

Summary
Upon encountering antigen in the context of antigen presenting cells, naïve CD4+ T cells undergo differentiation into effector T helper (Th) cells, which can secrete high levels of cytokines and other immunomodulators to mediate host defense and tissue inflammation. During the past three years, the immunology field has witnessed an explosion of research advances in the biology of Th17 cells, the most recently described subset of T helper cells, which play critical roles in host immunity and inflammation. Here we review emerging data on transcriptional regulatory networks that govern the differentiation program of Th17 cells, and focus on how the orphan nuclear receptor RORγt coordinates this process in concert with diverse cytokine-induced transcription factors.
Introduction
The T helper cell (Th) paradigm, introduced by Mosmann and Coffman more than two decades ago, has been used to explain how different adaptive immune responses are elicited in the host organism for the purpose of eradicating infections with diverse microbial pathogens [1]. Th1 and Th2 cells, described in the original studies, have now been joined by Th17 cells that make the signature cytokines IL-17, IL-17F and IL-22 [2••, 3•, 4•]. These cells are involved in clearance of extracellular pathogens, particularly at mucosal surfaces [5•, 6, 7], where IL-17 induces recruitment and differentiation of neutrophils and IL-22 is required for production of anti-microbial defensins.
Excessive or persistent effector T cell responses can drive the onset of inflammatory diseases. It is now clear that Th17 cells, along with Th1 cells, are often responsible for autoimmune disease, and may also contribute to tumor progression due to the role of their cytokines in inflammation and tissue repair [8]. Another subset of CD4+ T cells, the regulatory T cells (Tregs), suppress effector T cell responses and prevent their potentially pathogenic effects [9]. There are two major categories of Foxp3+ Tregs, the naturally occurring CD4+ CD25+ Tregs (nTregs) that arise in the thymus and the TGF-β-induced Tregs (iTregs) produced in the periphery. Both types of Tregs are likely to be important in maintaining peripheral tolerance and preventing autoimmunity, but their individual contributions have not yet been established in vivo [10]. The T helper cell differentiation program is largely controlled by cytokines produced in response to microbial products by innate immune cells. These include IL-12/IFNγ for Th1 differentiation, IL-4 for Th2 differentiation, and IL-23 which has been linked to pathogenesis ascribed to Th17 cells both in mice and humans [11]. However, differentiation of IL-17-producing murine T helper cells in vitro does not require IL-23, but is dependent instead on a unique cytokine combination, IL-6 plus TGF-β [5•,12••,13••]. Differentiation of human Th17 cells was initially thought to be independent of TGF-β, but has recently been shown to also require this cytokine [14-16].
The differentiation of each effector T cell subset requires the induction and/or function of a series of transcriptional regulators that interact with each other in complex networks and thus orchestrate the functional program of the cells. For each T helper cell differentiation program, single transcription factors have been identified as pivotal regulators, named by some “master regulators” because their overexpression can induce transcription of the relevant cytokine genes, presumably by directly binding to their cis-regulatory elements. These transcription factors include T-bet for Th1 cells and GATA3 for Th2 cells [17]. Foxp3 was identified as a specific regulator for Tregs and controls the expression of multiple genes that mediate Treg cell functions [18, 19]. Retinoid-related orphan receptor (ROR)γt, but not T-bet or GATA3, was identified as a Th17-specific transcription factor, further demonstrating that Th17 cells have an unique T effector program [20••]. However, RORγt does not function in isolation, but coordinates the activity of a series of other essential transcription factors in guiding the differentiation of Th17 cells. The roles of RORγt in the context of other transcriptional regulators will be discussed in this review.
