1 upregulation in the majority of cortical neurons in nondysplastic epileptic cortex (see Supplemental Fig 4a, A–C). However, unlike the low GluR2 levels in dysplastic neurons in tubers, neurons in the nondysplastic cortex showed only modest decreases in GluR2 (see Supplemental Fig 4a, D–F). The majority of cortical neurons in the spiking nondysplastic cortex expressed no obvious alterations in NR1 and NR2A subunits relative to control tissue (see Supplemental Fig 4b, A–C), unlike the low NR2A in dysplastic neurons in tubers. However, cortical neurons in epileptic non-dysplastic cortex showed increased NR2B (see Supplemental Fig 4b, D–F) and NR3A expression (see Supplemental Fig 4b, G–I), similar to dysplastic neurons in tubers.
Discussion
This study identifies a spectrum of abnormal cell types in TSC tubers based on morphological characteristics, markers of neuronal and glial differentiation, mTOR pathway activation, and iGluR subunit expression. Dysplastic cells in cortical tubers expressing high pS6 levels demonstrate unique iGluR expression patterns consistent with a hyperexcitable state and reminiscent of those present on immature neurons and astrocytes in normal human developing cortex.25 Differences in iGluR expression profiles between dysplastic and non-dysplastic epileptic tissue suggest that, in human cortical tubers, TSC1/TSC2 dysfunction may alter the developmental regulation of iGluRs and enhance glutamatergic function.
Cellular Abnormalities in Human Cortical Tubers
Cortical dysplasia, including TSC, is thought to be a malformation of cortical development, and the timing of the insult/dysfunction might explain the different manifestations of the disease.24,30 The TSC1/TSC2 proteins hamartin and tuberin are normally expressed at high levels in neurons and glia during early stages of corticogenesis and decline with age, suggesting an important role of both proteins in normal brain development and function.31 Indeed, we demonstrate several distinct subpopulations of tuber cells that resemble the neuronal and glial progenitor cells that are present in the normal human fetal brain25,26 (see Fig 5). Consistent with the central role of the mTOR pathway dysregulation in the pathogenesis of TSC,8 we found that all these dysplastic cell types demonstrate a marked increase in pS6 expression, suggestive of loss of TSC1/ TSC2 function and mTOR upregulation. Dysplastic neurons have characteristics of differentiated postmitotic neurons, being reactive for neuronal but not glial markers. Multinucleated giant cells express both neuronal and glial markers, suggesting that these hallmark cells of TSC may represent an arrested neuroglial progenitor cell, whereas dysplastic astroglial cells are negative for neuronal but positive for the immature glial markers, resembling an early radial glial phenotype.32
Previous electrophysiological studies in human dysplastic tissue have demonstrated that the dysplastic neurons are synaptically connected, whereas giant and balloon cells appear to lack synaptic inputs.30 Coexpression of c-Fos and synapsin in normal and dysplastic neurons, but not in giant cells, suggests that in human cortical tubers dysplastic neurons may be synaptically connected with surrounding tissue. The lack of GABA immunoreactivity in dysplastic neurons and giant cells suggests an excitatory phenotype, in agreement with prior reports of GAD65 negativity in these cell types.22 Consistent with recent reports by others, we observed scattered parvalbumin and calbindin-D (28k)–positive cytomegalic neurons in tubers, 21,34 but we found that these cells lack GABA expression, suggesting that they may lack inhibitory function.
Cell-Specific Abnormalities in Glutamate Receptor Expression in Human Cortical Tubers
AMPA and NMDA iGluR subtypes mediate excitatory neurotransmission and play a critical role in synaptogenesis, synaptic plasticity, and epileptogenesis.29 Experimental loss of Tsc1/Tsc2 in neurons results in increased glutamatergic function.16,19 To date, quantification of membrane-expressed AMPAR and NMDAR subunits in human cortical tubers and cell-specific expression patterns of individual subunits has not been directly examined, although multiple abnormalities in iGluR subunit messenger RNA have been reported previously.22 We found that GluR1 and GluR4 subunits were expressed at greater levels, whereas GluR2 and GluR3 levels were lower in tuber membrane protein extracts, relative to controls. In addition, NR1 and NR2A were not significantly altered, although there was a marked increase in NR2B and NR3A expression levels in tubers compared with controls. These differences in receptor subunit composition may substantially contribute to increased network excitability in cortical tubers.
