

Abstract
The indolequinone ES936 {5-methoxy-1,2-dimethyl-3-[(4-nitrophenoxy)methyl]indole-4,7-dione} was previously developed in our lab as an antitumor agent against pancreatic cancer. The objective of this study was to identify indolequinones with improved potency against pancreatic cancer and to define their mechanisms of action. Pancreatic cancer cell lines PANC-1, MIA PaCa-2, and BxPC-3 were used in in vitro assays [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) and clonogenic assays]; indolequinones displayed potent cytotoxicity against all three cell lines, and two specific classes of indolequinone were particularly potent agents. These indolequinones induced caspase-dependent apoptosis but no redox cycling or oxidative stress in MIA PaCa-2 and BxPC-3 cells. Selected indolequinones were also screened against the NCI-60 cell line panel and were found to be particularly effective against colon, renal, and melanoma cancer cells. A potential target of these indolequinones was identified as thioredoxin reductase. Indolequinones were found to be potent inhibitors of thioredoxin reductase activity both in pancreatic cancer cells and in cell-free systems. The mechanism of action of the indolequinones was shown to involve metabolic reduction, loss of a leaving group to generate a reactive electrophile resulting in alkylation of the selenocysteine residue in the active site of thioredoxin reductase. In vivo efficacy of the indolequinones was also tested in the MIA PaCa-2 pancreatic tumor xenograft in nude mice, and lead indolequinones demonstrated high efficacy and low toxicity. Inhibition of thioredoxin reductase represents a potential novel target in pancreatic cancer and may provide a biomarker of effect of lead indolequinones in this type of cancer.
Pancreatic cancer is the fourth leading cause of cancer death in the United States (Jemal et al., 2008), with a 5-year survival rate of <5%. Current treatment options of radiation therapy, chemotherapy, and surgery have been ineffective at improving the survival rate (Ghaneh et al., 2007). Development of novel targeted therapeutic approaches is desperately needed.
We have reported previously the development of an indolequinone, 5-methoxy-1,2-dimethyl-3-[(4-nitrophenoxy)methyl]-indole-4,7-dione (ES936, 1), that exhibited potent growth inhibition effects against human pancreatic cancer cell lines (Dehn et al., 2006). The antitumor activity of ES936 was originally attributed to its role as a mechanism-based inhibitor of human NQO1 [NAD(P)H:quinone oxidoreductase 1 (DT-diaphorase; EC 1.6.99.2)] (Winski et al., 2001). NQO1 inhibition by dicumarol, a nonspecific inhibitor, has been shown to be cytotoxic in human pancreatic cancer cells (Cullen et al., 2003; Lewis et al., 2004). However, when a series of indolequinone compounds based on ES936 was tested for structure-activity relationship, we found no correlation between the antitumor effects of these indolequinones and NQO1 inhibition in pancreatic cancer cells (Colucci et al., 2007; Reigan et al., 2007). Therefore, the search for molecular targets other than NQO1 is needed to better understand the mechanism of action of these indolequinones against pancreatic cancer.
In the present study, we have developed and examined a series of indolequinones (1-9; Table 1) based on the structure of ES936. In this article, we report the antitumor activity of this series of indolequinones against pancreatic cancer cells both in vitro and in vivo. The proposed mechanism of action of the indolequinones involves reduction, loss of a leaving group, and generation of an electrophile, leading to cell death. One potential target of quinone electrophiles is thioredoxin reductase (TrxR) (Powis et al., 2006; Chew et al., 2008), and we show in the present study that an established thioredoxin reductase inhibitor had a similar toxicity profile in the NCI-60 panel. Further work employing both purified enzyme and in cancer cells implicated thioredoxin reductase as a potential target of this series of indolequinones.
TABLE 1.
Structure of indolequinones
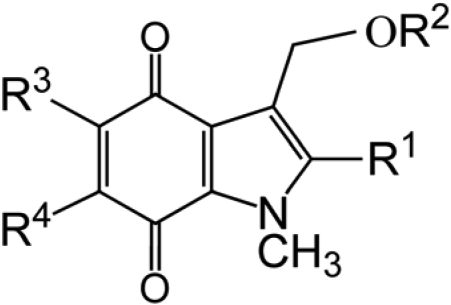
| Compound | R1 | R2 | R3 | R4 |
|---|---|---|---|---|
| 1 (ES936) | CH3 | 4-NO2-C6H4 | OCH3 | H |
| 2 | CH3 | 4-NO2-C6H4 | H | OCH3 |
| 3 | CH2OH | 4-NO2-C6H4 | OCH3 | H |
| 4 | CH2OH | 2,4,6-F3-C6H2 | OCH3 | H |
| 5 | CH2OH | 4-NO2-C6H4 | H | OCH3 |
| 6 | H | 4-NO2-C6H4 | H | OCH3 |
| 7 | H | 2,4,6-F3-C6H2 | H | OCH3 |
| 8 | H | 4-NO2-C6H4 | OCH3 | H |
| 9 | H | 2,4,6-F3-C6H2 | OCH3 | H |
Materials and Methods
Materials. The indolequinones ES936 [1 (Beall et al., 1998)], 6-methoxy-1,2-dimethyl-3-[(4-nitrophenoxy)methyl]indole-4,7-dione [2 (Colucci et al., 2007)], 2-hydroxymethyl-5-methoxy-1-methyl-3-[(4-nitrophenoxy)methyl]indole-4,7-dione [3 (Newsome, 2004)], 2-hydroxymethyl-5-methoxy-1-methyl-3-[(2,4,6-trifluorophenoxy)methyl]indole-4,7-dione [4 (A.C., unpublished results)], 2-hydroxymethyl-6-methoxy-1-methyl-3-[(4-nitrophenoxy)methyl]indole-4,7-dione [5 (Colucci, 2007)], 6-methoxy-1-methyl-3-[(4-nitrophenoxy)methyl]indole-4,7-dione [6 (Colucci, 2007)], 6-methoxy-1-methyl-3-[(2,4,6-trifluorophenoxy) methyl]indole-4,7-dione [7 (Colucci, 2007)], 5-methoxy-1-methyl-3-[(4-nitrophenoxy)methyl]indole-4,7-dione [8 (Everett et al., 2001)], 5-methoxy-1-methyl-3-[(2,4,6-trifluorophenoxy)methyl]indole-4,7-dione [9 (A.C., unpublished results)], and 5-methoxy-1,2-dimethyl-3-[1-oxo-2-(2,4,6-trifluorophenyl)ethyl]indole-4,7-dione [ACH983 (A.C., unpublished results)] were synthesized according to methods previously developed. Recombinant human NQO2 was obtained from Sigma (St. Louis, MO). Dihydronicotinamide riboside (NRH) was synthesized in our lab using published procedures (Friedlos et al., 1992; Yan et al., 2008). Recombinant rat thioredoxin reductase 1 was purchased from IMCO (Stockholm, Sweden). Rabbit polyclonal antibody against thioredoxin reductase 1 was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against cleaved caspase 3, 7, 8, and 9 are from Cell Signaling Technology (Danvers, MA). Annexin V was obtained from Invitrogen (Carlsbad, CA). Unless indicated, all other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Cell Lines. PANC-1 human pancreatic duct epithelioid carcinoma cells, MIA PaCa-2 human pancreatic carcinoma cells, and BxPC-3 human pancreatic adenocarcinoma cells were obtained from American Type Culture Collection (Manassas, VA). MIA PaCa-2 cells were grown in Dulbecco's modified Eagle's medium adjusted to contain 4 mM l-glutamine, 10% (v/v) fetal bovine serum, 2.5% (v/v) horse serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. PANC-1 and BxPC-3 cells were grown in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum, 2 mM l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. All cell lines were maintained in a humidified incubator containing 5% carbon dioxide at 37°C.