Unique cytokine environment for Th17 cell differentiation
Th17 cells were discovered after IL-23-deficient mice (lacking expression of the p19 polypeptide unique to this cytokine) were found to be resistant to multiple models of autoimmune disease and also to have markedly reduced expression of IL-17 by their activated T helper cells. It has been proposed that IL-23 is required for Th17 cell function in vivo through control of their expansion and/or maintenance, but this has not been demonstrated in experimental models [21, 22]. Moreover, recent reports have indicated that the frequency of IL-17+ T helper cells in the intestine at steady state or after infection with Citrobacter rodentium was not affected by the absence of IL-23 [5•, 23]. However, there is compelling evidence that production of IL-22 by T helper cells is dependent on IL-23 and survival of animals after C. rodentium infection requires both IL-23 and IL-22 [5•, 6]. Therefore, it will be important to evaluate the roles of both IL-17 and IL-22 in autoimmune diseases dependent on IL-23. It is also possible that IL-23 influences other effector functions that are critical to inflammatory diseases mediated by T helper cells [24]. Thus, the precise mechanisms underlying the contribution of IL-23 to Th17 cell differentiation and function in vivo remain to be elucidated.
Our current limited understanding of the roles of IL-23 and other cytokines in the differentiation of Th17 cells is largely derived from in vitro studies. In conjunction with T cell antigen receptor (TCR) activation, the proinflammatory cytokine IL-6 induces the expression of IL-21 in naïve CD4+ T cells. IL-21, in turn, induces its own expression in an autocrine manner, acting through the γc-family member IL-21 receptor that is induced by TCR stimulation. IL-21 (but not IL-6) also induces expression of the IL-23R. Together with TGF-β, IL-21, independently of IL-6, can induce IL-17 in mice and in humans [14, 25•, 26•, 27•]. In line with these findings, IL-21R-deficient cells expressed about three-fold less IL-17 upon stimulation of IL-6 plus TGF-β and IL-6-induced IL-23R expression was markedly reduced in IL-21R-deficient cells. These results suggested that IL-21 contributes to Th17 cell differentiation and serves as an intermediary mediator for IL-6-initiated signaling in the induction of IL-23R [25•]. Intriguingly, the absence of IL-21 signaling appears to have little effect on IL-17+ Th17 cell differentiation in vivo, suggesting that some factors (e.g. IL-6) may compensate for the loss of IL-21 signaling [23, 25•, 28•, 29•]. Since IL-21 is essential for IL-23R expression in vitro and IL-23 signaling is required for in vivo expression of IL-22, it will be important to determine whether IL-21R signaling is required for T cells to produce IL-22 in vivo [30].
Lack of IL-23R expression on naïve murine CD4+ T cells explains why IL-23 alone has no effect on Th17 induction in vitro. Once IL-23R is upregulated by cytokines such as IL-21, then IL-23 plus TGF-β are capable of inducing IL-17 expression [25•]. IL-23 further upregulates IL-23R expression, therefore imposing another amplifying loop and contributing to induction of Th17 cells [31••]. All together, these data suggest that IL-23 may function at a late stage of Th17 cell differentiation after initial induction by other proinflammatory cytokines (e.g. IL-6 and IL-21).
It was recently shown that mice deficient for gp130 (a shared receptor subunit for IL-6) mounted efficient Th17 responses and developed experimental autoimmune encephalomyelitis (EAE) after depletion of Treg cells, suggesting that IL-6 signaling was dispensable for the induction of pathogenic Th17 cells in vivo, at least in the absence of Treg cells [32]. These results highlight the complexity of the cytokine-induced Th17 differentiation program, suggesting that in vivo Th17 cell differentiation can be driven by multiple proinflammatory cytokines (IL-6, IL-21, IL-23 and/or other yet unidentified factors).
TGF-β has been extensively characterized as being required to maintain immunological tolerance, acting in both differentiation and maintenance of Foxp3+ Tregs that restrain effector T cell responses [33]. Why TGF-β is required for both proinflammatory Th17 cell and anti-inflammatory Treg differentiation remains paradoxical. Most recently, we showed that TGF-β orchestrates in vitro Th17 and Treg cell differentiation programs in a concentration-dependent manner (Figure 1) [31••]. At lower concentrations, together with IL-6 or IL-21, TGF-β synergistically induces IL-23R and thus promotes Th17 cell differentiation in the presence of IL-23. However, at higher concentrations, TGF-β inhibits IL-23R, IL-22 and IL-17 expression and favors induction of Foxp3 and, thus, Treg lineage differentiation. In spite of the absolute requirement of TGF-β, little is known about its precise signaling pathways in Th17 and Treg cell differentiation. Since Smad4 (a co-smad in the Smad-dependent TGF-β pathway) appears not to be required for Th17 cell differentiation [34], the involvement of Smad-dependent or –independent pathways in Th17 and Treg cell differentiation needs to be carefully examined. It will be important to determine if modulation of active TGF-β concentration or sensitivity to TGF-β in vivo results in differential loss of Treg versus Th17 cells. On this basis, polymorphisms in TGF-β signaling pathway genes may also contribute to human autoimmune diseases.