At cellular level, we found that all elements in human cortical tubers expressed altered AMPAR and NMDAR subunits, and the specific types of abnormal cells reliably expressed receptor subunit profiles resembling those present in normal developing neurons and glia (see Fig 5).
Dysplastic neurons expressed high levels of GluR1-containing, GluR2-lacking AMPARs and, therefore, likely permeable to Ca2+.35 They also expressed NMDARs containing predominantly NR2B and NR3A subunits, responsible for longer current decay times and insensitivity to Mg2+ block, respectively. 36,37 Altered expression of NR2B protein directly corroborates previous published results demonstrating increased NR2B messenger RNA expression in tubers and increased binding of ifenprodil, a highly specific ligand for NR2Bcontaining NMDARs.22 In addition, increased NR3A expression in tubers is consistent with electrophysiology data demonstrating the presence of NMDAR currents with decreased Mg2+ sensitivity in dysplastic tissue.38 Ca2+ influx through GluR2-deficient AMPARs may activate multiple signaling cascades involved in synaptogenesis, synaptic plasticity, and epileptogenesis.39,40 Expression of NR2B may contribute to increased excitability and synaptic plasticity by conferring slower kinetics and increased Ca2+ influx through the receptor, as well as by subunit-specific interactions with downstream signaling pathways such as calcium/calmodulin-dependent protein kinase II.41 Incorporation of the NR3A subunit may confer Mg2+ insensitivity to the NMDARs, so that the receptor may operate at a more negative membrane potential. 37 These unique iGluR expression profiles on dysplastic neurons are similar to the expression profiles observed in human normal immature cortical neurons25 and could be critical factors in the epileptogenicity of cortical tubers.
Normal-sized disoriented neurons and scattered normal-appearing neurons within tubers also showed altered patterns of iGluR expression, suggesting a more widespread neuronal dysfunction in TSC and the possible involvement of perituberal tissue in the predisposition to epilepsy. These neurons demonstrated greater levels of GluR1, NR2B, and NR3A subunits relative to control neurons, but unlike dysplastic neurons, they expressed high GluR2 and NR2A levels. Normal-sized neurons expressed low pS6 levels, suggesting that these alterations may not be directly related to TSC1/TSC2 dysfunction. The fact that these neurons expressed altered iGluRs and increased c-Fos suggests that seizures may have contributed to these changes.42 Because we observed similar iGluR expression patterns in nondys-plastic epileptic tissue further supports this hypothesis and is consistent with previous studies in human and rat models of chronic epilepsy.29,43 On the other hand, the nature of these changes is not the same as those observed in the TSC dysplastic neurons, suggesting that the alterations in iGluRs in tuber dysplastic cells cannot be a function of the seizures alone but may relate in part to the developmental dysregulation seen in this disorder.
iGluR expression was also altered in neuroglial and glial elements of the tubers. Giant cells predominantly expressed GluR4, NR2D, and NR3A. However, because they express both neuronal and glial markers, and no synaptic markers, their role in the epileptogenicity of TSC lesions appears unlikely. Dysplastic astroglial cells demonstrated high levels of GluR2-deficient (Ca2+ permeable) AMPARs, and increased expression of NR1, NR2A, and NR2C, that could enhance NMDAR activation. Notably, this expression pattern is common on radial glia and immature astrocytes in the midgestational human brain.25 The impact of these abnormal expression patterns in glial and neuroglial cells on network excitability is unclear, although iGluR activation in normal astrocytes can modulate glutamate uptake, potassium buffering, or gap-junctional coupling. 44 Indeed, Tsc1 null astrocytes demonstrate impaired extracellular glutamate and potassium uptake, 45,46 and astrocyte-specific Tsc1 conditional knock-out mice develop spontaneous seizures.17
The proportion of abnormal neuronal and glial cell types may vary between tubers, and the fact that some tubers are epileptogenic whereas others are quiescent may relate to their cellular composition. The high percentage of dysplastic neurons expressing abnormal iGluR subunits in the resected tissue used in this study suggests that tubers composed mainly of dysplastic neurons may be more epileptic than those primarily composed of giant cells or dysplastic astroglia. Further investigation directly comparing epileptogenic versus nonepileptogenic tubers will be necessary to determine the functional importance of cellular composition in a certain lesion.