Growth Inhibition Assay. Growth inhibition was measured using the MTT colorimetric assay (Mosmann, 1983). In these studies, cells in exponential growth phase were seeded at 2000 cells per well in 96-well plates in triplicate plates and allowed to attach for 16 h. Cells were then treated with indolequinones in complete medium (200 μl/well) for 72 h, or for 4 h followed by incubation in drug-free medium (200 μl/well) for an additional 72 h at 37°C. The medium was removed by aspiration, and MTT (50 μg) in complete medium (50 μl) was added to each well and incubated for a further 4 h. Cell viability was determined by measuring the cellular reduction of MTT to the crystalline formazan product that was dissolved by the addition of 100 μl of DMSO. Optical density was determined at 550 nm using a microplate reader (ThermoMax; Molecular Devices, Sunnyvale, CA). The IC50 values were defined as the concentration of indolequinone that resulted in 50% reduction in cell number compared with DMSO-treated controls.
Clonogenic Assays. The inhibition of the colony-forming ability of MIA PaCa-2 and BxPC-3 cells by indolequinones was assessed using the clonogenic assay. For these studies, 800 cells were seeded in complete medium into 100-mm tissue culture plates and allowed to attach for 16 h. Cells were then exposed to a concentration range of indolequinones in complete medium for 4 or 72 h. The medium was then replaced with fresh medium and cells incubated at 37°C for 10 days. Cells were then rinsed in PBS, fixed in 3% (v/v) acetic acid and 10% (v/v) methanol, and stained with 0.2% (w/v) crystal violet and 10% (v/v) formalin in PBS. Colonies were counted manually.
Flow Cytometry Analysis of Apoptosis. MIA PaCa-2 and BxPC-3 cells were seeded at 105 cells per well in six-well plates and treated with various concentrations of indolequinone 3 for 4 h followed by incubation in drug-free media for 24 h. Cells were collected, washed with PBS, resuspended in annexin binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2), and stained with annexin-V and PI according to manufacturers' instructions. Annexin-V and PI staining were determined using flow cytometry with a FAC-SCalibur (BD Biosciences, San Jose, CA). Cells with positive annexin-V staining were counted as apoptotic cells.
Immunoblot Analysis. Cells were seeded in 100-mm culture plates at 6 × 105 cells per plate and treated with various concentrations of indolequinone 3 for 4 h followed by incubation in drug-free media for 24 h. Cells were collected, washed with PBS, and resuspended in caspase lysis buffer [20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 250 mM sucrose, and 0.25 mM phenylmethyl sulfonyl fluoride with protease inhibitors cocktail (Roche Diagnostics, Indianapolis, IN)]. Cells were then sonicated and centrifuged (13,000 rpm × 15 min) and protein concentration of the supernatant was determined using the method of Lowry (Lowry et al., 1951). Cellular proteins (50 μg) were then separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride membrane. The membrane was then probed with primary antibodies against caspase-3, -7, -8, and -9 and β-actin (Cell Signaling Technology Inc., Danvers, MA) followed by horseradish peroxidase-conjugated goat anti-mouse/rabbit immunoglobulin G secondary antibodies (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Western signals were detected using chemiluminescence methods with ECL Western Blotting Detection Reagents (GE Healthcare).
Alkaline Comet Assay. DNA damage was evaluated using the single-cell gel electrophoresis method, commonly known as the alkaline comet assay, as described previously (Tice et al., 2000), including a modified version (Ward et al., 1997) to detect DNA cross-linking. In brief, cells were seeded in six-well plates at 1 × 105 cells/well and let attach for 16 h. After 1-h drug treatment, cells were harvested and 1 × 104 cells were then subjected to the comet assay. Comet slides were stained with PI and viewed using fluorescence microscopy under a Nikon invert microscope (Eclipse TE300; Nikon, Tokyo, Japan) at 20× magnification. Images were captured using an attached CoolSNAPES CCD camera. One hundred cells, 50 each on duplicate slides were captured and scored using a software package (Komet Version 5; Kinetic Imaging, Belfast, UK). The percentage of DNA in the comet tail was recorded for each comet as an indication of the extent of DNA single-strand breaks.
For measurement of DNA cross-linking, a fixed amount of single-strand breaks was induced after treatment into control and indolequinone-treated cells at each concentration point by incubating with 200 μM H2O2 for 20 min on ice. Cross-linked DNA is unable to migrate from the head of the comet, and the extent of DNA cross-linking can be indirectly measured by analyzing the relative reduction of DNA migration induced by H2O2 compared with untreated H2O2 controls.