Figure 1. TGF-β orchestrates in vitro Th17 and Treg cell differentiation in a concentration-dependent manner.

In the presence of TGF-β, TCR-activated CD4+ T cells express both RORγt and Foxp3, but RORγt function is antagonized by Foxp3. Such cells can differentiate into either Th17 or Treg cells dependent on the cytokine environment. In the presence of proinflammatory cytokines and low concentrations of TGF-β, RORγt expression is further upregulated, whereas Foxp3 expression and function are inhibited, thus tipping the balance in favor of the Th17 cell fate. RORγt-induced IL-23R expression on T cells confers responsiveness to IL-23, which further promotes Th17 cell differentiation. In contrast, in the absence of proinflammatory cytokines, high concentrations of TGF-β favor Foxp3 expression and result in Treg cell differentiation.
Th17 transcriptional regulatory networks
Th17 cell lineage specification requires RORγt, which was earlier identified as a critical transcription factor for early T cell and lymphoid organ development [35-37]. During a gene profiling analysis of Th17 cells, RORγt was identified as one of the most highly upregulated transcription factors. Accordingly, in the small intestinal lamina propria, RORγt+ T cells but not RORγt- T cells express IL-17. In vitro, IL-6 plus TGF-β treatment-induced IL-17 expression requires induction of RORγt, and forced expression of RORγt is sufficient to induce IL-17 expression in the absence of any exogenous cytokines. In line with its critical roles in Th17 cell differentiation, RORγt-deficient mice develop less severe autoimmune diseases and specifically lack Th17 cells in the inflammatory tissues [20••, 38]. Interestingly, another ROR family member, RORα, is also upregulated during in vitro Th17 cell differentiation [39••]. Although forced expression of RORα is sufficient to induce IL-17, lack of RORα has only a minor effect on Th17 cell differentiation (L. Zhou and D. R. Littman, unpublished). However, complete loss of lamina propria Th17 cells was observed in animals harboring compound mutations of RORγt and RORα, suggesting that these two closely related transcription factors, which presumably share the same DNA binding sequence, may have similar functions in Th17 cell differentiation (L. Zhou and D. R. Littman, unpublished) [39••]. Accordingly, chromatin immunoprecipitation assay (ChIP) has suggested that il17a is a direct target gene of RORγt [39••, 40••].
IL-6, IL-21 and IL-23 signaling all utilize the Jak-Stat pathway and activate Stat3 [41]. In Stat3-deficient CD4+ T cells, induction of IL-21 and IL-23R was barely detected. Accordingly, IL-17 expression induced either by IL-6 plus TGF-β or IL-21 plus TGF-β was also greatly reduced, suggesting that Stat3 plays an essential role in Th17 cell differentiation [25•, 27•, 42]. Stat3 binds to the il17a promoter directly as shown by ChIP [43]. Stat3 is also required for induction of RORγt by cytokines. Forced expression of RORγt can partially rescue IL-17 expression in Stat3-deficient cells, suggesting that RORγt may function downstream of Stat3 to induce IL-17 expression (L. Zhou and D. R. Littman, unpublished). Stat3 and RORγt can also act together to induce maximal IL-17 expression, suggesting that they may also form a complex and/or cooperatively bind to the cis-elements of the il17a locus [25•].
