

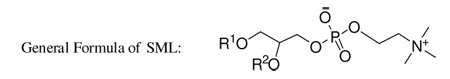
Abstract
We synthesized a family of sterol-modified glycerophospholipids (SML) in which the sn-1 or sn-2 position is covalently attached to cholesterol and the alternative position contains an aliphatic chain. The SML were used to explore how anchoring cholesterol to a phospholipid affects cholesterol behavior in a bilayer. Notably, cholesterol in the SML retains the membrane condensing properties of free cholesterol regardless of the chemistry or position of its attachment to the glycerol moiety of the phospholipid. SMLs by themselves formed liposomes upon hydration and in mixtures between an SML and diacylglycerophospholipids (C14 to C18 chain length) the thermotropic phase transition is eliminated at the SML equivalent of about 30 mole percent free cholesterol. Osmotic-induced contents leakage from SML (C14–C18) liposomes depends upon the linkage and position of cholesterol but in general is similar to that observed in diacylphosphatidylcholine/ cholesterol: 3/2 (mole ratio) liposomes. SML liposomes are exceptionally resistant to contents release in the presence of serum at 37 °C. This is probably due to fact that SML exchange between bilayers is more than 100 fold less than the exchange rate of free cholesterol in the same conditions. Importantly SML liposomes containing doxorubicin are as effective in treating the murine C26 colon carcinoma, as Doxil™ a commercial liposome doxorubicin formulation. SMLs stabilize bilayers but do not exchange hence provide a new tool for biophysical studies on membranes and they may improve liposomal drug delivery in organs predisposed to the extraction of free cholesterol from bilayers, such as; the skin, lung or blood.
Introduction
Cholesterol is an essential component of animal cell membranes and contributes to bilayer stability, bilayer fluidity and promotes the liquid condensed state in lipid mixtures containing unsaturated and saturated diacyl chains 1–3. When the cholesterol content of the bilayer is about 30 mole percent the thermotropic phase transition of synthetic diacylphospholipids is eliminated 4,5, the membrane permeability of hydrophilic molecules is reduced 6,7 and the insertion of hydrophobic compounds and proteins into the bilayer inhibited 8. Above 50 mole percent, cholesterol phase separates from the membrane into cholesterol-rich domains which can lead to the formation of cholesterol crystals 9. Thus, the mole ratio of cholesterol in biomembranes critically regulates their solvent properties, which in turn dynamically influences membrane protein organization and ultimately cell physiology 1,10. These profound effects on the physical characteristics of membranes require that the cholesterol membrane concentration be carefully regulated by a feedback system of proteins that monitor excess free cholesterol in cell membranes and ultimately modulate cholesterol synthesis and uptake at the transcriptional level 11,12. Needless to say the importance of cholesterol in health and disease has generated an enormous literature in the century since Windaus reported that atheromatous lesions had 6 times as much cholesterol as a normal arterial wall leading to his pioneering studies on cholesterol biosynthesis and metabolism 13.
In mammals, cholesterol is found as a free molecule, modified at the 3 position by sulfate or esterified at the 3 position to fatty acids. Neither cholesterol nor cholesterol esters can form bilayer membranes by themselves, whereas cholesterol derivatives with a hydrophilic head group at the 3 position such as; a sulfate, phosphate, phosphatidylcholine, ethylene oxide or hemisuccinate assemble into membranes as bilayer vesicles (liposomes) 14–19. Liposomes containing a high mole percentage of cholesterol are generally more stable and less leaky than those without cholesterol, and are widely used in the formulation of chemotherapeutic drugs 20. However, when liposomes composed of free cholesterol and phospholipids are placed in biological fluids in contact with biomembranes and proteins, free cholesterol rapidly transfers from the liposome into the biomembranes 21–23. This transfer of free cholesterol from the liposome results in the decrease in liposome stability and the subsequent loss of encapsulated contents from the liposome.
We were interested in learning the consequences on bilayer properties of an amphiphilic cholesterol-analog that could form bilayers but was unable to rapidly transfer between membranes. So we have designed and synthesized a new category of chimeric sterol-modified phospholipids (SMLs) consisting of both cholesterol and an aliphatic chain covalently linked to the glycerol backbone of phosphatidylcholine (Table 1). We hypothesize that the covalent hybridization of cholesterol and a phospholipid will confine cholesterol in the lipid bilayer and improve the bilayer cohesion properties 24. In this paper, we report the synthesis of SMLs, their biophysical properties and demonstrate their potential as an anticancer drug carrier.
Table 1.
Summary of sterol-modified phospholipids (SMLs)
 | |||
|---|---|---|---|
| Lipids | R1 | R2 | Method |
| 1a–c | Chol-OC(O) | CnH2n+1NHC(O)a | Route 1 |
| 2a–d | Chol-OC(O) | CnH2n+1b or C18H35 | Route 2 |
| 3a–d | Chol | CnH2n+1C(O)c or C17H33C(O) | Route 3 |
| 4a–d | CnH2n+1d or C18H35 | Chol-OC(O) | Route 4 |
| 5a–d | CnH2n+1C(O)e or C17H33C(O) | Chol-OC(O) | Route 5 |
| 6a–d | CnH2n+1C(O)e or C17H33C(O) | Chol-OC(O)CH2 CH2C(O) | Route 6 |
Chol-OH = cholesterol
n = 18, 16, 14
n = 17, 15, 13.
Results and Discussion
Chemistry of SMLs
The idea of a covalent chimera between a sterol and a phospholipid opens the way to a myriad of structures given the various combinations of: sterol, aliphatic chain length, aliphatic chain type, substitution position, linkage chemistry, stereochemistry, and head group. To test the concept we focused on lipids bearing a cholesterol, an aliphatic chain with length between C-14 and C-18 and phosphocholine as the head group. These constituents were selected due to their abundance and importance in mammalian membranes. A series of six SMLs (Table 1, and Scheme 1–6) were synthesized that include: ester, carbonate, carbamate, and ether linkages. These linkages were selected because the manner in which cholesterol and the aliphatic chain are linked to the glycerol backbone may significantly affect the properties of SMLs in bilayers and their fate in vivo. We adopted synthetic strategies that have been applied to the synthesis of glycerophospholipids 25 for the synthesis of SMLs. Semisynthetic approaches used commercially available precursors, such as glycerophosphocholine (Scheme 1) and lyso-lipids (Scheme 5–6) which were employed to prepare stereospecific molecules. For SMLs containing an ether linkage (Scheme 2–4), a longer synthetic route is required to obtain the specific 1,2-disubstituted glycerols. The conversion of 1,2-disubstituted glycerol to the corresponding phosphocholine was accomplished by the commonly used steps: phosphorylation with phosphorus oxychloride, followed by the coupling with choline tetraphenyl borate using 2,4,6-triisopropylbenzysulfonyl chloride as the condensing reagent. This is a practical and reliable way to convert 1,2-diradyl glycerol to phosphocholine in our lab and providing an overall yield of above 70%. It should be noted that some SMLs are diastereomers when racemic starting material were used. The packing of diastereomers in the membrane may be quite different from the optical pure SMLs.
Scheme 1.
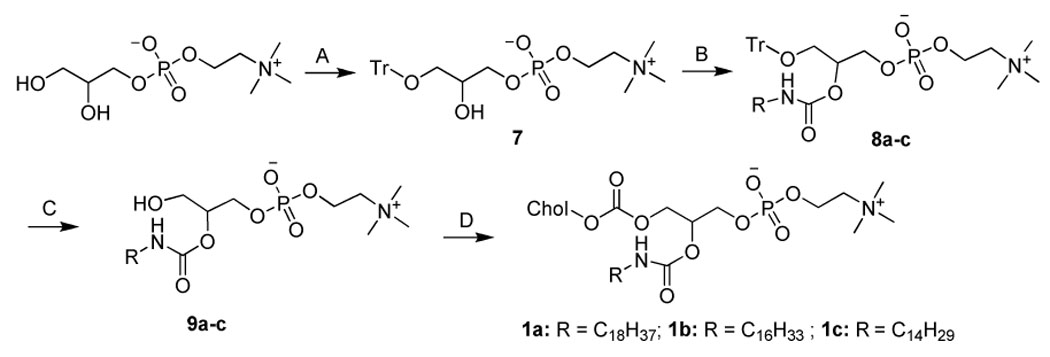
Synthesisa of 1a–c
aReagents and conditions. (A) Trityl chloride (1.03 equiv.), ZnCl2 (0.95 equiv.), DMF, 4 °C, 10 h; (B) Alkyl isocyanate (1.0 equiv.), DMSO, 100 °C, 24 h; (C) TFA in CHCl3 (16.7%), r.t., 4 h; (D) cholesteryl chloroformate (5 equiv.), DIPEA (5.2 equiv.), CHCl3, r.t., 16 h.
Scheme 6.

Synthesisa of 6a–d
aReagents and conditions. (A) Cholesteryl hemisuccinate (1.2 equiv.), DMAP (1 equiv.), DCC (1.2 equiv.), CHCl3, r.t., 24 h.
Scheme 5.