The regulation of iGluR function and signaling in human cortical tubers is not understood. Recently, it has been shown that mTOR-dependent protein synthesis plays an important role in controlling long-lasting synaptic strength,47 and inhibition of mTOR by rapamycin results in a reduction of late-phase long-term potentiation,48 suggesting that the mTOR pathway might play an important role in normal synaptic plasticity. We show that mTOR activation correlates on a cellular basis with specific alterations in iGluR expression, raising the possibility that dysregulation of this pathway may result in abnormal protein synthesis at synapses that could enhance glutamatergic transmission. 16 Recent articles on focal cortical dysplasia report an upregulation of different components of the mTOR pathway in dysplastic cells,11,12,49 and there is increasing evidence for activity-induced mTOR activation in experimental models.47,48,50 We report here modest increases of pS6 levels in nondysplastic epileptic tissue, although at much lower levels compared with TSC or cortical dysplasia, raising the possibility that mTOR, independent of the route of its activation, may play a role in long-lasting synaptic plasticity and epileptogenesis in human nondysplastic epileptic tissue.
Despite inherent potential confounding variables when using human tissue (age, brain region, prior antiepileptic medication, effect of seizures), these data justify future studies evaluating the interaction between cell-autonomous events and the effects of seizures and maturation on the nature and epileptogenicity of human cortical tubers. Therapies directed at abnormally expressed iGluR subtypes might be of potential use in seizure suppression in TSC as an alternative to surgical resection.
Supplementary Material
Acknowledgments
This work was supported by the Epilepsy Foundation (D.M.T.), Tuberous Sclerosis Alliance (D.M.T., D.J.K., F.E.J.), Lombroso Trust (D.M.T.), NIH (National Institute of Neurological Disorders and Stroke, NS31718, F.E.J.; P01 NS24279, D.J.K.; National Institute of Child Health and Human Development, Mental Retardation Research Center Grant, P30 HD18655, F.E.J.), Brain and Tissue Bank for Developmental Disorders, University of Maryland, Baltimore, MD (National Institute of Child Health and Human Development, N01HD43368). The role of the Brain and Tissue Bank is to distribute tissue, and therefore it cannot endorse the studies performed or the interpretation of the results.
We are most grateful to Drs Howard Weiner and Rebecca Folkerth for their contributions and assistance with tissue collection. We thank Dr T. Diefenbach and the MRDDRC (Mental Retardation and Developmental Disabilities Research Center) Imaging Core for assistance with confocal microscopy.
Footnotes
This article includes supplementary materials available via the Internet at http://www.interscience.wiley.com/jpages/0364-5134/suppmat
References
- 1.van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805–808. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- 2.Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–1315. doi: 10.1016/0092-8674(93)90618-z. [DOI] [PubMed] [Google Scholar]
- 3.DiMario FJ., Jr Brain abnormalities in tuberous sclerosis complex. J Child Neurol. 2004;19:650–657. doi: 10.1177/08830738040190090401. [DOI] [PubMed] [Google Scholar]
- 4.Huttenlocher PR, Wollmann RL. Cellular neuropathology of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:140–148. doi: 10.1111/j.1749-6632.1991.tb37756.x. [DOI] [PubMed] [Google Scholar]
- 5.Curatolo P, Verdecchia M, Bombardieri R. Tuberous sclerosis complex: a review of neurological aspects. Eur J Paediatr Neurol. 2002;6:15–23. doi: 10.1053/ejpn.2001.0538. [DOI] [PubMed] [Google Scholar]
- 6.Luat AF, Makki M, Chugani HT. Neuroimaging in tuberous sclerosis complex. Curr Opin Neurol. 2007;20:142–150. doi: 10.1097/WCO.0b013e3280895d93. [DOI] [PubMed] [Google Scholar]