In the course of IL-6-dependent Th17 cell polarization, there is a strong correlation between downregulation of Foxp3 and upregulation of IL-17 [13••]. This finding can be explained by the ability of Foxp3 to suppress Th17 cell differentiation through antagonism of RORγt activity [31••]. TGF-β induces both RORγt and Foxp3, but is unable to induce IL-17 unless combined with proinflammatory cytokines (IL-6, IL-21 or IL-23) (Figure 1) [31••]. Foxp3 has the potential for physical interaction with RORγt and RORα and can thus inhibit their transcriptional activities [31••, 44]. Accordingly, murine Foxp3 lacking amino acids encoded by exon 2 (Foxp3ΔExon2) cannot inhibit RORγt function due to a loss of interaction with RORγt. The interaction between RORγt and Foxp3 has been confirmed by several other groups [34, 40••, 45, 46], but it remains unclear if it is direct or in the context of a larger complex. Intriguingly, the inhibitory effect of Foxp3 on IL-17 induction was largely circumvented in the presence of IL-6 or IL-21, even though the levels of Foxp3 and RORγt proteins were not affected [31••]. This result suggests that the inhibition of RORγt by Foxp3 was relieved by proinflammtory cytokines through a post-translational mechanism. Although it is clear that the interaction between RORγt and Foxp3 is critical for in vitro Th17 cell differentiation, in vivo relevance of this interaction is yet to be defined. Notably, neither the natural function of Foxp3ΔEx2, an isoform found only in humans, nor the precise mechanism underlying Foxp3–mediated inhibition of RORγt is known. Most recently, RORγt was shown to interact with Runx1, a transcription factor upregulated during TCR stimulation and required both for differentiation of Th17 cells and for Foxp3 function [40••, 47] (M. Chong and D. R. Littman, unpublished). Binding of RORγt and Runx1 together to the il17a locus leads to increased expression of IL-17, whereas Foxp3 inhibits both Runx1 and RORγt activity [40••]. It is possible that Runx1 can modify RORγt/Foxp3 complexes in the presence of proinflammatory cytokines, therefore relieving the inhibition of RORγt activity by Foxp3. Alternatively, Runx1 may differentially associate with RORγt or Foxp3, and thus participate in mediating their respective transcriptional activating or repressing activities, according to the cytokine-initiated signals.
IRF-4, a transcription factor previously shown to be important for Th2 cell differentiation, was also discovered to be essential for Th17 cell differentiation. IRF4-deficient mice were protected from EAE and T cells from these animals failed to differentiate into Th17 cells. IRF-4, which regulates expression of IL-21 and IL-23R, is in turn inhibited by IRF-4-binding protein (IBP) [48••, 49, 50•]. RORγt and RORα induction were impaired in IRF4-deficient T cells, but their forced expression could partially restore induction of IL-17, suggesting that IRF-4 may function upstream of the nuclear receptors [48••, 49]. Since rescue was only partial, it is likely that a complex transcriptional network, rather than a linear process, governs the Th17 cell differentiation program. Furthermore, IRF4-deficient T cells had increased Foxp3 expression, highlighting the importance of the RORγt-Foxp3 balance in Th17/Treg cell differentiation. Th17 cells have also been shown to express c-Maf, a transcription factor involved in regulation of Th2 cell differentiation. Genetic loss of c-Maf resulted in a defect in IL-21 production, IL-23R expression, and, consequently, in fewer Th17 cells [51].
The discovery of the involvement of aryl hydrocarbon receptor (AhR) in the regulation of transcription of Th17 cytokines has added another layer of complexity to this field [52••, 53••]. AhR, a mediator of the effects of environmental toxins (e.g. dioxin, a polycyclic aromatic hydrocarbon xenobiotic compound), is a ligand-dependent transcription factor that is structurally distinct from the nuclear receptor superfamily. Upon binding to a ligand (such as dioxin or FICZ, a UV photoproduct of tryptophan), cytosolic AhR translocates into the nucleus, heterodimerizes with its partner aryl hydrocarbon receptor nuclear translocator (ARNT), and turns on transcription of its target genes [54]. The findings of the involvement of AhR in Th17 cell differentiation suggest a potential link between environmental pollution and inflammation. However, the precise contribution of AhR to Th17 cell differentiation is unclear. Analysis of AhR-deficient cells has shown that it is required for IL-22 and, to a lesser extent, IL-17 expression in Th17 polarizing conditions in the presence of either dioxin or FICZ [52••] (L. Zhou and D. R. Littman, unpublished). However, a requirement for AhR in IL-17 expression is still controversial [55]. Thus, one study suggested that different AhR ligands (dioxin versus FICZ) exert opposite effects on Th17 and Treg cell differentiation [53], whereas another showed no difference between individual AhR ligands [55]. Thus, dioxin was found to suppress progression of EAE [53••], while FICZ exacerbated EAE [52••, 53••]. The reasons behind these discrepancies remain unclear and may be due to different culture conditions and animal housing environments [56]. We have found that AhR cooperates with RORγt to induce maximal amounts of IL-17 and IL-22 and also inhibits TGF-β-induced Foxp3 expression, thus highlighting the antagonism between Th17 and Treg cell differentiation (L. Zhou and D. R. Littman, unpublished). Together, these results suggest that RORγt may function as a node in the Th17 cell transcriptional network and may interact either positively or negatively with other factors to influence lineage specification.