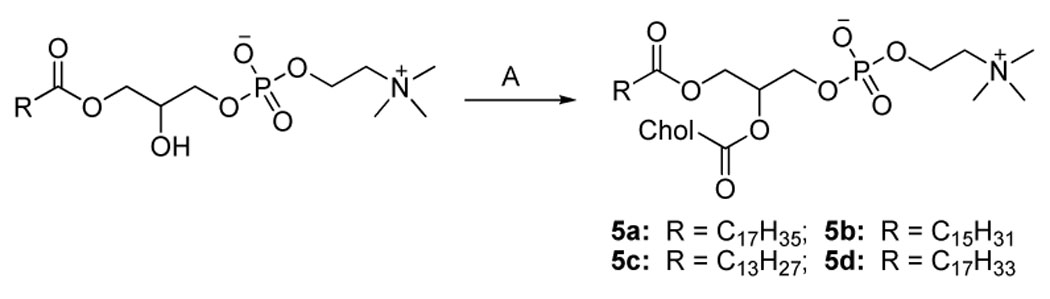
Synthesisa of 5a–d
aReagents and conditions. (A) Cholesteryl chloroformate (2.3 equiv.), DMAP (4 equiv.), CHCl3, r.t., 16 h.
Scheme 2.
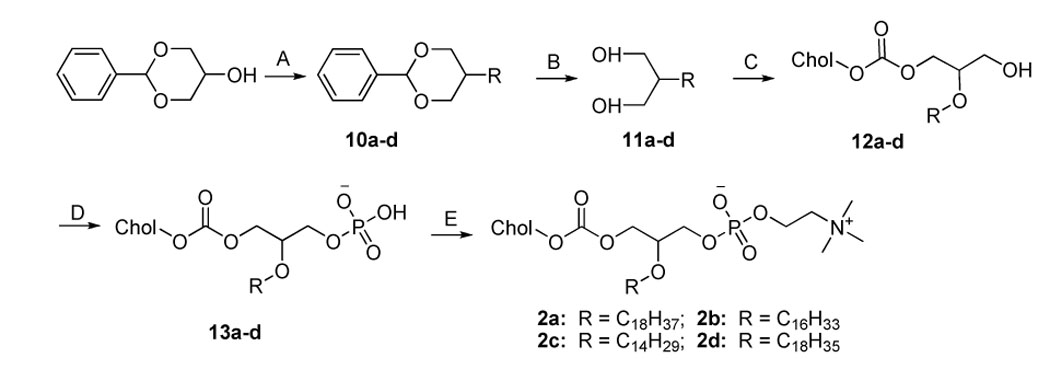
Synthesisa of 2a–d
aReagents and conditions. (A) 1) NaH (1.2 equiv.), toluene, r.t., 30 min; 2) iodoalkane (1.25 equiv.), reflux, overnight; (B) HCl (conc.) in MeOH (10%), reflux, 5 h; (C) Cholesteryl chloroformate (1.05 equiv.), DIPEA (1.4 equiv.), DMAP (0.5 equiv.), CHCl3, 0 °C, 0.5 h then r.t., overnight; (D) POCl3 (1.1 equiv.), pyridine (2 equiv.), THF, 0 °C, 2–3 h; (E) Choline tetraphenyl borate (2 equiv.), TPS (2.5 equiv.), pyridine, 70 °C, 1 h, then r.t., 3 h.
Scheme 4.
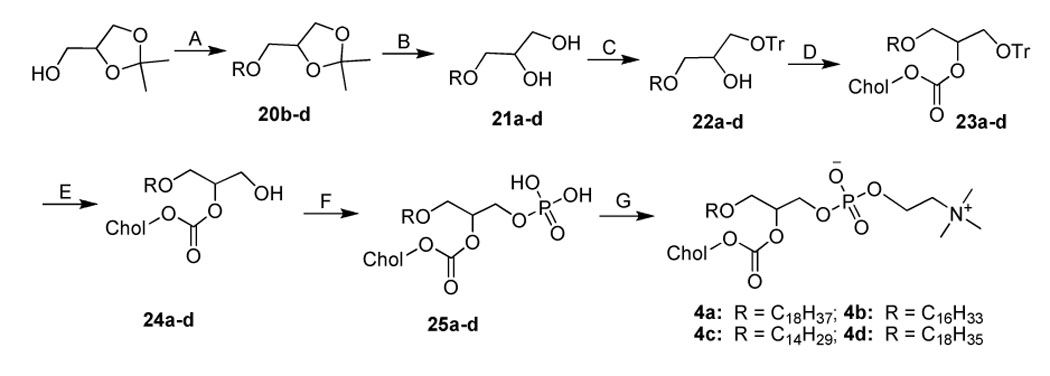
Synthesisa of 4a–d
aReagents and conditions. (A) NaH (1.7 equiv.), bromoalkane (0.9 equiv.), toluene, 120 °C, overnight; (B) HCl in MeOH (2 M), reflux, 1.5 h; (C) Trityl chloride (1.5 equiv.), pyridine, 50 °C, 18 h; (D) Cholesteryl chloroformate (1.2 equiv.), DMAP (1.2 equiv.), CHCl3, r.t., overnight; (E) BF3.Et2O (4 equiv.), CHCl3, 0 °C, 3 h; (F) POCl3 (1.1 equiv.), pyridine (2 equiv.), THF, 0 °C, 2–3 h; (G) Choline tetraphenyl borate (2 equiv.), TPS (2.5 equiv.), pyridine, 70 °C, 1 h, then r.t., 3 h.
In the synthesis of 1a–c (Scheme 1), the yield-limiting step was the introduction of alkylcarbamoyl chain at the C2 position through the reaction of isocyanate and the 2-hydroxy group. The low yield (ca. 30%) of this reaction may be due to the side reaction of isocyanate and the poor solubility of 7. The presence of the carbamate bond at the C2 position may favor the formation of strong hydrogen bonding in the water-oil surface region and alter the properties of 1a–c. The synthesis of 2a–d started with the selective alkylation of the C2 position of 1,3-protected glycerol (Scheme 2) followed by the removal of the protective group through acid hydrolysis. The reaction of 11a–d and cholesteryl chloroformate with controlled stoichiometry led mainly to the desired product 12a–d with slight 1,3-disubstitued side product which was easily removed by high performance flash chromatography (HPFC). The conversion of 12a–d to 2a–d was achieved by the common procedure. Cholesteryl glycerol (15), the key intermediate of 3a–d, was obtained by the treatment of cholesteryl tosylate with excessive soketal at 90 °C followed by acid hydrolysis (Scheme 3). Our effort to repeat the previously reported method 26 ended only with side product from the elimination reaction of cholesteryl tosylate due to the harsh conditions: the presence of strong base and high reaction temperature (120 °C). After the protection of the C3 hydroxyl group with trityl group, the acyl group was selectively coupled to the C2 position. The removal of the trityl group from 17a–d at 0 °C with boron trifluoride etherate led to 18a–d with no detectable isomers as judged by TLC. Nevertheless, they were immediately converted to 3a–d using the standard procedure to avoid acyl migration. The synthesis of 4a–d (Scheme 4) was similar to that of 3a–d except that the position of cholesterol and aliphatic chain was switched. The synthesis of 21a–d was carried out under higher temperature due to the stability of the aliphatic chain. After selective attachment of cholesterol at the C2 position through a carbonate linkage, trityl group was removed and the 1,2-diradyl glycerols were transformed to 4a–d. The synthesis of 5a–d (Scheme 5) was achieved by direct coupling of cholesteryl chloroformate and the corresponding lyso-phosphocholine in high yield. Similarly, direct conjugation of cholesteryl hemisuccinate and lyso-phosphatidylcholine led to 6a–d (Scheme 6). It should be noted that the lyso-lipid should be completely dissolved in the dry ethanol-free chloroform to obtain a high yield.
Scheme 3.
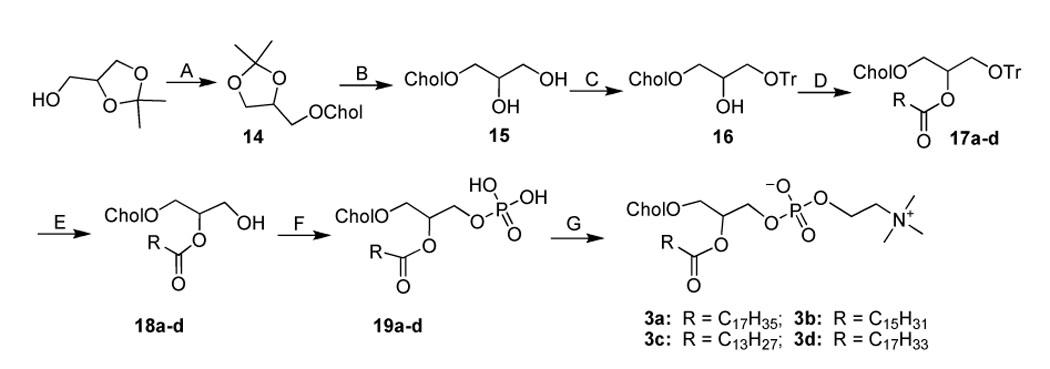
Synthesisa of 3a–d
aReagents and conditions. (A) Cholesteryl tosylate, toluene, 90 °C, 4 h; (B) TFA-HCl (conc.) 2:1, THF, r.t., 4 h; (C) Trityl chloride (1.5 equiv.), pyridine, 50 °C, 18 h; (D) Aliphatic acid (1.05 equiv.), DCC (1.05 equiv.), DMAP (0.3 equiv.), CHCl3, r.t., overnight; (E) BF3.Et2O (4 equiv.), CHCl3, 0 °C, 3 h; (F) POCl3 (1.1 equiv.), pyridine (2 equiv.), THF, 0 °C, 2–3 h; (G) Choline tetraphenyl borate (2 equiv.), TPS (2.5 equiv.), pyridine, 70 °C, 1 h, then r.t., 3 h.