- 7.Thiele EA. Managing epilepsy in tuberous sclerosis complex. J Child Neurol. 2004;19:680–686. doi: 10.1177/08830738040190090801. [DOI] [PubMed] [Google Scholar]
- 8.Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14:R251–R258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 9.Lamb RF, Roy C, Diefenbach TJ, et al. The TSC1 tumour suppressor hamartin regulates cell adhesion through ERM proteins and the GTPase Rho. Nat Cell Biol. 2000;2:281–287. doi: 10.1038/35010550. [DOI] [PubMed] [Google Scholar]
- 10.el Hashemite N, Zhang H, Henske EP, et al. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet. 2003;361:1348–1349. doi: 10.1016/S0140-6736(03)13044-9. [DOI] [PubMed] [Google Scholar]
- 11.Miyata H, Chiang AC, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol. 2004;56:510–519. doi: 10.1002/ana.20234. [DOI] [PubMed] [Google Scholar]
- 12.Baybis M, Yu J, Lee A, et al. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478–487. doi: 10.1002/ana.20211. [DOI] [PubMed] [Google Scholar]
- 13.Chan JA, Zhang H, Roberts PS, et al. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol. 2004;63:1236–1242. doi: 10.1093/jnen/63.12.1236. [DOI] [PubMed] [Google Scholar]
- 14.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 15.Jensen FE. Developmental factors regulating susceptibility to perinatal brain injury and seizures. Curr Opin Pediatr. 2006;18:628–633. doi: 10.1097/MOP.0b013e328010c536. [DOI] [PubMed] [Google Scholar]
- 16.Tavazoie SF, Alvarez VA, Ridenour DA, et al. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- 17.Uhlmann EJ, Wong M, Baldwin RL, et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- 18.Meikle L, Talos DM, Onda H, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Greenwood JS, Calcagnotto ME, et al. Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann Neurol. 2007;61:139–152. doi: 10.1002/ana.21058. [DOI] [PubMed] [Google Scholar]
- 20.Arseni C, Alexianu M, Horvat L, et al. Fine structure of atypical cells in tuberous sclerosis. Acta Neuropathol (Berl) 1972;21:185–193. doi: 10.1007/BF00688497. [DOI] [PubMed] [Google Scholar]
- 21.Valencia I, Legido A, Yelin K, et al. Anomalous inhibitory circuits in cortical tubers of human tuberous sclerosis complex associated with refractory epilepsy: aberrant expression of parvalbumin and calbindin-D28k in dysplastic cortex. J Child Neurol. 2006;21:1058–1063. doi: 10.1177/7010.2006.00242. [DOI] [PubMed] [Google Scholar]
- 22.White R, Hua Y, Scheithauer B, et al. Selective alterations in glutamate and GABA receptor subunit mRNA expression in dysplastic neurons and giant cells of cortical tubers. Ann Neurol. 2001;49:67–78. doi: 10.1002/1531-8249(200101)49:1<67::aid-ana10>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 23.Gomez M, Sampson J, Whittemore V, et al. The tuberous sclerosis complex. 3rd ed. Oxford: Oxford University Press; 1999. [Google Scholar]
- 24.Palmini A, Najm I, Avanzini G, et al. Terminology and classification of the cortical dysplasias. Neurology. 2004;62:S2–S8. doi: 10.1212/01.wnl.0000114507.30388.7e. [DOI] [PubMed] [Google Scholar]
- 25.Talos DM, Follett PL, Folkerth RD, et al. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral. J Comp Neurol. 2006;497:61–77. doi: 10.1002/cne.20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weissman T, Noctor SC, Clinton BK, et al. Neurogenic radial glial cells in reptile, rodent and human: from mitosis to migration. Cereb Cortex. 2003;13:550–559. doi: 10.1093/cercor/13.6.550. [DOI] [PubMed] [Google Scholar]
- 27.Sarnat HB, Nochlin D, Born DE. Neuronal nuclear antigen (NeuN): a marker of neuronal maturation in early human fetal nervous system. Brain Dev. 1998;20:88–94. doi: 10.1016/s0387-7604(97)00111-3. [DOI] [PubMed] [Google Scholar]
- 28.Dragunow M, Robertson HA. Generalized seizures induce c-fos protein(s) in mammalian neurons. Neurosci Lett. 1987;82:157–161. doi: 10.1016/0304-3940(87)90121-2. [DOI] [PubMed] [Google Scholar]