In vivo relevance and implications
The local cytokine environment greatly influences immune system homeostasis through regulation of various transcription factors. Local TGF-β concentration, together with proinflammatory cytokines, may determine the balance between Foxp3 and RORγt (and probably other Th17 transcription factors as well), leading to different T cell fates. This is especially relevant in the gut environment where, upon encountering commensal bacteria, dendritic cells secrete proinflammatory cytokines. High concentrations of TGF-β, together with retinoic acid (RA), may be required for induction of Foxp3+Tregs to suppress potentially detrimental inflammatory Th17 cell responses [57-62]. The inhibition of IL-6 receptor (IL-6Rα) expression by RA, suggested by a recent study, provides at least in part an explanation for how RA antagonizes Th17 cell differentiation [63]. Foxp3 inhibits RORγt-directed IL-17 expression in mouse T cells, but it remains to be determined whether this also occurs in humans. This is relevant because FOXP3ΔExon2 is a natural spliced isoform that was only identified in humans. Thus, the balance between full-length FOXP3 versus FOXP3ΔExon2 may influence the frequency of Th17 and Treg cells and susceptibility to autoimmunity in humans. Modulation of the interaction between RORγt (together with other Th17 transcription factors) and Foxp3 may thus maintain immune homeostasis and permit control of inflammatory responses in different disease settings.
Conclusions
Over the past three years, there have been remarkable advances in our understanding of Th17 cell differentiation and function. Transcriptional regulatory networks that specify Th17 differentiation program have begun to be elucidated, but our understanding is likely still superficial (Figure 2). Little is known about precise molecular mechanisms whereby these interactions determine the Th17 differentiation program. Furthermore, aside from several effector cytokine genes, the targets of these transcriptional regulators in the Th17 program are unclear. In addition, the mechanism by which RORγt is regulated by a yet unknown ligand remains to be characterized. RORγt function and its interaction with Foxp3 may be manipulated pharmacologically using small molecule compounds. Our knowledge of the interplay among different T helper cell lineages at the molecular level will shed light on human disease pathogenesis and may eventually provide novel means for treating inflammatory and autoimmune diseases.
Figure 2. Transcriptional regulatory network that governs the Th17 cell differentiation program.
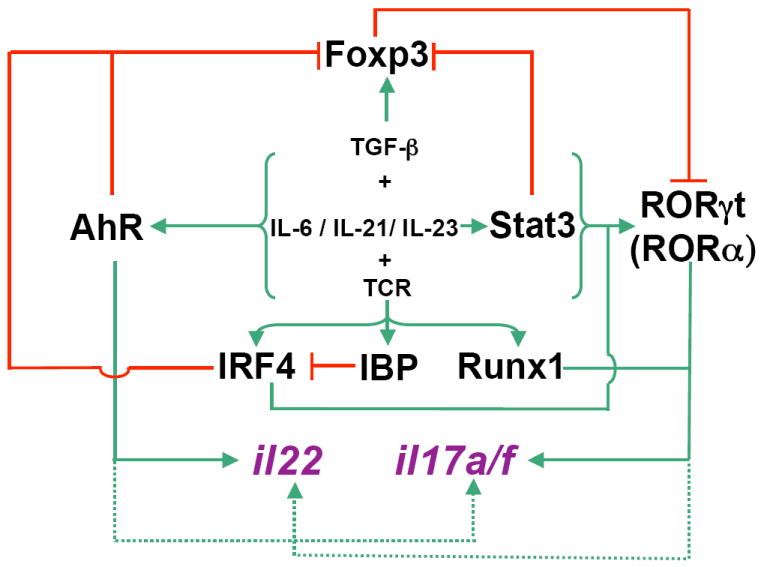
The inflammatory cytokine (IL-6)-initiated signaling cascade, together with TGF-β signaling, induces RORγt expression in a Stat3-dependent manner. In the absence of proinflammatory cytokines, TGF-β-induced Foxp3 inhibits RORγt (and RORα) activity, and thus promotes Treg cell differentiation. AhR is also induced during Th17 cell polarization and it downregulates Foxp3 expression. Ligand activated AhR cooperates with RORγt to induce maximal amounts of IL-17 and IL-22 and also to inhibit TGF-β-induced Foxp3 expression, ensuring full progression of Th17 cell differentiation. TCR-induced IRF-4 upregulates RORγt expression and inhibits Foxp3 expression, and thus promotes Th17 cell differentiation and antagonizes Treg cell differentiation. TCR-activated IBP inhibits IRF-4 function by sequestering IRF-4. TCR-induced Runx1 influences Th17 cell differentiation by inducing RORγt expression and by binding to and acting together with RORγt to direct il17 transcription.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest published within the period of review have been highlighted as:
-
•
of special interest
-
••
of outstanding interest
- 1.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- •• 2.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257.This was the first paper to suggest the existence of distinct IL-17-producing cells that are involved in organ-specific autoimmunity.
- • 3.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254.This study showed inhibitory effects of IFNγ and IL-4 on Th17 cell differentiation, and demonstrated that STAT1, T-bet, STAT4 and STAT6 were dispensable for development of IL-17- producing effectors in vitro, establishing an independent developmental lineage for Th17 cells.
- • 4.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261.This report provided independent evidence of a distinct developmental lineage for Th17 cells and observations for in vivo function of IL-17.
- • 5.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754.This study showed that IL-23 is important for IL-6/TGF-β-induced Th17 cells to protect the host from infection with a mucosal pathogen.
- 6.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 7.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 9.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity. 2008;28:445–453. doi: 10.1016/j.immuni.2008.03.001. [DOI] [PubMed] [Google Scholar]
- •• 12.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001.This was the first study to show that treatment of IL-6 plus TGF-β induces Th17 cell differentiation in vitro.
- •• 13.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753.This paper first highlighted the reciprocal relationship between TGF-β-induced Foxp3+ Treg cells and IL-17-producing T cells.
- 14.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 16.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 18.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 19.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- •• 20.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035.This paper described a Th17-specific transcription factor and validated that IL-17-producing cells are indeed a unique T cell subset.
- 21.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 22.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 25.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- • 26.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 27.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969.References 25, 26 and 27 showed that IL-21, together with TGF-β, could program Th17 cell differentiation in an autocrine manner.
- • 28.Sonderegger I, Kisielow J, Meier R, King C, Kopf M. IL-21 and IL-21R are not required for development of Th17 cells and autoimmunity in vivo. Eur J Immunol. 2008;38:1833–1838. doi: 10.1002/eji.200838511. [DOI] [PubMed] [Google Scholar]
- • 29.Coquet JM, Chakravarti S, Smyth MJ, Godfrey DI. Cutting edge: IL-21 is not essential for Th17 differentiation or experimental autoimmune encephalomyelitis. J Immunol. 2008;180:7097–7101. doi: 10.4049/jimmunol.180.11.7097.References 28 and 29 provided evidence that IL-21 deficiency had little effects on in vivo differentiation of Th17 cells and autoimmunity, suggesting potential redundancy of proinflammatory cytokines.
- 30.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- •• 31.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878.This study was the first to show that RORγt activity is inhibited by Foxp3 and that TGF-β concentration may have a significant impact on Th17/Treg cell differentiation, suggesting that the cytokine microenvironment dictates whether progenitor cells adopt either fate.