Since this new category of lipids do not fall within any of the categories identified in the most recent lipid classification recommendations 27, we designate the chimeric phospholipids abbreviations according to the rules for the common names of glycerolphospholipids. For example, 1-cholesterylcarbonoyl-2-palmityl-glycero-3-phosphatidylcholine is abbreviated as ChcPePC, wherein: 1) groups at the sn-1/sn-2 positions are represented by capitalized abbreviations (for example, “Ch” for cholesterol, “P” for palmitoyl) in the sequence of their substitution positions; 2) the subscript letter or lowercase letter is used to indicate the linkage type such as: “c” for carbonate, “e” for ether, “a” for carbamate, and the blank (or no subscript letter) for ester according to the existing convention; 3) the head groups are named according to the convention such as “PC” for phosphocholine.
Thermotropic Phase Behavior of Bilayers Composed of SML and Synthetic Diacylphosphatidylcholines
The addition of free cholesterol into a bilayer composed of saturated phospholipids will alter the thermotropic phase behavior of the bilayer resulting in the elimination of the phase transition at about 33 mole percent cholesterol 4. We employed differential scanning calorimetry (DSC) to quantify the effect an SML exerts on the phase transition of synthetic lipids when the aliphatic chain length in the SML is the same as the aliphatic chain length in the diacyl phospholipids. Accordingly, SMLs containing C-16 chain with various linkages (1b–5b) and SMLs of one linkage type group with different acyl chains (5a–5d) were chosen for the DSC study. All of the SML’s used in the following studies were readily dispersed in water from the dry state and formed lipid vesicles as evidenced by dynamic light scattering, freeze fracture electron microscopy and negative stain transmission electron microscopy (SI, Fig. 9).
When a SML is mixed with a diacyl phospholipid, there is an additional acyl chain in SML that must be accounted for when calculating the total moles of aliphatic chains so that the computation of cholesterol mole percentage is consistent with that used in the conventional free cholesterol-diacyl lipid mixtures. The equivalent free cholesterol mole percentage in a mixture of SML with a diacyl lipid is calculated according to the following formula:
nSML refers to the moles of SML; ndiacyl refers to the moles of diacyl lipids.
A pure SML has one cholesterol and one acyl chain, thus the equivalent free cholesterol percentage is 1/1.5 × 100 = 67. Liposomes composed of the SML alone, have a greater mole fraction of cholesterol than the maximum that can be achieved by mixing free Chol with diacyl chain lipids. In a 1:1 mole mixture of a SML and a diacyl lipid, the equivalent free Chol percentage is 1/(1.5+1) × 100 = 40. The same calculation method was applied to all other SML formulations.
The DSC thermogram (Fig. 1A) of SChcPC and its mixtures with 1,2-distearoyl-sn-glycero-phosphatidylcholine (DSPC) was the typical result observed for most of the SMLs; SChcPC at about 35 mole percent of cholesterol eliminated the phase transition of DSPC (Fig. 1A). Replacing the C-18 acyl chain with a C-14 acyl chain did not reduce the ability of MChcPC to eliminate the phase transition of DSPC (Fig. 1B). Thus a cholesterol-like effect was observed rather than a phase separation of the MChcPC and the longer chain length DSPC (Fig. 1B). (See SI Fig. 1–7 for the DSC thermograms of other SMLs).
Figure 1.

Differential scanning calorimetry study of SML liposomes. (A) Thermograms of SChcPC with various amounts of equivalent cholesterol. (B) Thermograms of MChcPC/DSPC. (C) Effect of cholesterol on enthalpy. (D) Effect of cholesterol on transition temperature. (See Table 1 in Supporting Information for the tabulated data.
The major transition temperature (Tm) and enthalpy (ΔH) were plotted against the percentage of the cholesterol in the lipid mixture (Fig. 1C and 1D, SI Table 1). Not surprisingly, there was no detectable phase transition in the temperature range of 10–80 °C for all the pure SMLs tested regardless of the chemical structure of the SMLs. Thus, a homogeneous liquid ordered bilayer of high cholesterol concentration (67%) is achievable if cholesterol and the aliphatic chain are positioned close to each other. In mammalian cells, a similar domain could be formed in the plasma membrane if the membrane protein has a strong affinity for both the aliphatic lipid chain and free cholesterol. The addition of SML to the corresponding diacyl lipids broadened the transition peak in a similar manner as does free cholesterol 4,5,28, and decreased both the transition temperature and transition enthalpy (Fig. 1C and 1D, SI Table 1). This effect was dependent on the mole fraction of the SML and eventually led to the elimination of the phase transition when the equivalent free cholesterol reached a certain mole fraction.
The condensing effects of SMLs of different linkages are similar, although there are noticeable differences on Tm and ΔH especially between the isomers ChcPePC and PeChcPC. ChcPePC was able to rapidly lower the Tm and ΔH and eliminate the phase transition completely at 30% equivalent cholesterol while other SMLs with same chain length need at least 35% cholesterol. It seems that the 2-ether linkage of ChcPePC is responsible for the difference since ChcPaPC and PeChcPC are similar in effect on Tm and ΔH but differ from the effect of ChcPePC on Tm and ΔH. The addition of OChcPC to DSPC resulted in very broad transition peaks (SI Fig. 7) at less than 20% cholesterol. This is significantly different from the effect of SChcPC on DSPC (Fig. 1A), and may be the result of mismatch in chain length or chain packing due to the kinked unsaturated oleoyl chain and the saturated stearoyl chain. A similar result has been observed in the effects of different side-chain analogs of cholesterol on the thermotropic phase behavior of stearoyloleoylphosphatidylcholine 29.
Leakage of SML Liposomes
The inclusion of cholesterol in a lipid composition used to make a lipid vesicle will generally make the liposome less leaky in vitro 6,7 and in vivo 30,31. Measuring the leakage of contents from liposomes under an osmotic gradient is an effective way to evaluate the elastic deformation and critical failure of lipid membranes 32,33. When liposomes are subjected to high outwardly-directed transbilayer osmotic pressure, the membrane will swell and burst at the critical point to rapidly release a portion of the contents. Vesicle will then reseal into a mechanically stable structure once sufficient contents have been expelled to bring the system into osmotic equilibrium. The leakage of calcein from selected liposomes under an osmotic gradient was monitored at 37 °C (Fig. 2A, 2B). The leakage profiles of SML liposomes with different chain length but the same linkage have been measured along with the traditional diacyl lipid/cholesterol liposomes (Fig. 2A). SML liposomes showed the stability trend of C18 > C16 > C14 for the specific linkage tested. Liposomes containing C-14 chain showed different leakage profiles depending on the linkage type (Fig. 2B). Liposome of ChcMaPC exhibited similar leakage profiles to that of 1,2-dimyristoyl-sn-glycero-phosphatidylcholine (DMPC)/Cholesterol (3:2) liposomes and better stability than CheMPC and MChcPC. The cholesterol-free DMPC liposome was the leakiest composition. The influence of chain length on the liposome stability may change when the linkage type varies. For example, ChcMaPC liposome is less leaky than SChcPC liposome. This may be due to the stabilizing effect of hydrogen bonding from the carbamate linkage. Generally, SML liposomes of appropriate linkage maintain their contents at least as well as the corresponding cholesterol/diacyl lipid mixtures under the osmotic gradients tested.
Figure 2.

Leakage profile of calcein loaded SML liposomes. (A) Effect of chain length on osmotic stress-induced leakage. (B) Effect of linkage and formulation on osmotic stress-induced leakage. The fraction of calcein remaining in the liposome was calculated and plotted versus the osmotic gradient. The error of data is within 0.5%. (C) Contents leakage in 30% fetal bovine serum at 37 °C was monitored by measuring the fluorescence intensity change. The fraction of calcein remaining in the liposome was calculated and plotted versus the time of incubation. The control formulations have 40 mol % cholesterol.
The physiological environment is a significant barrier for in vivo liposome drug delivery due to the propensity of serum protein and biological membranes to extract free cholesterol from the liposome bilayer, resulting in contents leakage. The retention of contents by SML liposomes was tested in 30% fetal bovine serum at 37 °C and compared to liposomes with conventional compositions of DSPC/Chol (3:2) or DMPC/Chol (3:2). A striking observation is that the ChcMaPC liposomes retained their contents while the DSPC/Chol (3:2) liposome gradually released its contents over a 4 week period (Fig. 2C). Liposomes composed of SChcPC or CheMPC showed similar leakage profiles to that of ChcMaPC with less than a 10% contents loss in 4 weeks. The prolonged retention of contents of the SML liposomes stands in stark contrast to the control liposome: DMPC/Chol (3:2) released 70% contents in the first week and DSPC/Chol liposome had a 60% contents released after the four week incubation. The contents release rate of SML liposomes may be controlled by the structure of the SML. For example, compared with SChcPC, MChcPC liposomes released 50% of the contents in about 18 days. The results of SML liposomes leakage in serum are consistent with the osmotic leakage profiles. For liposome of DMPC/Chol, despite its high resistance to the osmotic pressure, it is the least stable formulation in the presence of 30% serum that may be caused by the transfer of cholesterol from the lipid bilayer to serum proteins.
Cholesterol Exchange
The desorption of free cholesterol from the donor lipid-water interface is rate-limiting for the overall transfer process and the rate of this step is influenced by interactions of free cholesterol molecules with neighboring phospholipid molecules 21,22. Thus, we predicted that cholesterol covalently bound to the phospholipid would have a reduced exchange rate between membranes. To investigate the cholesterol exchange rate between donor and acceptor liposomes, we formulated SML liposomes and control liposomes with 10% negatively charged phosphatidylglycerol and 40% cholesterol (or the equivalent from SML) and 50% diacyl lipid. Ten fold mole excess of neutral 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) liposome was used as the acceptor. All SML liposomes exhibited very little exchange of cholesterol, particularly compared with control liposomes of Chol/1,2-dipalmitoyl-sn-glycero-phosphatidylcholine (DPPC)/1,2-dipalmitoyl-sn-glycero-phosphatidylglycerol (DPPG) or Chol/DMPC/1,2-dimyristoyl-sn-glycero-phosphatidylglycerol (DMPG) (Fig. 3). These data confirm that covalently linked cholesterol in SML compounds does not transfer between bilayers at a significant rate, while the exchange of free cholesterol from a conventional liposome has a half time of approximate 2 hours. This result provides a plausible explanation for why SML liposomes are more stable than liposomes containing free cholesterol in biological fluids.