- 29.McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006:re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- 30.Cepeda C, Andre VM, Levine MS. Epileptogenesis in pediatric cortical dysplasia: the dysmature cerebral developmental hypothesis. Epilepsy Behav. 2006;9:219–235. doi: 10.1016/j.yebeh.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 31.Murthy V, Stemmer-Rachamimov AO, Haddad LA, et al. Developmental expression of the tuberous sclerosis proteins tuberin and hamartin. Acta Neuropathol (Berl) 2001;101:202–210. doi: 10.1007/s004010000269. [DOI] [PubMed] [Google Scholar]
- 32.Noctor SC, Martinez-Cerdeno V, Ivic L, et al. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- 33.Cepeda C, Hurst RS, Flores-Hernandez J, et al. Morphological and electrophysiological characterization of abnormal cell types in pediatric cortical dysplasia. J Neurosci Res. 2003;72:472–486. doi: 10.1002/jnr.10604. [DOI] [PubMed] [Google Scholar]
- 34.Andre VM, Wu N, Yamazaki I, et al. Cytomegalic interneurons: a new abnormal cell type in severe pediatric cortical dysplasia. J Neuropathol Exp Neurol. 2007;66:491–504. doi: 10.1097/01.jnen.0000240473.50661.d8. [DOI] [PubMed] [Google Scholar]
- 35.Burnashev N, Monyer H, Seeburg PH, et al. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 36.Monyer H, Burnashev N, Laurie DJ, et al. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 37.Sasaki YF, Rothe T, Premkumar LS, et al. Characterization and comparison of the NR3A subunit of the NMDA receptor in recombinant systems and primary cortical neurons. J Neurophysiol. 2002;87:2052–2063. doi: 10.1152/jn.00531.2001. [DOI] [PubMed] [Google Scholar]
- 38.Andre VM, Flores-Hernandez J, Cepeda C, et al. NMDA receptor alterations in neurons from pediatric cortical dysplasia tissue. Cereb Cortex. 2004;14:634–646. doi: 10.1093/cercor/bhh024. [DOI] [PubMed] [Google Scholar]
- 39.Gu JG, Albuquerque CJ, Lee CJ, et al. Synaptic strengthening through activation of Ca2+-permeable AMPA receptors. Nature. 1996;381:793–796. doi: 10.1038/381793a0. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez RM, Dai W, Levada RE, et al. AMPA/kainate receptor-mediated downregulation of GABAergic synaptic transmission by calcineurin after seizures in the developing rat brain. J Neurosci. 2005;25:3442–3451. doi: 10.1523/JNEUROSCI.0204-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 42.Rakhade SN, Yao B, Ahmed S, et al. A common pattern of persistent gene activation in human neocortical epileptic foci. Ann Neurol. 2005;58:736–747. doi: 10.1002/ana.20633. [DOI] [PubMed] [Google Scholar]
- 43.Zilles K, Qu MS, Kohling R, et al. Ionotropic glutamate and GABA receptors in human epileptic neocortical tissue: quantitative in vitro receptor autoradiography. Neuroscience. 1999;94:1051–1061. doi: 10.1016/s0306-4522(99)00392-9. [DOI] [PubMed] [Google Scholar]
- 44.Lopez-Bayghen E, Espinoza-Rojo M, Ortega A. Glutamate down-regulates GLAST expression through AMPA receptors in Bergmann glial cells. Brain Res Mol Brain Res. 2003;115:1–9. doi: 10.1016/s0169-328x(03)00136-0. [DOI] [PubMed] [Google Scholar]
- 45.Wong M, Ess KC, Uhlmann EJ, et al. Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann Neurol. 2003;54:251–256. doi: 10.1002/ana.10648. [DOI] [PubMed] [Google Scholar]
- 46.Jansen LA, Uhlmann EJ, Crino PB, et al. Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia. 2005;46:1871–1880. doi: 10.1111/j.1528-1167.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 47.Tsokas P, Ma T, Iyengar R, et al. Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci. 2007;27:5885–5894. doi: 10.1523/JNEUROSCI.4548-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang SJ, Reis G, Kang H, et al. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ljungberg MC, Bhattacharjee MB, Lu Y, et al. Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol. 2006;60:420–429. doi: 10.1002/ana.20949. [DOI] [PubMed] [Google Scholar]
- 50.Tsokas P, Grace EA, Chan P, et al. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci. 2005;25:5833–5843. doi: 10.1523/JNEUROSCI.0599-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.