Thioredoxin Reductase Activity in Cells. Cells were seeded in 100-mm culture plates at 6 × 105 cells per plate and treated with various concentrations of indolequinones for 4 h; cells were then collected in radioimmunoprecipitation assay buffer, sonicated, and centrifuged (13,000 rpm × 15 min) and protein concentration in supernatant was determined using the method of Lowry. Thioredoxin reductase activity assay was then performed in 96-well plates using an endpoint insulin reduction assay as described previously (Fang et al., 2005). In brief, reactions (50 μl) contained 50 mM Tris-HCl, pH 7.4, 2 mM EDTA, 200 μM NADPH, 1.5 mg/ml insulin, 20 μM Escherichia coli thioredoxin (Trx), and 40 μg of protein from each cell extract. After incubation for 20 min at 37°C, the reaction was terminated by adding 200 μl of 1 mM DTNB in 6 M guanidine hydrochloride (dissolved in 50 mM Tris-HCl, pH 8.0). The free thiols of the reduced insulin were determined by DTNB reduction, measured by the absorbance at 412 nm, where 1 mol of disulfide give rise to 2 mol of free 2-nitro-5-thiobenzoic acid with the extinction coefficient 13.6 mM-1 · cm-1. A blank measurement for each sample, containing everything except Trx, was treated in the same manner, and the blank value was subtracted from the corresponding absorbance value of the sample. The activity of TrxR was expressed as the percentage of DMSO-treated control.
Inhibition of Thioredoxin Reductase in Cell-Free System. Inhibition reaction was carried out in 100 mM potassium phosphate buffer, pH 7.4, containing 2 mM EDTA and 1 mg/ml BSA. A mixture of 0.5 μM recombinant rat TrxR, 250 μM NADPH, 2 μM NQO2, and 200 μM NRH were incubated in the above buffer at room temperature for 5 min. Then various concentrations of indolequinone were added (the final volume of the mixture was 150 μl) and a 20-μl sample was removed every 5 min up to 30 min and measured for TrxR activity using the DTNB reduction assay, as described previously (Fang et al., 2005). The TrxR activity assay mixture contains 100 mM potassium phosphate buffer, pH 7.4, 2 mM EDTA, 1 mg/ml bovine serum albumin, 250 μM NADPH, and 2.5 mM DTNB. The release of 2-nitro-5-thiobenzoic acid from DTNB was monitored at 412 nm for 1 min after sample addition, the rate was calculated, and a blank reading without samples was subtracted from every sample. Results were expressed as percentage of control (20-μl sample removed before addition of IQs) and were representative of three separate experiments.
Detection of Indolequinone-Modified Residues in TrxR using Biotin-Conjugated Iodoacetamide. The free selenocysteine in TrxR was detected as reported previously (Chew et al., 2008). TrxR was incubated with increasing concentrations of indolequinone 3 under identical conditions as in the pure enzyme inhibition assay. After a 5-min incubation at room temperature, 1 μl of reaction mixture was taken out and mixed with 19 μl of 100 μM biotin-conjugated iodoacetamide (BIAM; pH 6.5) and incubated at 37°C for another 30 min to alkylate the free -SeH groups in the enzyme. The products were then separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane. Proteins labeled with BIAM were detected with horseradish peroxidase-conjugated streptavidin and enhanced chemiluminescence detection.
Indolequinone Antitumor Activity in Human Pancreatic Xenograft Tumors. All experiments were approved by the University of Colorado Denver Animal Care and Use Committee and were carried out according to approved protocols. Female athymic nude mice (Ncr nu/nu; National Cancer Institute, Frederick, MD) were received at 5 to 6 weeks of age and were allowed to acclimate for 2 weeks in sterile microisolator cages with constant temperature and humidity. Mice had free access to food and water. MIA PaCa-2 cells in log-phase growth were harvested on the day of use. Cells were suspended in 75:25 unsupplemented medium/Matrigel and 0.1 ml (2 × 107 cells) was injected subcutaneously into the right flank of each animal. After inoculation of tumor cells, mice were monitored daily and weighed twice weekly; digital caliper measurements were begun when tumors became visible. When tumors had grown to ∼200 mm3 (∼12 days after implantation), tumor-bearing mice were randomized into control and drug treatment groups. For these studies, the indolequinones (dissolved in 100% sterile DMSO) were injected every other day for 20 days at the dose of 1.0 or 2.5 mg/kg i.p. No obvious toxicities or weight loss were observed in the control (DMSO) or indolequinone-treated animals during treatment. Tumor volume was calculated by the formula (L × W2)/2, where L is the longer measurement of the tumor and W is the smaller tumor measurement. Ratios of tumor volumes of treated and control tumors as an indicator of drug efficacy were calculated as described previously (Dehn et al., 2006).
Statistical Analysis. Statistical analysis was performed using one-way analysis of variance followed by appropriate post hoc tests: Dunnett test for comparison of multiple observations to a single control; Student's t test for pairwise comparisons. Data are represented as mean ± S.D. of at least three replicate experiments.
Results
Growth Inhibitory Activity of the Indolequinones in Human Pancreatic Cancer Cell Lines. The effect of the indolequinones on the growth of human pancreatic cancer cells was assessed using the MTT assay in PANC-1, MIA PaCa-2, and BxPC-3 pancreatic cancer cell lines after 4- or 72-h treatment with the indolequinones, and the IC50 values in each cell line are listed in Table 2. In all three cell lines, the indolequinones exhibited marked growth inhibitory activity. A structure-activity relationship was observed and this series of indolequinones can be subdivided into three classes by the substituent on the 2-position of the indole ring. The order of potency of the indolequinones was 2-unsubstituted class (6-9) > 2-hydroxymethyl class (3-5) > 2-methyl class (1, 2).
TABLE 2.
IC50 values for indolequinones in pancreatic cancer cell lines
Calculated IC50 values are the mean ± S.D. of three independent determinations using the MTT growth inhibition assay.
|
IC50
|
||||||
|---|---|---|---|---|---|---|
| Compound |
PANC-1
|
MIA PaCa-2
|
BxPC-3
|
|||
| 4 h | 72 h | 4 h | 72 h | 4 h | 72 h | |
| nM | ||||||
| 1 (ES936) | 788 ± 51 | 88 ± 13 | 629 ± 73 | 507 ± 35 | 408 ± 53 | 305 ± 24 |
| 2 | 476 ± 50 | 194 ± 26 | 638 ± 54 | 354 ± 48 | 476 ± 40 | 285 ± 29 |
| 3 | 120 ± 9 | 88 ± 3 | 96 ± 11 | 74 ± 7 | 163 ± 25 | 103 ± 12 |
| 4 | 181 ± 12 | 80 ± 5 | 87 ± 9 | 52 ± 5 | 329 ± 17 | 179 ± 16 |
| 5 | 110 ± 15 | 56 ± 8 | 72 ± 3 | 59 ± 10 | N.D. | N.D. |
| 6 | 49 ± 4 | 24 ± 3 | 32 ± 9 | 18 ± 5 | N.D. | N.D. |
| 7 | 242 ± 13 | 115 ± 11 | 26 ± 7 | 19 ± 4 | N.D. | N.D. |
| 8 | 89 ± 7 | 37 ± 5 | 39 ± 13 | 24 ± 6 | 190 ± 8 | 79 ± 5 |
| 9 | N.D. | N.D. | 34 ± 9 | 18 ± 6 | 100 ± 11 | 43 ± 10 |
N.D., not determined.