Figure 3.

Relative rates of cholesterol exchange at 37 °C. All formulations have 40% free cholesterol or the equivalent cholesterol in the SML.
Cytotoxicity of SML Liposomes in Culture Cells
The unique structures of the SML raised the prospect that they may have toxic effects on cells due to their potential for interfering with lipid degradation pathways 34. As a first toxicity screen, we examined the effects of SML liposomes on the metabolic activity of C26 colon carcinoma cultured cells using the MTT assay. Most SMLs showed no detectable cytotoxic effects at concentration as high as 1 mM although the C16 and C18 SMLs containing the carbamate linkage (ChcPaPC and ChcSaPC) inhibited metabolic activity at 100 µM concentration (SI Fig. 8). All other SMLs tested were as well tolerated as liposome dispersions prepared from free cholesterol and a diacylphosphatidylcholine 35.
Comparison of the Effect on Tumor Progression and Animal Survival of Doxorubicin Encapsulated in SML Liposomes
Optimal therapeutic outcome of an anticancer liposome formulation requires the precise control of the cholesterol concentration 36,37. The SML lipids enable this precise control. We encapsulated doxorubicin in a liposome composed of PChcPC/PEG-DSPE/α-tocopherol (α-T): 94.8/5.0/0.2 and compared the effect of this formulation on tumor progression and animal survival in the BALB/C mice tumored with C-26 colon carcinoma against the effect of non-encapsulated doxorubicin or Doxil™. The Doxil™ formulation is a commercial cholesterol-containing liposome formulation of doxorubicin that is approved by the Food and Drug Administration and was originally tested in the C-26 model 38.
In this model, animals treated with the non-drug containing vehicle PBS had a median survival time of 20 days and no animals survived to 22 days (Fig. 4). Animals treated with the maximum tolerated dose of non-encapsulated doxorubicin (10 mg/kg body weight) had a median survival time of 24 days and all animals died by day 26. Animals treated with Doxil™ or doxorubicin encapsulated in the SML liposome at 15 mg/kg had a median survival time greater than 90 days and 4 of 5 animals survived greater than day 90. The effect on animal survival comparing the liposome formulations to either the vehicle control or to the non-encapsulated drug was significant (log-rank test, P < 0.01) for both SML liposome and Doxil™. There was no significant difference on animal survival between the two liposome formulations. Animals treated by the liposomal doxorubicin (Doxil™ or SML-Dox) have significantly greater tumor growth delay 39 than the control groups (PBS or free doxorubicin) (P < 0.001, Student-Newman-Keuls pairwise comparison). Actually, four out of five mice treated by SML-Dox or Doxil™ were cured with the complete elimination of the tumor. We further compared the biodistribution and blood circulation of the SML-Dox and Doxil™. Although SML-Dox has a shorter circulation time than Doxil™, the amount of doxorubicin accumulated in C-26 tumor 48 h after the i.v. injection of SML-Dox is about 70% that of Doxil™ (Fig. 4). The SML formulation might not seem like a big advantage compared with the current Doxil™ formulation, but there is one less lipid component in the SML formulation which could improve the manufacturing and quality control stage in the preparation of the encapsulated drug.
Figure 4.

Therapeutic data and biodistribution of doxorubicin-loaded SML liposome on BALB/c mice bearing C-26 tumor. A single dose of 15 mg/kg (10 mg/kg for free doxorubicin) was administered i.v. on day 8 after the tumor inoculation (A) Survival curve. SML-Dox: doxorubicin encapsulated in liposome composed of PChcPC/PEGDSPE/α-tocopherol (94.8/5/0.2). Log-rank analysis of the paired survival data: p = 0.0025 for SML-Dox vs PBS and Doxil vs PBS, p = 0.0018 for SML-Dox vs Free Dox and Doxil vs Free Dox. No significant difference between SML-Dox and Doxil (p = 0.94). (B) Tumor growth curve. The tumor growth delays among the treatment groups (SML-Dox and Doxil) are significantly greater than the control group (PBS) based on Student-Newman-Keuls method, p < 0.001. (C) Liposomal doxorubicin biodistribution 48 h after i.v. injection at 15 mg/kg. (D) Blood concentration of doxorubicin after the i.v. injection of liposomal doxorubicin.
After an extensive review of the literature we have been unable to find examples of phospholipids where a sterol is attached to either the glycerol or sphingosine 40–44. Indeed, the recently introduced lipid classification system 27 does not have a category for the SML and to the best of our knowledge such lipids have not as yet been identified or isolated from biological systems. This is perplexing; it would seem incorporation into biomembranes of a rigid amphipathic lipid with a slow transfer would provide a consistent permeability barrier for cells. The disadvantage of SML would be the inability to rapidly respond to changes in the environment by physical mechanisms, i.e. the rapid transfer of the sterol from the membrane 12,45. Unlike cholesterol, the SML do not transfer rapidly, are unlikely to undergo a facile transbilayer flip 23, and exhibit a similar membrane condensing effect on membranes composed of diacylphospholipids regardless of the nature of the aliphatic group paired with the sterol. Thus to respond to environment changes, a cell having two classes of lipids with distinctively different properties (phospholipids and sterols) would be able to employ a combinatorial approach to fine tune membrane properties 12,45. This could be thermodynamically more efficient than having to degrade and resynthesize lipids to meet the demands on the cell to maintain internal homeostasis in the face of a changing environment.
Conclusions
We have synthesized six series of SMLs created from biocompatible components and demonstrated that the attachment of cholesterol to the glycerol backbone does not interfere with cholesterol’s membrane condensing properties. We also demonstrated that the cholesterol exchange rate was greatly reduced when cholesterol was attached to the phospholipid glycerol backbone. Finally, we documented that SMLs are biocompatible and liposome prepared from SML can be used as a drug carrier. The SMLs library could be expanded to have thousands of new lipids by varying the combination of chain length, position of substitution, type of linkage, and head group. SMLs provide a rich supplement to the existing lipid world. They contribute new properties to expand the possible uses lipids have in biology and commerce.
Materials and Methods
Chemistry
Representative procedures are described here. Detailed procedures of synthesis and characterization of the lipids of different chain lengths are provided in Supporting Information. Glycerophosphocholine was from BACHEM (Torrance, CA). Lyso-phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL). Other reagents were from Aldrich (Milwaukee, WI). Solvents were used either directly or purified and dried before use according to the standard protocol. TLC analyses were performed on 0.25-mm silica gel F254 plates using a variety of developing systems: (A) CHCl3/MeOH/NH4OH (65/25/4), (B) CHCl3/MeOH/NH4OH (65/35/5), (C) CHCl3/MeOH/H2O (65/25/4), (D) hexane/EtOAc (2/1), (E) hexane/EtOAc (10/1), (F) hexane/EtOAc (5/1), (G) toluene/ether (9/1), (H) toluene/ether (1/1). High performance flash chromatography (HPFC) was carried out on a Biotage (Charlottesville, VA) Horizon™ HPFC™ system with pre-packed silica gel columns (60 Ǻ, 40–63 µm). Unless noted otherwise, the ratios describing the composition of solvent mixtures represent relative volumes. 1H NMR spectra were acquired on a Varian 400 MHz instrument. Chemical shifts are expressed as parts per million using tetramethylsilane as internal standard. J values are in Hertz. MALDI-TOF mass spectra were obtained at the Mass Spectrometry Facility, University of California San Francisco. The general procedures used in the synthesis were described below.
Protection of 3-Hydroxy Group of 1-Substituted Glycerol
A mixture of 1-substituted glycerol and triphenyl chloride (1.5 equiv.) in anhydrous pyridine was stirred at 50 °C for 18 h under anhydrous condition. After cooling to room temperature (r.t., ca. 23 °C), the mixture was poured into ice-cold water, and extracted with 3 portions of hexane. Undissolved triphenylmethanol was removed by filtration. The filtrate was washed 3 times with water and dried over anhydrous sodium sulfate. The solvent was evaporated and the residue was dissolved in minimum amount of hexane. Additional triphenyl methanol was precipitated from the solution by standing overnight at 4 °C. The solid was filtered off, and the filtrate was evaporated to dryness. The residue was dried over high vacuum and used directly for next step reaction. The reaction was monitored by TLC and yield was generally 80–90%.
Removal of Trityl Group from 1, 2-Substituted-3-Trityl Glycerol
1,2-substituted-3-trityl glycerol in chloroform was treated with boron trifluoride diethyl etherate (4 equiv.) at 0 °C for 3 h. The solution was washed with water/chloroform/methanol (2:2:1). The organic layer was dried over sodium sulfate and evaporated. The residue was dried and used directly for next step reaction. The reaction was monitored by TLC and yield was above 90%.
Phosphorylation of 1,2-Subsitituted-Glycerol
A solution of 1,2-substituted glycerol and anhydrous pyridine (2 equiv.) in anhydrous tetrahydrofuran (THF) was added dropwise to the freshly distilled phosphorus oxychloride (1.1 equiv.) in THF with stirring at 0 °C. Stirring was continued for 2–3 h at 0 °C. Then 10% sodium bicarbonate (ca. 5 equiv.) was added, and the mixture was stirred for 15 min at 0 °C. The solution was then poured on ice water, acidified with HCl (pH ca. 2), and extracted with diethyl ether. The product in aqueous layer was precipitated by adding acetone into water. The precipitate was combined with product from the ether extract, azeotropically dried with toluene, and used directly for next step reaction. Yield was generally above 90%.