- 32.Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, Vollmar P, Stritesky GL, Kaplan MH, Waisman A, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, Littman DR. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science. 2000;288:2369–2373. doi: 10.1126/science.288.5475.2369. [DOI] [PubMed] [Google Scholar]
- 36.Egawa T, Eberl G, Taniuchi I, Benlagha K, Geissmann F, Hennighausen L, Bendelac A, Littman DR. Genetic evidence supporting selection of the Valpha14i NKT cell lineage from double-positive thymocyte precursors. Immunity. 2005;22:705–716. doi: 10.1016/j.immuni.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 37.Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 38.Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD, et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology. 2009;136:257–267. doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- •• 39.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016.The paper reported that Th17 cells express the RORγt-related nuclear receptor, RORα. Double deficiencies in RORγt and RORα impaired Th17 generation and protected mice against autoimmunity.
- •• 40.Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. 2008;9:1297–1306. doi: 10.1038/ni.1663.This paper provided independent confirmation of Foxp3 inhibition of RORγt activity, and showed that Runx1 influences Th17 cell differentiation by inducing RORγt expression and by binding to and acting together with RORγt during il17 transcription.
- 41.O’Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei L, Laurence A, Elias KM, O’Shea JJ. IL-21 is produced by TH17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007 doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O’Shea JJ. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du J, Huang C, Zhou B, Ziegler SF. Isoform-specific inhibition of ROR alpha-mediated transcriptional ctivation by human FOXP3. J Immunol. 2008;180:4785–4792. doi: 10.4049/jimmunol.180.7.4785. [DOI] [PubMed] [Google Scholar]
- 45.Lochner M, Peduto L, Cherrier M, Sawa S, Langa F, Varona R, Riethmacher D, Si-Tahar M, Di Santo JP, Eberl G. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J Exp Med. 2008;205:1381–1393. doi: 10.1084/jem.20080034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ichiyama K, Yoshida H, Wakabayashi Y, Chinen T, Saeki K, Nakaya M, Takaesu G, Hori S, Yoshimura A, Kobayashi T. Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. J Biol Chem. 2008;283:17003–17008. doi: 10.1074/jbc.M801286200. [DOI] [PubMed] [Google Scholar]
- 47.Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- •• 48.Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, Arpaia E, Mak TW, Kamradt T, Lohoff M. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol. 2007;8:958–966. doi: 10.1038/ni1500.This paper showed that IRF-4-deficient mice did not develop experimental autoimmune encephalomyelitis. CD4+ T cells from such mice failed to differentiate into Th17 cells and had less expression of RORγt and more expression of Foxp3.
- 49.Huber M, Brustle A, Reinhard K, Guralnik A, Walter G, Mahiny A, von Low E, Lohoff M. IRF4 is essential for IL-21-mediated induction, amplification, and stabilization of the Th17 phenotype. Proc Natl Acad Sci U S A. 2008;105:20846–20851. doi: 10.1073/pnas.0809077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 50.Chen Q, Yang W, Gupta S, Biswas P, Smith P, Bhagat G, Pernis AB. IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity. 2008;29:899–911. doi: 10.1016/j.immuni.2008.10.011.This paper demonstrated that the specific interaction of IRF-4 and IBP prevented IRF-4 from targeting the ill17 and il21 genes. Absence of IBP led to enhanced expression of IL-21 and IL-17 and to the development of autoimmunity.
- 51.Bauquet AT, Jin H, Paterson AM, Mitsdoerffer M, Ho IC, Sharpe AH, Kuchroo VK. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol. 2009;10:167–175. doi: 10.1038/ni.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •• 52.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- •• 53.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880.References 52 and 53 identified AhR as an important transcription factor for Th17 cell differentiation. Reference 51 further suggested that different AhR ligands may have opposite functions in Treg/Th17 differentiation, but this notion is controversial as suggested by reference 55.
- 54.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008;105:9721–9726. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–49. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 60.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schambach F, Schupp M, Lazar MA, Reiner SL. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur J Immunol. 2007;37:2396–2399. doi: 10.1002/eji.200737621. [DOI] [PubMed] [Google Scholar]
- 62.Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, O’Shea JJ. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hill JA, Hall JA, Sun CM, Cai Q, Ghyselinck N, Chambon P, Belkaid Y, Mathis D, Benoist C. Retinoic Acid Enhances Foxp3 Induction Indirectly by Relieving Inhibition from CD4(+)CD44(hi) Cells. Immunity. 2008;29:758–770. doi: 10.1016/j.immuni.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]