Cytotoxic Activity of the Indolequinones in Pancreatic Cancer Cell Lines. To confirm the growth inhibition data obtained using the mitochondria-based MTT assay, the effect of the indolequinones in human pancreatic cancer cell lines was also assessed using the clonogenic assay. The colony-forming ability was measured in MIA PaCa-2 (indolequinones 1-8) and BxPC-3 (1-4, 8, 9) cells after 4- or 72-h treatment with the indolequinones (Fig. 1). A similar order of potency, as in the MTT assay, was observed for the three classes of indolequinones.
Fig. 1.

Effect of indolequinone treatment on colony formation in human pancreatic cancer cells. Colony-forming ability was measured in MIA PaCa-2 and BxPC-3 cells after 4- or 72-h indolequinone treatment. Data represent mean ± S.D. of three independent determinations.
Induction of Caspase-Dependent Apoptosis by the Indolequinones in Pancreatic Cancer Cell Lines. The ability of the indolequinones to induce apoptotic cell death was measured in MIA PaCa-2 and BxPC-3 cells using flow cytometry analysis. A dose-dependent increase in apoptosis was observed in both MIA PaCa-2 and BxPC-3 cells (Fig. 2A) after indolequinone 3 treatment (24 h), at concentration ranges around the MTT IC50 value. The indolequinone 3-induced apoptosis could be completely blocked by the pancaspase inhibitor z-VAD-fmk (Fig. 2B), suggesting that apoptosis is caspase-dependent. In fact, caspase-3, -7, -8, and -9 activation was detected in both cell lines after 24-h drug treatment using immunoblot analysis (Fig. 2C). There was a dose-dependent increase in the amount of the cleaved form of caspase 3 (17/19 kDa), caspase 7 (20 kDa), caspase 8 (18 kDa), and caspase 9 (35/37 kDa) in both cell lines.
Fig. 2.

Induction of caspase-dependent apoptosis by indolequinone 3 in pancreatic cancer cells. A, effect of indolequinone 3 treatment on apoptotic cell death. MIA PaCa-2 and BxPC-3 cells were treated with indolequinone 3 at various concentrations. Apoptosis was measured 24 h after 4-h drug treatment using annexin-PI staining in combination with flow cytometry analysis. B, effect of pretreatment with the pan-caspase inhibitor z-VAD-fmk on indolequinone 3-induced apoptosis. BxPC-3 cells were pretreated with 100 μM z-VAD-fmk for 30 min before indolequinone 3 treatment. Data represent mean ± S.D. of three independent determinations. C, induction of caspase activation by indolequinone 3 in MIA PaCa-2 and BxPC-3 pancreatic cancer cells. The cleaved forms of caspase 3, 7, 8, and 9 were detected in both cell lines using immunoblot analysis 24 h after 4-h drug treatment. Staurosporine (STAU) treatment (500 nM) was included as positive control for caspase activation. Immunoblot shown was representative of at least three independent experiments.
Effect of Indolequinone Treatment on DNA Single Strand Breaks and DNA Cross-Links in Pancreatic Cancer Cell Lines. The ability of the indolequinones to induce DNA damage, including both DNA single-strand breaks and DNA cross-linking, was measured in MIA PaCa-2 and BxPC-3 cell lines using the comet assay. In Fig. 3A, the percentage of DNA in the comet tail was recorded as an indication of the extent of DNA single-strand breaks. H2O2 treatment as a positive control induced significant amounts of DNA single strand breaks in both cell lines; no significant DNA single-strand breaks could be observed in either cell line after 1 h of indolequinone 3 treatment. In Fig. 3B, indolequinone-induced DNA single-strand breaks were compared with two known redox cycling quinones β-lapachone and streptonigrin. The lack of DNA single-strand breaks relative to the redox-cycling quinones suggest that the indolequinone compounds do not induce significant redox cycling and oxidative stress. In Fig. 3C, the extent of DNA cross-linking caused by indolequinone treatment was indirectly measured by analyzing the relative reduction of H2O2-induced DNA migration into the comet tail compared with drug-untreated H2O2 control. The fact that there was no significant difference in percentage tail DNA among all the treatment groups indicated that indolequinone treatment did not result in measurable DNA cross-links in either cell line. Indolequinone-induced DNA damage was also monitored at later time points (up to 4 h); DNA strand breaks were detectable concomitant with apoptosis using both COMET and γH2AX histone blotting (data not shown), using high concentrations of compounds (>500 nM) above the MTT and clonogenic IC50 values.
Fig. 3.

Effect of indolequinone 3 treatment on DNA single-strand breaks and DNA cross-links in MIA PaCa-2 and BxPC-3 cells. DNA damage was determined using the comet assay after treating cells with varying concentrations of indolequinone 3 (IQ3) for 1 h. A, DNA single-strand breaks were expressed as percentage of DNA in the comet tail. H2O2 treatment (200 μM for 20 min) was included as a positive control. B, comparison of drug-induced DNA single strand breaks in MIA PaCa-2 cells between indolequinone 3 and known redox cycling quinones β-lapachone (β-lap) and streptonigrin (SN). Cells were treated for 1 h. The dose range used for β-lapachone was 1 to 10 μM. C, no DNA cross-links were observed after 1-h indolequinone 3 treatment because there was no decrease in percentage tail DNA in drug-treated cells compared with untreated H2O2 control. Results are expressed as the mean ± S.D. of three separate determinations. **, significantly different from nontreatment control cells, p < 0.01.