1,2-Substituted-Glycero-Phosphocholine
1,2-Substituted-glycero phosphate, choline tetraphenyl borate (2 equiv.) and 2,4,6-triisoproylbenzene sulfonyl chloride (TPS) (2.5 equiv.) were dissolved in anhydrous pyridine with brief warming, then stirred for 1h at 70 °C and 3 h at room temperature. After the addition of water, the solvents are removed by rotary evaporation. The residue was extracted with diethyl ether twice. The extract was combined and evaporated. The crude product was purified by HPFC. Yield of this step is generally 80–90%.
Synthetic route 1 (1a–c)
1-O-Trityl-sn-glycero-3-phosphocholine (7)
Zinc chloride powder (anhydrous, 25 g, 175 mmol) was added to the suspension of glycerophosphocholine (50 g, 185 mmol) in anhydrous DMF (500 mL). The mixture was stirred at r.t. for 30 min, and trityl chloride (53g, 190 mmol) was added at 4 °C. The reaction was kept at 4 °C for 10 h. Then, the crude product was precipitated by the addition of 1 L diethyl ether. The oily product was dissolved in 1L chloroform/isobutanol (2:1), washed with 300 ml 4% aqueous ammonia, dried over anhydrous sodium sulfate. After the evaporation of the volatiles, the crude product was azeotropically dried with toluene. The residue was triturated with acetonitrile for 5 h at r.t.. The white precipitate was then collected and dried over high vacuum. Yield: 45.2 g (49% wrt glycerophosphocholine). TLC: Rf = 0.08 (eluent A). 1H NMR (MeOH-d4), δ 3.12 (m, 2H); 3.15 (s, 9H); 3.56 (m, 2H); 3.90 (m, 2H); 4.01 (m, 1H); 4.20 (m, 2H); 7.27 (m, 9H); 7.42 (m, 6H). MALDI-MS calcd for C27H35NO6P+ [M + H]+ 500.22, found 500.31.
1-O-Trityl-2-stearylcarbamoyl-sn-glycero-3-phosphocholine (8a)
To a solution of 7 (1 g, 2 mmol) in dimethylsulfoxide (anhydrous, 10 mL) was added octadecylisocyanate (0.6 g, 2 mmol). The reaction mixture was stirred at 100 °C under N2 for 24 h. After the evaporation of the solvent, the residue was extracted with methanol. The white solid was filtered off and the filtrate was evaporated to dryness. The Crude product was purified by HPFC (CHCl3/MeOH/H2O, 35/13/2). Yield: 450 mg (28.3% wrt 7). TLC: Rf = 0.25 (eluent A). 1H NMR (CDCl3), δ 0.89 (t, J = 6.4, 3H); 1.25–1.31 (m, 30H); 1.47 (m, 2H); 3.01 (m, 2H); 3.20 (s, 9H); 3.27 (m, 2H); 3.72 (m, 2H); 4.03–4.12 (m, 3H); 4.24 (m, 2H); 5.07 (br, 1H); 7.20 (m, 9H); 7.41 (m, 6H). MALDI-MS calcd for C46H72N2O7P+ [M + H]+ 795.51, found 795.52.
1-Hydroxy-2-stearylcarbamoyl-sn-glycero-3-phosphocholine (9a)
Compound 8a (420 mg, 0.52 mmol) was treated with trifluoroacetic acid (TFA, 1mL) in chloroform (5 mL) at r.t. for 4 h. The volatiles were evaporated and the residue was purified by HPFC (CHCl3/MeOH/H2O, 10/5/1). Yield: 320 mg (99% wrt 8a). TLC: Rf = 0.05 (eluent B). 1H NMR (CDCl3), δ 0.88 (t, J = 6.4, 3H); 1.21–1.31 (br, 30H); 1.47 (m, 2H); 3.03 (m, 1H); 3.11 (m, 1H); 3.29 (s, 9H); 3.66 (m, 2H); 3.76 (m, 2H); 3.99 (m, 2H); 4.30 (m, 2H); 4.78 (m, 1H); 6.51 (br, 1H). MALDI-MS calcd for C27H58N2O7P+ [M + H]+ 553.40, found 553.38.
1-Cholesterylcarbonoyl-2-stearylcarbamoyl-sn-glycero-3-phosphocholine (1a, ChcSaPC)
To a solution of solution of 9a (0.3 g, 0.54 mmol) and diisopropylethylamine (DIPEA, 0.5 mL, 2.8 mmol) in dry ethanol-free chloroform (10 mL), was added dropwise the solution of cholesteryl chloroformate (1.21 g, 2.7 mmol) in ethanol-free chloroform (5 mL) at r.t.. After 16 h reaction at r.t., volatiles were evaporated and the residue was purified by HPFC (CHCl3/MeOH/H2O, 40/18/3). Yield: 438 mg (84% wrt 9a). TLC: Rf = 0.28 (eluent A). 1H NMR (CDCl3), δ 0.69 (s, 3H); 0.85–1.65 (m, 68H); 1.78–2.01 (m, 5H); 2.38 (m, 2H); 3.03 (m, 1H); 3.17 (m, 1H); 3.38 (s, 9H); 3.88 (m, 2H); 4.0 (m, 2H); 4.25 (m, 1H); 4.36 (m, 4H); 5.08 (m, 1H); 5.39 (1H, d, J = 4.4); 6.02 (br, 1H). MALDI-MS calcd for C55H102N2O9P+ [M + H]+ 965.73, found 965.68.
Synthetic route 2 (2a–d)
1,3-Benzylidene-2-stearyl-glycerol (10a)
A solution of 1,3-benzylidene glycerol (7.2 g, 40 mmol) in toluene (100 mL) was added to NaH (60% in mineral oil, 1.92 g, 48 mmol, washed with hexane) suspension in toluene (30 mL) at r.t. with stirring. Then 1-iodo-octadecane (20g, 50 mmol) in toluene (40 mL) was added dropwise into the reaction mixture. After the addition, the mixture was refluxed under nitrogen overnight, and cooled to r.t.. Excessive NaH was destroyed by careful addition of water into the mixture. The reaction mixture was then washed with water (100 mL × 2). The organic layer was collected, dried over sodium sulfate. Solvent was evaporated and the residue was used directly for next step reaction.
2-Stearyl-glycerol (11a)
The crude product of 10a was hydrolyzed by refluxing in the mixed solution of HCl (conc., 30 mL) and methanol (270 mL) for 5 h. The reaction mixture was cooled to r.t. and evaporated under reduced pressure. The residue was dissolved in diethyl ether (300 mL) and washed consecutively with sodium hydroxide solution (0.5 M, 100 mL) and water (150 mL × 2). The ether layer was then dried and evaporated. The crude product was purified by HPFC (30–80% ethyl acetate in hexane). Yield: 11.3 g (82% wrt 1,3-benzylidene glycerol). TLC: Rf = 0.17 (eluent D). 1H NMR (CDCl3), δ 0.86 (t, J = 6.4, 3H); 1.29 (br, 30H); 1.58 (m, 2H); 3.44–3.78 (m, 7H). MALDI-MS calcd for C21H45O3+ [M + H]+ 345.34, found 345.33.
1-Cholesterylcarbonoyl-2-stearyl-glycerol (12a)
To a solution of 11a (0.7 g, 2 mmol), DIPEA (0.5 mL, 2.8 mmol) and DMAP (0.12 g, 1 mmol) in dry ethanol-free chloroform (10 mL), was added dropwise cholesteryl chloroformate (0.94 g, 2.1 mmol) chloroform solution (10 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 0.5 h, then at r.t. overnight. The volatiles were evaporated, and the crude product was purified by HPFC (5–15% ethyl acetate in hexane). TLC: Rf = 0.41 (eluent E). 1H NMR (CDCl3), δ 0.69 (s, 3H); 0.85–1.65 (m, 68H); 1.78–2.01 (m, 5H); 2.40 (m, 2H); 3.52 (m, 2H); 3.60 (m, 2H); 3.68 (m, 1H); 4.22 (m, 2H); 4.43 (m, 1H); 5.40 (1H, d, J = 4.4); MALDI-MS calcd for C49H89O5+ [M + H]+ 757.67, found 757.68.
1-Cholesterylcarbonoyl-2-stearyl-rac-glycero-3-phosphate (13a)
This compound was synthesized according to the general procedure of phosphorylation. TLC: Rf = 0.05 (eluent A). MALDI-MS calcd for C49H89NaO8P+ [M + Na]+ 859.62, found 859.60.
1-Cholesterylcarbonoyl-2-stearyl-rac-glycero-3-phosphocholine (2a, ChcSePC)
This compound was synthesized according to the general procedure of phosphocholine synthesis. TLC: Rf = 0.3 (eluent A). δ 0.69 (s, 3H); 0.85–1.65 (m, 68H); 1.78–2.04 (m, 5H); 2.38 (m, 2H); 3.41 (s, 9H); 3.52 (m, 2H); 3.68 (m, 1H); 3.88 (m, 4H); 4.19 (m, 1H); 4.35 (m, 4H); 5.39 (1H, d, J = 4.4); MALDI-MS calcd for C54H101NO8P+ [M + H]+ 922.73, found 922.74.
Synthetic route 3 (3a–d)
3-(2,3-Isopropylidene-1-glyceryl) cholesterol (14)
A mixture of cholesteryl tosylate (50 g, 90 mmol) and solketal (250 mL, 2 mol) in toluene (50 mL) was stirred at 80–90 °C for 4 h under nitrogen. After cooling to r.t., toluene (300 mL) was added to the mixture. The mixture was washed with brine (300 mL). After separation, additional 200 mL toluene was added to the organic layer. The organic layer was then washed with brine (300 mL), dried, and evaporated to dryness. The crude product was used directly for next step reaction. TLC: Rf = 0.55 (eluent G).
1-Glyceryl cholesterol (15)
The crude product of 14 was dissolved in the mixed solvents of THF (130 mL)-TFA (40 mL)-HCl (conc., 20 mL). The mixture was kept at r.t. for 4 h. The volatiles were evaporated under reduced pressure. The residue was dissolved in CHCl3/MeOH (400 mL/100 mL), and washed with water (100 mL). The organic layer was then dried over sodium sulfate, filtered, and evaporated. The crude product was purified by recrystallization from ethanol at − 20 °C. TLC: Rf = 0.12 (eluent H). 1H NMR (CDCl3), δ 0.69 (s, 3H); 0.85–1.65 (m, 33H); 1.78–2.31 (m, 7H); 3.10 (m, 1H); 3.45–3.70 (m, 4H); 3.79 (m, 1H); 5.34 (1H, d, J = 4.4); MALDI-MS calcd for C30H53O3+ [M + H]+ 461.40, found 461.44.
1-Cholesteryl-3-trityl glycerol (16)
The protection of 3-hydroxy group of 15 with trityl group was carried out according to the general procedure. Product was purified by HPFC (9%–25% ethyl acetate in hexane). TLC: Rf = 0.38 (eluent F). 1H NMR (CDCl3), δ 0.69 (s, 3H); 0.85–1.65 (m, 33H); 1.86 (m, 3H); 2.0 (m, 2H); 2.18 (m, 1H); 2.32 (m, 1H); 2.42 (br, 1H); 3.11–3.22 (m, 3H); 3.57 (m, 2H); 3.93 (m, 1H); 5.35 (1H, d, J = 4.4); 7.28 (m, 9H); 7.43 (m, 6H). MALDI-MS calcd for C49H67O3+ [M + H]+ 703.51, found 703.53.
1-Cholesteryl-2-stearoyl-3-trityl glycerol (17a)
To a solution of 16 (2.11 g, 3 mmol), stearic acid (0.94 g, 3.15 mmol), and 4-dimethylaminopyridine (DMAP, 0.13 g) in dry ethanol-free chloroform (20 mL), was added DCC (0.65 g, 3.15 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 30 min, then r.t. overnight. White precipitate was filtered off, and the filtrate was evaporated to dryness. The crude product was purified by HPFC (1%–10% ethyl acetate in hexane). TLC: Rf = 0.5 (eluent E). 1H NMR (CDCl3), δ 0.69 (s, 3H); 0.85–1.65 (m, 66H); 1.81 (m, 3H); 2.0 (m, 2H); 2.19 (m, 1H); 2.26 (m, 1H); 2.35 (t, J = 7.2, 2H); 3.12 (m, 1H); 3.25 (m, 2H); 3.67 (m, 2H); 5.17 (m, 1H); 5.32 (d, J = 4.4, 1H); 7.27 (m, 9H); 7.44 (m, 6H). MALDI-MS calcd for C67H101O4+ [M + H]+ 969.77, found 969.73.
1-Cholesteryl-2-stearoyl glycerol (18a)
The removal of trityl group was carried out according to the general procedure. The crude product was used directly for next step reaction. TLC: Rf = 0.08 (eluent E).
1-Cholesteryl-2-stearoyl-rac-glycero-3-phosphate (19a)
This compound was synthesized according to the general procedure of phosphorylation. TLC: Rf = 0.05 (eluent A).
1-Cholesteryl-2-stearoyl-rac-glycero-3-phosphocholine (3a, CheSPC)
This compound was synthesized according to the general procedure of phosphocholine synthesis. TLC: Rf = 0.31 (eluent A). 1H NMR (CDCl3), δ 0.68 (s, 3H); 0.85–1.65 (m, 66H); 1.84 (m, 3H); 2.0 (m, 2H); 2.12 (m, 1H); 2.30 (m, 3H); 3.15 (m, 1H); 3.39 (s, 9H); 3.63 (m, 2H); 3.81 (m, 2H); 4.25–4.45 (m, 5H); 5.33 (d, J = 4.4, 1H). MALDI-MS calcd for C53H99NO7P+ [M + H]+ 892.72, found 892.73.
Synthetic route 4 (4a–d)
1-Stearyl-3-trityl glycerol (22a)
The 3-trityl group was introduced according to the general procedure. TLC: Rf = 0.11 (eluent E). 1H NMR (CDCl3), δ 0.87 (t, J = 7.2, 3H); 1.27 (br, 30H); 1.55 (m, 2H); 2.40 (br, 1H); 3.18 (m, 2H); 3.32–3.53 (m, 4H). 3.94 (m, 1H); 7.23 (m, 9H); 7.43 (m, 6H). MALDI-MS calcd for C40H59O3+ [M + H]+ 587.45, found 587.44.
1-Stearyl-2-cholesterylcarbonoyl-3-trityl glycerol (23a)
To a solution of 22a (2.4 g, 4 mmol) and DMAP (0.6 g) in dry ethanol-free chloroform (10 mL), was added dropwise the solution of cholesteryl chloroformate (2.2 g, 4.8 mmol) in chloroform (5 mL) at r.t.. The reaction mixture was stirred at r.t. overnight. Then a mixture solvent of CHCl3/MeOH/H2O (40 mL/ 20 mL/ 30 mL) was added to the reaction mixture. The organic layer was dried over sodium sulfate, filtered, and evaporated. The crude product was purified by HPFC (0–10% ethyl acetate in hexane). TLC: Rf = 0.43 (eluent E). 1H NMR (CDCl3), δ 0.69 (s, 3H); 0.85–1.65 (m, 68H); 1.79–2.02 (m, 5H); 2.42 (m, 2H); 3.25 (m, 2H); 3.38 (m, 19 2H); 3.60 (m, 2H); 4.48 (m, 1H); 5.04 (m, 1H); 5.39 (d, J = 4.4, 1H); 7.27 (m, 9H); 7.43 (m, 6H). MALDI-MS calcd for C68H103O5+ [M + H]+ 999.78, found 999.75.
1-Stearyl-2-cholesterylcarbonoyl glycerol (24a)
This compound was synthesized according to the general procedure of removal of trityl group. TLC: Rf = 0.63 (eluent D).
1-Stearyl-2-cholesterylcarbonoyl-rac-glycero-3-phosphate (25a)
This compound was synthesized according to the general procedure of phosphorylation. TLC: Rf = 0.57 (eluent C).
1-Stearyl-2-cholesterylcarbonoyl-rac-glycero-3-phosphocholine (4a, SeChcPC)
This compound was synthesized according to the general procedure of phosphocholine synthesis. TLC: Rf = 0.53 (eluent A). 1H NMR (CDCl3), δ 0.68 (s, 3H); 0.85–1.65 (m, 66H); 1.84–2.05 (m, 5H); 2.36 (m, 2H); 3.39 (s, 9H); 3.44 (m, 2H); 3.61 (m, 2H); 3.83 (m, 2H); 4.01 (m, 2H); 4.35 (m, 2H); 4.44 (m, 1H); 4.98 (m, 1H); 5.39 (d, J = 4.4, 1H). MALDI-MS calcd for C54H101NO8P+ [M + H]+ 922.73, found 922.73.
Synthetic route 5 (5a–d)
1-Stearoyl-2-cholesterylcarbonoyl-sn-glycero-3-phosphocholine (5a, SChcPC)
To a solution of 1-stearoyl-2-hydroxy-sn-glycero-phosphocholine (1 g, 1.91 mmol) and DMAP (1 g) in ethanol-free dry chloroform (50 mL), was added dropwise the chloroform solution (10 mL) of cholesteryl chloroformate (2 g, 4.45 mmol) at r.t.. After 16 h reaction at r.t., solvent was evaporated and the residue was purified by HPFC (CHCl3 to CHCl3-MeOH-H2O 65/25/4). Yield, 1.57 g, 88%. TLC: Rf = 0.54 (eluent C). 1H NMR (CDCl3/MeOH-d4/pyridine-d5, 10:2:1), δ 0.68 (s, 3H); 0.85–1.65 (m, 66H); 1.84–2.05 (m, 5H); 2.33 (t, J = 7.6 Hz, 2H); 2.39 (m, 2H); 3.28 (s, 9H); 3.67 (m, 2H); 4.10 (m, 2H); 4.24 (m, 1H); 4.32 (m, 2H); 4.47 (m, 2H); 5.11 (m, 1H); 5.41 (d, J = 4.4, 1H). MALDI-MS calcd for C54H99NO9P+ [M + H]+ 937.61, found 937.67.