Antitumor Activity Profile of the Indolequinones in the NCI-60 Tumor Cell Line Panel. Selected indolequinone compounds were screened in the Molecular Cancer Therapeutics Program NCI-60 tumor cell line panel. Indolequinones 2, 3, and 6, representative of the 2-methyl, 2-hydroxymethyl, and 2-unsubstituted indolequinone classes, respectively, were found to have potent antiproliferative activities particularly against renal, colorectal, and melanoma cancers (Fig. 4; the results for 3 and 6 are shown, the result for 2 is similar). It is noteworthy that this unique pattern of antitumor activity is very similar to that of AW464 (Fig. 4), an antitumor quinol compound (Bradshaw et al., 2005) that has been proven to be a potent inhibitor of the thioredoxin reductase/thioredoxin system. In recent work, AW464 and its derivatives were shown to preferentially target thioredoxin reductase rather than thioredoxin (Chew et al., 2008). The similarity in the pattern of antitumor activity (NCI-60) between our compounds and AW464 suggested that thioredoxin reductase might be a critical target of these indolequinones.
Fig. 4.

Antitumor activity of indolequinones in the NCI-60 cell line panel. Growth inhibition screening in the NCI-60 cell line panel was performed by the NCI/National Institutes of Health developmental therapeutics program. The LC50 mean graph was compared side-by-side for compound 3 (left), 6 (middle), and AW464 (right), a recently established thioredoxin reductase inhibitor. The original graph for compound 3 and 6 were obtained from the National Cancer Institute. [The AW464 part of this figure was adapted from Bradshaw TD, Matthews CS, Cookson J, Chew EH, Shah M, Bailey K, Monks A, Harris E, Westwell AD, Wells G, Laughton CA, and Stevens MF (2005) Elucidation of thioredoxin as a molecular target for antitumor quinols. Cancer Res 65:3911-3919. Copyright © 2005 American Association for Cancer Research. Used with permission.]
Inhibition of Thioredoxin Reductase by Indolequinones in Human Tumor Cell Lines. The ability of indolequinone 3 (2-hydroymethyl class) and 9 (2-unsubstituted class) to inhibit thioredoxin reductase activity was tested in the human pancreatic cancer cell line MIA PaCa-2 after 4-h drug treatment. Thioredoxin reductase activity was measured in these cells using the endpoint insulin reduction assay (Fang et al., 2005). A dose-dependent inhibition of thioredoxin reductase activity was observed for both IQ 3 and IQ 9 (Fig. 5A). Consistent with the cytotoxicity data, IQ 9, which was more toxic to pancreatic cancer cells than IQ 3, was more potent at inhibiting TrxR activity in cells. The I50 values for TrxR inhibition were 146 and 82 nM for IQs 3 and 9, respectively. In contrast, an indolequinone analog (designated ACH983; structure shown in Fig. 5A) that is incapable of expelling a leaving group after reduction and does not induce growth inhibition in MIA PaCa-2 cells was unable to inhibit TrxR in MIA PaCa-2 cells. A growth inhibitory IC50 (MTT) for ACH983 could not be determined because of a lack of growth inhibitory effect at concentrations up to 5 μM.
Fig. 5.

Inhibition of thioredoxin reductase activity by indolequinone 3. A, inhibition of thioredoxin reductase activity in MIA PaCa-2 cells by indolequinone 3 (▴), indolequinone 9 (▿), and a nontoxic indolequinone analog, ACH983 (▾). Thioredoxin reductase activity in MIA PaCa-2 cells was measured using the endpoint insulin reduction assay after 4 h of drug treatment. Data were expressed as percent of DMSO-treated control. Data represent mean ± S.D. of three independent determinations. B, inhibition of thioredoxin reductase activity in cell-free system by indolequinone 3. Recombinant rat TrxR (0.5 μM) was preincubated for 5 min with 250 μM NADPH in the presence of NQO2/NRH, then indolequinone 3 was added and incubated for 5 min (maximum inhibition was achieved at 5 min). A 20-μl aliquot was taken out for measurement of TrxR activity using DTNB as substrate. □, reaction system contained every component; ▵, nonreduced TrxR (-NADPH) was not inhibited; ▾, nonreduced indolequinone 3 (-NQO2/NRH) resulted in no inhibition. C, alkylation of the C-terminal selenocysteine of recombinant rat TrxR by indolquinone 3 after reduction by NQO2/NRH inhibited subsequent biotinylation of the C-terminal selenocysteine by biotinylated iodoacetamide. Top, biotinylation of TrxR was detected using streptavidin-conjugated horseradish peroxidase in combination with ECL; bottom, the membrane was stripped and reprobed for total TrxR.
Inhibition of Thioredoxin Reductase by Indolequinone 3 in Cell-Free System. The ability of indolequinone 3 to inhibit recombinant rat TrxR was tested in cell-free system using NQO2/NRH as the reductive activation step (indolequinones tested did not inhibit NQO2 activity under these conditions). When NADPH-reduced TrxR was incubated with NQO2/NRH reduced IQ 3, a dose-dependent inhibition of TrxR activity was observed (Fig. 5B, □). The indolequinone-induced inactivation of TrxR was fast; maximum inhibition was achieved 5 min after addition of compound. Inhibition was NADPH-dependent because the activity of nonreduced TrxR (no preincubation with NADPH) was not affected by NQO2/NRH-reduced IQ 3 (Fig. 5B, ▵), suggesting that the selenocysteine active site of the enzyme was involved in the inhibition by indolequinones. The indolequinone compound also has to be reduced for the inhibition to occur because the enzyme activity of TrxR was unaffected in the absence of NQO2/NRH (Fig. 5B, ▴), indicating that the toxic species responsible for TrxR inhibition was generated after reduction and ejection of the leaving group. The inhibition of TrxR seemed to be irreversible, because desalting of the inhibited TrxR enzyme did not result in recovery of enzyme activity up to 24 h after initial inhibition.
Detection of the TrxR Selenocysteine Redox-Active Site as the Potential Target for Indolequinone Modification. To test whether the IQs were inhibiting TrxR through modification of the selenocysteine active site, the BIAM alkylation assay was carried out to probe the amount of free -SeH groups in the enzyme after inhibition by indolequinone 3. BIAM has been shown to preferentially alkylate with selenocysteine at pH 6.5 (Chew et al., 2008). A dose-dependent decrease in the amount of free selenocysteine in TrxR was observed after incubation with reduced indolequinone (Fig. 5C), suggesting that the indolequinones attack the selenocysteine in the active site, resulting in irreversible inhibition of TrxR.