Synthetic route 6 (6a–d)
1-Stearoyl-2-cholesterylhemisuccinoyl-sn-glycero-3-phosphocholine (6a, SChemsPC)
To a solution of 1-stearoyl-2-hydroxy-sn-glycero-phosphocholine (2 g, 3.82 mmol) and cholesterylhemisuccinate (2.68 g, 4.58 mmol ) in ethanol-free dry chloroform (60 mL) at room temperature, were added DMAP (0.47 g, 3.82 mmol) and DCC (0.95 g, 4.58 mmol). The reaction mixture was stirred at r.t. for 24 h. The mixture was filtered and the filtrate was diluted with 100 mL mixture of chloroform and methanol (2/1, v/v), washed with 30 mL 1 M hydrochloride. The organic layer was dried over anhydrous sodium sulfate, concentrated by rotary evaporation. The residue was applied to HPFC for purification (CHCl3 to CHCl3-MeOH-H2O 65/25/4). Yield, 3.41 g, 90%. TLC: Rf = 0.48 (eluent C). 1H NMR (CDCl3), δ 0.68 (s, 3H); 0.85–1.65 (m, 66H); 1.84–2.05 (m, 5H); 2.29–2.31 (m, 4H); 2.55–2.62 (m, 4H); 3.31 (s, 9H); 3.78 (m, 2H); 4.04 (m, 2H); 4.24 (m, 1H); 4.41 (m, 3H); 4.55 (m, 1H); 5.21 (m, 1H); 5.40 (d, J = 4.4, 1H). MALDI-MS calcd for C57H103NO10P+ [M + H]+ 992.73, found 992.65.
Differential Scanning Calorimetry
Differential scanning calorimetric (DSC) measurements were carried out by using a high-temperature MC-DSC 4100 calorimeter (Calorimetry Sciences Corp., Lindon, UT) with three reusable Hastelloy sample ampoules and a reference ampoule. Data was collected typically over a range of 5 °C−85 °C at 0.5 °C/min with Milli-Q® water as the reference. The CpCalc 2.1 software package from Calorimetry Sciences Corp was used to convert the raw data into molar heat capacity (MHC). Data were then imported into Origin 6.0 (Microcal, Northampton, MA) for further processing and calculation. Liposomes used for DSC measurement were prepared by hydrating the lipid film (10 µmol) in Milli-Q® water (200 µL) at 65 °C under argon for 15 min with intermittent vortex. Samples were then cooled to room temperature, degassed, and loaded into the sample ampoule using gas-tight Hamilton® syringe (100 µL per sample). Samples were scanned through a heatingcooling- heating cycle and the second heating scan data was used for analysis.
Encapsulation of Calcein in Liposome
Calcein (2.49 g, 4 mmol) was dissolved in Tris-HCl buffer (10 mM, pH 7.5, 6 mL) after the addition of 50% sodium hydroxide (695 µL, 13.2 mmol). This stock solution was then loaded on a Sephadex LH-20 column (2.5 cm × 40 cm) and eluted with Tris-HCl buffer (10 mM, pH 7.5). The concentration of pooled fraction of calcein was determined by measuring the absorbance (494 nm) of diluted sample at pH 9. The purified calcein (56 mM) was then encapsulated into the liposomes for the leakage assay. Generally, the dry lipid film(10 µmol) of given formulation was hydrated in 1 mL calcein containing buffer at 60 °C under argon for 15 min with intermittent vortex. Then the sample was extruded through a polycarbonate membrane 11 times at 60 °C followed y passing a Sephadex G-50 column with the corresponding isosmotic eluent. The pooled liposome fractions were then analyzed to determine the calcein concentration, and diluted to the linear fluorescence range for leakage study.
Osmotic Stress Induced Leakage
Liposomes used for this study were prepared according to the above mentioned method with high concentration content (56 mM calcein, 10 mM Tris, 711 mM NaCl), extruded through 100 nm membrane, and eluted with the isosmotic buffer (50 mM HEPES, 775 mM NaCl). Liposomes containing 40% (mole) free cholesterol such as DMPC/Chol (3:2), DSPC/Chol (3:2) and DPPC/Chol (3:2) were used as the positive control in the leakage study of SML liposomes. Solutions of various osmotic concentrations were prepared by mixing the calcein free isosmotic buffer (1600 mOsm) and a 50 mOsm dilution buffer (50 mM HEPES). Liposomes were then exposed to solutions of various osmotic concentrations by mixing 10 µL of liposome with 990 µL testing buffer at 37 °C. Fluorescence signal at 517 nm (ext.: 494 nm) was read after 5 min equilibration by using a Quantech™ fluorometer (Barnstead/Thermolyne, Dubuque, IA). Liposomes were then lysed by adding 100 µL 10% Triton X-100 to release calcein completely. The fluorescence of the total calcein was measured and used as 100% signal (F100%). The fraction of calcein remaining in the liposome before lysis was defined as 1-(Fsignal-Fblank)/(F100%-Fblank), where Fsignal is the fluorescence intensity of the sample and Fblank is the fluorescence intensity of liposome in the isosmotic buffer.
Leakage in 30% Fetal Bovine Serum
Calcein was encapsulated into the liposome by the method described above. Liposomes were extruded through 200 nm membrane and the free calcein was removed by passing the liposomes through the Sephadex G-50 column using HEPES buffer (10 mM HEPES, 140 mM NaCl, pH 7.4) as the isosmotic eluent. Conventional liposome formulations containing 40% cholesterol were used as the control in the long term leakage assay. An aliquot of liposome sample (20–50 µL) was diluted by 30% fetal bovine serum to a total volume of 2 mL. Samples were then sealed in the glass tube and incubated at 37 °C. Fluorescence intensities of samples were monitored at different time points and the fraction of calcein remaining in the liposome was determined by the similar method of osmotic stress induced leakage.
Cholesterol Exchange Experiment
Unilamellar liposomes were prepared by the extrusion method 46. The donor liposomes consisted of 40% cholesterol (or equivalent from SML), 10% negatively charged corresponding phosphatidylglycerol (PG), and 50% 1,2-diacyl-sn-glycero-3-phosphocholine (PC) (or the equivalent from SML). Specifically, the five donor liposomes were formulated at the following molar ratios: 1) PChcPC/DPPC/DPPG (5/4/1), 2) MChcPC/DMPC/DMPG (5/4/1), 3) ChcMaPC/DMPC/DMPG (5/4/1), 4) Chol/DPPC/DPPG(4/5/1), Chol/DMPC/DMPG(4/5/1). A ten-fold molar excess of neutral POPC liposome was used as the acceptor liposome. After extrusion, the diameter of the donor liposomes ranged from 97 nm to 129 nm with a narrow size distribution (SI, Table 2), and 115 nm for the acceptor liposome. The unilamellarity of the liposomes were confirmed by 31P-NMR 47 (SI Fig. 10). For the exchange experiments, 1 mL donor liposomes (10 mM) and 1 mL acceptor liposomes (100 mM, 10 fold) were warmed at 37 °C first, then mixed and incubated at 37 °C. An aliquot (250 µL) of mixture was sampled at given time point and applied to a small (ca. 2 cm in length) anion exchange column (Q-Sepharose XL). The column was pretreated with 0.1 mL 10 mM POPC before the loading of the exchange sample to reduce the nonspecific binding of the neutral liposome. The column was eluted with 1 mL pH 7.4 10 mM NaCl, 10 mM HEPES buffer. The eluate was lyophilized and analyzed by the cholesterol assay 48 to quantify the amount of cholesterol exchanged. Briefly, the lyophilized lipid powder was dissolved in 100 µL distilled water and 50 µL was transferred to screw-capped glass tube (13mm ×100mm). After the addition of 5 mL cholesterol assay reagent (339 mg ferric perchlorate hexahydrate in 300 mL ethyl acetate mixed with 200 mL concentrated sulfuric acid at 4 °C), the mixture was heated at 100 °C for 90 seconds, immersed immediately into ice-water. The absorbance at 610 nm was measured and the amount of cholesterol was calculated according to the standard curve.
Cytotoxicity Determination
The cytotoxicity of the SML lipids was evaluated on C26 cells. After 3 days incubation, cell viability was measured with the standard MTT (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) assay method 49 as described 50.
C26 Murine Adenocarcinoma Model
All animal experiments were performed in compliance with the NIH guidelines for animal research under a protocol approved by the Committee on Animal Research at the University of California, San Francisco. For all chemotherapy experiments, on day 0, BALB/c mice were given subcutaneous injections of C-26 tumor cells (4 × 105 cells per mouse) in the right flank and were then randomized with 5 mice per group and numbered. On day 8 post-tumoring mice received 0.2 mL via a single tail vein injection of either phosphate buffered saline, doxorubicin at 10 mg/kg, Doxil™ at 15 mg/kg or doxorubicin at 15 mg/kg encapsulated in PChcPC-PEG-DSPE-αT: 94.8:5.0:0.2 mole ratio. Mice were weighed and tumor sizes were monitored daily during the experimental period. The tumor volume was estimated by measuring three orthogonal diameters (a, b, and c) with calipers; the volume was calculated as (a × b × c) × 0.5 cm3. Tumors that were just palpable were defined as 1 mm × 1 mm × 1 mm. In each experiment the mice were monitored for up to 90 days post-inoculation or until one of the following conditions for euthanasia was met: 1) their body weight dropped below 15% of their initial mass; 2) their tumor was greater than 2.0 cm across in any dimension; 3) they became lethargic or sick and unable to feed; or 4) they were found dead. On day 90, all surviving mice were euthanized. All animals that survived 60 days also survived until day 90.
Statistical Analysis
Statistical analysis was performed using SigmaPlot version 11 (San Jose, CA). To evaluate the therapeutic efficacy, one-way ANOVA and Student-Newman-Keuls pairwise comparisons were performed using tumor growth delay data 39 derived from the tumor growth curve. Survival data was analyzed by the log-rank test.
Supplementary Material
Synthesis of SMLs, DSC thermograms and the tabulated data of Tm and ΔH, cytotoxicity of SMLs, electron microscope of SML liposomes, size and 31P NMR of liposomes, biodistribution study of liposomal doxorubicin, and complete Ref. 27. This information is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgement
We thank the NIH EB003008 and GM61851 for supporting this research. We acknowledge the excellent technical assistance of Dr. Mahmoud Reza Jaafari and Nichole Macaraeg. Thanks are also extended to the UCSF Mass Spectrometry Facility (A.L. Burlingame, Director) supported by NIH NCRR RR0161.