Antitumor Effects of the Indolequinones in Pancreatic Tumor Xenografts. The in vivo antitumor activity of the indolequinones against pancreatic cancer was tested using MIA PaCa-2 xenograft tumors implanted into nude mice. For these studies, the most active indolequinones in cellular assays compounds 8 and 9 were dosed at 1.0 and 2.5 mg/kg i.p. every other day for 20 days. Tumor volumes were calculated from control and indolequinone-treated mice during drug treatment. Compounds 8 and 9 induced marked growth inhibition of the MIA PaCa-2 xenograft in a dose-dependent manner (Fig. 6). The optimal ratio of volumes of treated and control tumors, based on tumor volume analysis, was 25.2% for indolequinone 9 in the 2.5 mg/kg group.
Fig. 6.

Inhibition of pancreatic xenograft tumor growth in nude mice after treatment with indolequinones. MIA PaCa-2 xenograft tumors were grown on the flanks of nude mice and then treated with selected indolequinones (1.0 and 2.5 mg/kg i.p.) every other day for 20 days. Mice were weighed twice weekly, and neither the control mice nor the treatment groups suffered significant weight loss or any apparent toxicity. Tumor volume was calculated by the formula (L × W2)/2, where L is the longer measurement of the tumor and W is the smaller tumor measurement. Data represents the mean ± S.E.M. of six mice. *, tumor volume in the treatment group was statistically different from controls as determined by the Dunnett's test.
Discussion
ES936 and analogs were developed in our lab originally as mechanism-based inhibitors of NQO1. After reduction to the hydroquinone form by NQO1, ES936 loses the p-nitrophenol leaving group from the indole 3-position, and the iminium ion generated subsequently reacts with tyrosine residues in the active site of NQO1, resulting in the inhibition of enzymatic activity (Winski et al., 2001). ES936 and analogs were found to be active against human pancreatic cancer (Dehn et al., 2006); however, the antitumor activity of ES936 and analogs did not correlate with their ability to inhibit NQO1 (Colucci et al., 2007; Reigan et al., 2007). We corroborated this finding in the current study using three different classes of indolequinone analogs. The 2-hydroxymethyl and 2-unsubstituted indolequinones, which are much poorer inhibitors of NQO1 compared with the 2-methyl indolequinone ES936 (D. Siegel, P. Reigan, M. Colucci, A. Chilloux, C. Moody, and D. Ross, unpublished data), were markedly more toxic to pancreatic cancer cell lines in vitro; and the 2-unsubstituted derivatives that were tested in xenografts were more active than ES936, which was tested in a previous study (Dehn et al., 2006). The dissociation of antitumor activity from NQO1 inhibition suggests that other molecular targets are involved in the antitumor activity of this indolequinone series.
A clear structure-activity relationship was observed among the indolequinones tested. Variation in the substituent on the 2-position of the indole ring had a marked effect on the antitumor potency of the indolequinones. The ES936 class (2-methyl) is the least active, the 2-hydroxymethyl class is severalfold more potent, and the 2-unsubstituted class is the most active at inducing growth inhibition of pancreatic tumor cells. Indolequinones of the 2-hydroxymethyl class (compound 3 as prototype) are more water-soluble than either the 2-methyl class or the 2-unsubstituted classes, which may be an advantage in future drug development.
Indolequinone-induced cytotoxicity in pancreatic cancer cell lines was mediated by apoptotic cell death. A dose-dependent increase in apoptosis was induced by indolequinone 3 treatment in both MIA PaCa-2 and BxPC-3 cells at drug concentrations around the IC50 values (Fig. 2, A and B). This was confirmed by the detection of cleaved caspase 3 and 7 (executioner caspases) in both cell lines (Fig. 2C). The activation of both caspase 8 (extrinsic pathway of apoptosis) and caspase 9 (intrinsic pathway of apoptosis) implies a cross-talk between the two major signaling pathways of apoptosis. However, treatment with the indolequinones did not induce increased levels of superoxide in pancreatic cancer cells (data not shown), suggesting that the indolequinones do not induce oxidative stress. In addition, no significant DNA damage, either single-strand breaks or DNA cross-links (COMET assay), was observed in MIA PaCa-2 or BxPC-3 pancreatic cancer cell lines after 1-h indolequinone treatment (Fig. 3A). The lack of single-strand break induction relative to known redox cycling quinones such as β-lapachone and streptonigrin (Fig. 3B) further confirmed the absence of indolequinone-induced oxidative stress. However, DNA strand breaks were detectable at later time points concomitant with apoptosis using both COMET and γH2AX histone blotting, but only at high drug concentrations above MTT and clonogenic IC50 values. These observations suggested that the cytotoxicity and caspase-dependent apoptosis observed in our study were not caused by redox cycling and oxidative stress.
In an attempt to elucidate potential molecular targets of the indolequinones, three indolequinone compounds, 2, 3, and 6, one from each of the three 2-substitued classes of compound, were screened against the NCI-60 cell line panel, and a distinct pattern of antitumor activity was revealed (Fig. 4). The indolequinones tested were particularly effective against colon, renal, and melanoma cancer cells (the NCI-60 does not contain a pancreatic subpanel). It is noteworthy that this pattern of activity is very similar to that of AW464, an antitumor quinol compound that has recently been established as a thioredoxin reductase inhibitor (Bradshaw et al., 2005; Chew et al., 2008). The thioredoxin system, consisting of Trx, TrxR, and NADPH, plays an essential role in maintaining the redox state of thiols in cellular proteins (Arnér and Holmgren, 2006). The mammalian thioredoxin reductases are selenium-containing flavoproteins with a C-terminal Cys-selenocysteine active site essential for its redox activity (Gromer et al., 1998). Thioredoxin is reduced at the free selenocysteine site of TrxR that is generated when TrxR is reduced by NADPH. Reduced thioredoxin can promote proliferation, inhibit apoptosis, and protect cells against oxidative stress (Powis and Montfort, 2001). TrxR has emerged as an important target for cancer chemotherapy recently, because the level of TrxR and thioredoxin has been shown to be elevated in a variety of human cancer types (Urig and Becker, 2006). TrxR and Trx overexpression have been associated with enhanced tumor proliferation, decreased apoptosis, increased angiogenesis, increased resistance to chemotherapeutic drugs, and reduced survival.
Several antitumor compounds known to generate electrophiles have been shown to be inhibitors of TrxR (Kirkpatrick et al., 1998; Nordberg et al., 1998; Wipf et al., 2004; Bradshaw et al., 2005; Fang et al., 2005; Witte et al., 2005; Lu et al., 2007; Chew et al., 2008). The selenocysteine of thioredoxin reductase has been shown to be very vulnerable to electrophilic attack because of its low pKa and marked reactivity relative to cysteine residues (Arnér and Holmgren, 2006). Alkylation and inhibition of thioredoxin reductase by electrophiles could result in the induction of cell death by affecting the redox state of thioredoxin and other downstream cellular proteins (Lu et al., 2007). Alternatively, alkylated thioredoxin reductase has been shown to have an altered functionality in cancer cells and can drive apoptosis (Anestål and Arnér, 2003; Cassidy et al., 2006). The reduction of the indolequinones by cellular reductases results in the ejection of the leaving group and the generation of an electrophilic alkylating species. Several pieces of data support thioredoxin reductase as a potential target of the indolequinones. First, indolequinone 3 and 9 were found to inhibit TrxR activity in MIA PaCa-2 cells in a dose-dependent manner, suggesting that the indolequinones were capable of inhibiting thioredoxin reductase in tumor cells at drug concentrations approximating IC50 values. It is noteworthy that the more toxic compound IQ 9 is more potent than IQ 3 at inhibiting TrxR in cells. Second, the indolequinone ACH983, which does not have a leaving group and is nontoxic, had no effect on TrxR activity in cells, suggesting that inhibition of TrxR activity is related to the growth-inhibitory effect of the indolequinones and that the ejection of the leaving group is a prerequisite for activity of the indolequinones. Third, indolequinone 3 was also shown to irreversibly inhibit thioredoxin reductase activity in cell-free systems, but only after bioreductive activation by NQO2/NRH, suggesting that a reduction step is needed and that the toxic species are generated after reduction and ejection of the leaving group. The observation that inhibition of TrxR was NADPH-dependent and irreversible suggested that the free selenocysteine active site of TrxR generated via NADPH-mediated reduction was the potential target of indolequinone-derived electrophiles. This was further confirmed by the BIAM alkylation method, which showed the loss of the free selenocysteine residue in the active site of recombinant TrxR after incubation with reduced IQs. A proposed mechanism of TrxR inhibition by the indolequinones is summarized in Fig. 7.
Fig. 7.

A proposed mechanism for the inhibition of TrxR by indolequinones. A, indolequinone activation by two-electron reduction to generate a reactive iminium electrophile. B, reduction of oxidized TrxR by NADPH to generate the reduced C-terminal selenocysteine. C, inhibition of TrxR via alkylation of the reduced C-terminal selenocysteine by the reactive indolequinone iminium electrophile.
In conclusion, we have examined three classes of indolequinone compounds that differ in antitumor potency. All three classes of indolequinones displayed potent antitumor activity against pancreatic tumor cells in vitro; compounds in the most active 2-unsubstituted class demonstrated marked activity against the MIA PaCa-2 xenograft in vivo. The cytotoxicity of the indolequinones in pancreatic cancer cells was mediated by caspase activation and apoptotic cell death. It is noteworthy that the indolequinones were found to inhibit thioredoxin reductase activity both in cells and in cell-free systems, providing a possible explanation for the molecular mechanism of action of the indolequinone series. Validation of thioredoxin reductase as the molecular target of these indolequinone compounds is currently under way.
This research was supported by the National Institutes of Health National Cancer Institute [Grant R01-CA114441] (to D.R.); by FORCE (Friends of the Oncology and Radiotherapy Centre, Exeter, UK); and by the Association for International Cancer Research (to C.J.M.). C.J.M., D.R., and D.S. are scientific cofounders and stockholders in QGenta Inc., which holds an option to license molecules described in this article.
ABBREVIATIONS: ES936, 5-methoxy-1,2-dimethyl-3-[(4-nitrophenoxy)methyl]indole-4,7-dione; NQO1, NAD(P)H:quinone oxidoreductase 1; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Trx, thioredoxin; TrxR, thioredoxin reductase; ACH983, 5-methoxy-1,2-dimethyl-3-[1-oxo-2-(2,4,6-trifluorophenyl)ethyl]indole-4,7-dione; NRH, dihydronicotinamide riboside; NQO2, NRH:quinone oxidoreductase2; DMSO, dimethyl sulfoxide; DTNB, 5,5′-dithiobis(2-nitrobenzoic acid); IQ, indolequinone; BIAM, biotin-conjugated iodoacetamide; z-VAD-fmk, N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone.
References
- Anestål K and Arnér ES (2003) Rapid induction of cell death by selenium-compromised thioredoxin reductase 1 but not by the fully active enzyme containing selenocysteine. J Biol Chem 278 15966-15972. [DOI] [PubMed] [Google Scholar]
- Arnér ES and Holmgren A (2006) The thioredoxin system in cancer. Semin Cancer Biol 16 420-426. [DOI] [PubMed] [Google Scholar]
- Beall HD, Winski S, Swann E, Hudnott AR, Cotterill AS, O'Sullivan N, Green SJ, Bien R, Siegel D, Ross D, et al. (1998) Indolequinone antitumor agents: correlation between quinone structure, rate of metabolism by recombinant human NAD(P)H: quinone oxidoreductase, and in vitro cytotoxicity. J Med Chem 41 4755-4766. [DOI] [PubMed] [Google Scholar]
- Bradshaw TD, Matthews CS, Cookson J, Chew EH, Shah M, Bailey K, Monks A, Harris E, Westwell AD, Wells G, et al. (2005) Elucidation of thioredoxin as a molecular target for antitumor quinols. Cancer Res 65 3911-3919. [DOI] [PubMed] [Google Scholar]
- Cassidy PB, Edes K, Nelson CC, Parsawar K, Fitzpatrick FA, and Moos PJ (2006) Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis 27 2538-2549. [DOI] [PubMed] [Google Scholar]
- Chew EH, Lu J, Bradshaw TD, and Holmgren A (2008) Thioredoxin reductase inhibition by antitumor quinols: a quinol pharmacophore effect correlating to antiproliferative activity. FASEB J 22 2072-2083. [DOI] [PubMed] [Google Scholar]
- Colucci MA (2007) Quinine-based inhibitors of NQO1. PhD Thesis, University of Nottingham, Nottingham, UK.