References
- 1.McIntosh TJ, Simon SA. Annu. Rev. Biophys. Biomol. Struct. 2006;35:177–198. doi: 10.1146/annurev.biophys.35.040405.102022. [DOI] [PubMed] [Google Scholar]
- 2.Sugahara M, Uragami M, Yan X, Regen SL. J. Am. Chem. Soc. 2001;123:7939–7940. doi: 10.1021/ja016199c. [DOI] [PubMed] [Google Scholar]
- 3.McConnell HM, Vrljic M. Annu. Rev. Biophys. Biomol. Struct. 2003;32:469–492. doi: 10.1146/annurev.biophys.32.110601.141704. [DOI] [PubMed] [Google Scholar]
- 4.Mabrey S, Mateo PL, Sturtevant JM. Biochemistry. 1978;17:2464–2468. doi: 10.1021/bi00605a034. [DOI] [PubMed] [Google Scholar]
- 5.McMullen TP, Lewis RN, McElhaney RN. Biochemistry. 1993;32:516–522. doi: 10.1021/bi00053a016. [DOI] [PubMed] [Google Scholar]
- 6.Demel RA, Bruckdorfer KR, van Deenen LL. Biochim. Biophys. Acta. 1972;255:321–330. doi: 10.1016/0005-2736(72)90031-4. [DOI] [PubMed] [Google Scholar]
- 7.Papahadjopoulos D, Nir S, Oki S. Biochim. Biophys. Acta. 1972;266:561–583. doi: 10.1016/0006-3002(72)90001-7. [DOI] [PubMed] [Google Scholar]
- 8.Haines TH. Prog. Lipid Res. 2001;40:299–324. doi: 10.1016/s0163-7827(01)00009-1. [DOI] [PubMed] [Google Scholar]
- 9.Bach D, Wachtel E. Biochim. Biophys. Acta. 2003;1610:187–197. doi: 10.1016/s0005-2736(03)00017-8. [DOI] [PubMed] [Google Scholar]
- 10.Anderson RG. Trends Cell. Biol. 2003;13:534–539. doi: 10.1016/j.tcb.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Brown MS, Goldstein JL. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 12.Goldstein JL, DeBose-Boyd RA, Brown MS. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 13.Windaus A. Nobel Lectures Chemistry 1922–1941. New York: Elsevier Science Publishing Co., Inc.; 1996. pp. 105–121. [Google Scholar]
- 14.Brockerhoff H, Ramsammy LS. Biochim. Biophys. Acta. 1982;691:227–232. doi: 10.1016/0005-2736(84)90139-1. [DOI] [PubMed] [Google Scholar]
- 15.Demel RA, Lala AK, Kumari SN, Vandeenen LLM. Biochim. Biophys. Acta. 1984;771:142–150. doi: 10.1016/0005-2736(84)90526-1. [DOI] [PubMed] [Google Scholar]
- 16.Patel KR, Li MP, Schuh JR, Baldeschwieler JD. Biochim. Biophys. Acta. 1984;797:20–26. doi: 10.1016/0304-4165(84)90377-5. [DOI] [PubMed] [Google Scholar]
- 17.Lai MZ, Duzgunes N, Szoka FC. Biochemistry. 1985;24:1646–1653. doi: 10.1021/bi00328a012. [DOI] [PubMed] [Google Scholar]
- 18.Epand RM, Bottega R, Robinson K. Chem. Phys. Lipids. 1990;55:49–53. [Google Scholar]
- 19.Gotoh M, Ribeiro N, Michels B, Elhabiri M, Albrecht-Gary AM, Yamashita J, Hato M, Ourisson G, Nakatani Y. Chem. Biodivers. 2006;3:198–209. doi: 10.1002/cbdv.200690023. [DOI] [PubMed] [Google Scholar]
- 20.Torchilin VP. Nat. Rev. Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 21.Phillips MC, Johnson WJ, Rothblat GH. Biochim. Biophys. Acta. 1987;906:223–276. doi: 10.1016/0304-4157(87)90013-x. [DOI] [PubMed] [Google Scholar]
- 22.Hamilton JA. Curr. Opin. Lipidol. 2003;14:263–271. doi: 10.1097/00041433-200306000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Kan CC, Yan J, Bittman R. Biochemistry. 1992;31:1866–1874. doi: 10.1021/bi00121a040. [DOI] [PubMed] [Google Scholar]
- 24.Needham D, Evans E. Biochemistry. 1988;27:8261–8269. doi: 10.1021/bi00421a041. [DOI] [PubMed] [Google Scholar]
- 25.Paltauf F, Hermetter A. Prog. Lipid Res. 1994;33:239–328. doi: 10.1016/0163-7827(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 26.MacKellar C, Graham D, Will DW, Burgess S, Brown T. Nucleic Acids Res. 1992;20:3411–3417. doi: 10.1093/nar/20.13.3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fahy E, et al. J. Lipid Res. 2005;46:839–861. doi: 10.1194/jlr.E400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Lai MZ, Vail WJ, Szoka FC. Biochemistry. 1985;24:1654–1661. doi: 10.1021/bi00328a013. [DOI] [PubMed] [Google Scholar]
- 29.Vilcheze C, McMullen TP, McElhaney RN, Bittman R. Biochim. Biophys. Acta. 1996;1279:235–242. doi: 10.1016/0005-2736(95)00258-8. [DOI] [PubMed] [Google Scholar]
- 30.Gregoriadis G, Davis C. Biochemical and Biophysical Research Communications. 1979;89:1287–1293. doi: 10.1016/0006-291x(79)92148-x. [DOI] [PubMed] [Google Scholar]
- 31.Mayhew E, Rustum YM, Szoka F, Papahadjopoulos D. Cancer Treat. Rep. 1979;63:1923–1928. [PubMed] [Google Scholar]
- 32.Mui BLS, Cullis PR, Evans EA, Madden TD. Biophys. J. 1993;64:443–453. doi: 10.1016/S0006-3495(93)81385-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shoemaker SD, Vanderlick TK. Ind. Eng. Chem. Res. 2002;41:324–329. [Google Scholar]
- 34.Allen TM, Murray L, MacKeigan S, Shah M. J. Pharmacol. Exp. Ther. 1984;229:267–275. [PubMed] [Google Scholar]
- 35.Parnham MJ, Wetzig H. Chem. Phys. Lipids. 1993;64:263–274. doi: 10.1016/0009-3084(93)90070-j. [DOI] [PubMed] [Google Scholar]
- 36.Mayer LD, Tai LCL, Ko DSC, Masin D, Ginsberg RS, Cullis PR, Bally MB. Cancer Res. 1989;49:5922–5930. [PubMed] [Google Scholar]
- 37.Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. J. Pharm. Sci. 2008 doi: 10.1002/jps.21358. [DOI] [PubMed] [Google Scholar]
- 38.Huang SK, Mayhew E, Gilani S, Lasic DD, Martin FJ, Papahadjopoulos D. Cancer Res. 1992;52:6774–6781. [PubMed] [Google Scholar]
- 39.Schluep T, Hwang J, Cheng JJ, Heidel JD, Bartlett DW, Hollister B, Davis ME. Clin. Cancer Res. 2006;12:1606–1614. doi: 10.1158/1078-0432.CCR-05-1566. [DOI] [PubMed] [Google Scholar]
- 40.Urata K, Takaishi N. Eur. J. Lipid Sci. Tech. 2001;103:29–39. [Google Scholar]
- 41.Salunke DB, Hazra BG, Pore VS. Curr. Med. Chem. 2006;13:813–847. doi: 10.2174/092986706776055562. [DOI] [PubMed] [Google Scholar]
- 42.Guo X, Szoka FC. Acc.Chem. Res. 2003;36:335–341. doi: 10.1021/ar9703241. [DOI] [PubMed] [Google Scholar]
- 43.Bhattacharya S, Bajaj A. Curr. Opin. Chem. Biol. 2005;9:647–655. doi: 10.1016/j.cbpa.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 44.Huang Z, Szoka FC. In: Liposome technology. 3rd Ed. Gregoriadis G, editor. Vol. 1. New York: Informa Healthcare; 2007. pp. 165–196. [Google Scholar]
- 45.Lange Y, Ye J, Steck TL. J. Biol. Chem. 2005;280:36126–36131. doi: 10.1074/jbc.M507149200. [DOI] [PubMed] [Google Scholar]
- 46.Szoka F, Olson F, Heath T, Vail W, Mayhew E, Papahadjopoulos D. Biochim. Biophys. Acta. 1980;601:559–571. doi: 10.1016/0005-2736(80)90558-1. [DOI] [PubMed] [Google Scholar]
- 47.Frohlich M, Brecht V, Peschka-Suss R. Chem. Phys. Lipids. 2001;109:103–112. doi: 10.1016/s0009-3084(00)00220-6. [DOI] [PubMed] [Google Scholar]
- 48.Wybenga DR, Pileggi VJ, Dirstine PH, J D. Clin. Chem. 1970;16:980–984. [PubMed] [Google Scholar]
- 49.Mosmann T. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 50.Huang Z, Li W, MacKay JA, Szoka FC., Jr Mol. Ther. 2005;11:409–417. doi: 10.1016/j.ymthe.2004.10.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthesis of SMLs, DSC thermograms and the tabulated data of Tm and ΔH, cytotoxicity of SMLs, electron microscope of SML liposomes, size and 31P NMR of liposomes, biodistribution study of liposomal doxorubicin, and complete Ref. 27. This information is available free of charge via the Internet at http://pubs.acs.org.