- Colucci MA, Reigan P, Siegel D, Chilloux A, Ross D, and Moody CJ (2007) Synthesis and evaluation of 3-aryloxymethyl-1,2-dimethylindole-4,7-diones as mechanism-based inhibitors of NAD(P)H:quinone oxidoreductase 1 (NQO1) activity. J Med Chem 50 5780-5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen JJ, Hinkhouse MM, Grady M, Gaut AW, Liu J, Zhang YP, Weydert CJ, Domann FE, and Oberley LW (2003) Dicumarol inhibition of NADPH:quinone oxidoreductase induces growth inhibition of pancreatic cancer via a superoxide-mediated mechanism. Cancer Res 63 5513-5520. [PubMed] [Google Scholar]
- Dehn DL, Siegel D, Zafar KS, Reigan P, Swann E, Moody CJ, and Ross D (2006) 5-Methoxy-1,2-dimethyl-3-[(4-nitrophenoxy)methyl]indole-4,7-dione, a mechanism-based inhibitor of NAD(P)H:quinone oxidoreductase 1, exhibits activity against human pancreatic cancer in vitro and in vivo. Mol Cancer Ther 5 1702-1709. [DOI] [PubMed] [Google Scholar]
- Everett SA, Naylor MA, Barraja P, Swann E, Patel KB, Stratford MR, Hudnott AR, Vojnovic B, Locke RJ, Wardman P, et al. (2001) Controlling the rates of reductively activated elimination from the (indol-3-yl)methyl position of indolequinones. J Chem Soc Perkin Trans 2 843-860. doi: 10.1039/b009652k [DOI]
- Fang J, Lu J, and Holmgren A (2005) Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J Biol Chem 280 25284-25290. [DOI] [PubMed] [Google Scholar]
- Friedlos F, Jarman M, Davies LC, Boland MP, Knox RJ (1992) Identification of novel reduced pyridinium derivatives as synthetic co-factors for the enzyme DT diaphorase (NAD(P)H dehydrogenase (quinone), EC 1.6.99.2). Biochem Pharmacol. 44 25-31. [DOI] [PubMed] [Google Scholar]
- Ghaneh P, Costello E, and Neoptolemos JP (2007) Biology and management of pancreatic cancer. Gut 56 1134-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromer S, Arscott LD, Williams CH Jr, Schirmer RH, and Becker K (1998) Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J Biol Chem 273 20096-20101. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, and Thun MJ (2008) Cancer statistics, 2008. CA Cancer J Clin 58 71-96. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick DL, Kuperus M, Dowdeswell M, Potier N, Donald LJ, Kunkel M, Berggren M, Angulo M, and Powis G (1998) Mechanisms of inhibition of the thioredoxin growth factor system by antitumor 2-imidazolyl disulfides. Biochem Pharmacol 55 987-994. [DOI] [PubMed] [Google Scholar]
- Lewis A, Ough M, Li L, Hinkhouse MM, Ritchie JM, Spitz DR, and Cullen JJ (2004) Treatment of pancreatic cancer cells with dicumarol induces cytotoxicity and oxidative stress. Clin Cancer Res 10 4550-4558. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr Al, and Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193 265-275. [PubMed] [Google Scholar]
- Lu J, Chew EH, and Holmgren A (2007) Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A 104 12288-12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65 55-63. [DOI] [PubMed] [Google Scholar]
- Newsome JJ (2004) Heterocyclic quinones as new bioreductive delivery systems. PhD Thesis, University of Exeter, Exeter, UK.
- Nordberg J, Zhong L, Holmgren A, and Arnér ES (1998) Mammalian thioredoxin reductase is irreversibly inhibited by dinitrohalobenzenes by alkylation of both the redox active selenocysteine and its neighboring cysteine residue. J Biol Chem 273 10835-10842. [DOI] [PubMed] [Google Scholar]
- Powis G and Montfort WR (2001) Properties and biological activities of thioredoxins. Annu Rev Pharmacol Toxicol 41 261-295. [DOI] [PubMed] [Google Scholar]
- Powis G, Wipf P, Lynch SM, Birmingham A, and Kirkpatrick DL (2006) Molecular pharmacology and antitumor activity of palmarumycin-based inhibitors of thioredoxin reductase. Mol Cancer Ther 5 630-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reigan P, Colucci MA, Siegel D, Chilloux A, Moody CJ, and Ross D (2007) Development of indolequinone mechanism-based inhibitors of NAD(P)H:quinone oxidoreductase 1 (NQO1): NQO1 inhibition and growth inhibitory activity in human pancreatic MIA PaCa-2 cancer cells. Biochemistry 46 5941-5950. [DOI] [PubMed] [Google Scholar]
- Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC, and Sasaki YF (2000) Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen 35 206-221. [DOI] [PubMed] [Google Scholar]
- Urig S and Becker K (2006) On the potential of thioredoxin reductase inhibitors for cancer therapy. Semin Cancer Biol 16 452-465. [DOI] [PubMed] [Google Scholar]
- Ward TH, Butler J, Shahbakhti H, and Richards JT (1997) Comet assay studies on the activation of two diaziridinylbenzoquinones in K562 cells. Biochem Pharmacol 53 1115-1121. [DOI] [PubMed] [Google Scholar]
- Winski SL, Faig M, Bianchet MA, Siegel D, Swann E, Fung K, Duncan MW, Moody CJ, Amzel LM, and Ross D (2001) Characterization of a mechanism-based inhibitor of NAD(P)H:quinone oxidoreductase 1 by biochemical, X-ray crystallographic, and mass spectrometric approaches. Biochemistry 40 15135-15142. [DOI] [PubMed] [Google Scholar]
- Wipf P, Lynch SM, Birmingham A, Tamayo G, Jiménez A, Campos N, and Powis G (2004) Natural product based inhibitors of the thioredoxin-thioredoxin reductase system. Org Biomol Chem 2 1651-1658. [DOI] [PubMed] [Google Scholar]
- Witte AB, Anestål K, Jerremalm E, Ehrsson H, and Arnér ES (2005) Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic Biol Med 39 696-703. [DOI] [PubMed] [Google Scholar]
- Yan C, Kepa JK, Siegel D, Stratford IJ, and Ross D (2008) Dissecting the role of multiple reductases in bioactivation and cytotoxicity of the antitumor agent 2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1). Mol Pharmacol 74 1657-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]