

Abstract
p70 S6 kinase (p70S6K) plays an important role in protein translation and cell cycle progression. Increased levels of p70S6K have been associated with drug resistance. In the present study, we have investigated the involvement of p70S6K in DNA damage-induced apoptosis. The DNA damaging agent cisplatin caused a concentration-dependent decrease in full-length p70S6K in small cell lung cancer H69 and non-small cell lung cancer A549 cells with concomitant increase in an approximately 45-kDa fragment. The proteolytic cleavage of p70S6K was inhibited by a broad specificity caspase inhibitor but not by the proteosome or calpain inhibitor. Cell-permeable peptide inhibitor and siRNA against capsase-3 inhibited cisplatin-induced proteolytic cleavage of p70S6K. In vitro-translated p70S6K was cleaved by human recombinant caspase-3. Cisplatin failed to induce cleavage of p70S6K in MCF-7 cells that lack functional caspase-3 but ectopic expression of caspase-3 in MCF-7 cells resulted in the cleavage of p70S6K. p70S6K was primarily cleaved at a noncanonical recognition site Thr-Pro-Val-Asp, after Asp-393. Site-directed mutagenesis of Asp-393 to Ala resulted in protection against cisplatin-mediated apoptosis whereas introduction of the N-terminal cleaved fragment resulted in potentiation of cisplatin-induced apoptosis. These results suggest that p70S6K is a novel substrate for caspase-3 and that the proteolytic cleavage of p70S6K is important for cisplatin-induced apoptosis.
Keywords: p70S6K, caspase, apoptosis
p70 ribosomal S6 kinase (p70S6K) is a serine/threonine protein kinase which phosphorylates ribosomal S6 protein (S6) and promotes translation of a subset of mRNAs necessary for cell cycle progression (1). An elevation/activation of p70S6K has been associated with several cancers and resistance to chemotherapeutic drugs (2-6). Many anticancer drugs kill cancer cells by inducing apoptosis and a failure to induce apoptosis leads to treatment failure. While several reports implicated the involvement of p70S6K in DNA damage-induced apoptosis (3, 5), little is known about how p70S6K regulates apoptosis induced by DNA damaging agents.
Apoptosis is initiated by the activation of caspases, a family of cysteine proteases that cleave after Asp residues (7-9). Caspases are present in most healthy cells as inactive precursors known as procaspases, which undergo proteolytic processing to generate the active enzyme when an apoptotic signal is received (8). While caspase-8 and −9 participate in the initiation phase of apoptosis, caspase-3, −6 and −7 are involved in the execution phase of apoptosis (7-9). Caspase-2 can function both as an initiator as well as an effector caspase (10-12). Proteolytic cleavage of critical cellular proteins, such as poly(ADP-ribose) polymerase (PARP), DNA-dependent protein kinase, lamin B and protein kinase C-δ by executioner caspases has been associated with cell death (7, 13, 14).
Since p70S6K acts downstream of mammalian target of rapamycin (mTOR), most of the studies have utilized rapamycin to investigate the role of p70S6K in apoptosis (15-20). Rapamycin is a pharmacological inhibitor of mTOR and it may act on other targets (21, 22). Since p70S6K is elevated in lung cancer, we recently examined its involvement in cisplatin resistance (3). We inadvertently found that p70S6K was downregulated following treatment with cisplatin. In the present study, we have examined the mechanism of p70S6K downregulation and the impact of p70S6K downregulation on cisplatin-induced apoptosis. We made a novel observation that p70S6K is a substrate for caspase-3. Our results demonstrate that p70S6K is cleaved by caspase-3 at a non-canonical site. Furthermore, proteolytic cleavage of p70S6K was important for cisplatin-induced apoptosis.
EXPERIMENTAL PROCEDURES
Reagents
Polyclonal antibodies to p70S6K was purchased from Cell Signaling Technology, Inc. (Danvers, MA). Monoclonal antibody to GAPDH was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Monoclonal antibody to PARP and caspase-2 were purchased from Pharmingen (San Diego, CA). Polyclonal antibody to caspase-3 was obtained from BioSource/Invitrogen (Carlsbad, CA). Active human recombinant caspase-3 was obtained from Axora (San Diego, CA). MG132 and calpeptin were obtained from Calbiochem (San Diego, CA). Monoclonal antibody to actin was obtained from Amersham (Piscataway, NJ). siRNA SMARTpool against caspases, and non-targeting SMARTpool siRNA were obtained from Dharmacon (Lafayette, CO). Horseradish peroxidase-conjugated goat anti-mouse and donkey anti-rabbit antibodies were obtained from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Polyvinylidene difluoride membrane was from Millipore (Bedford, MA), and enhanced chemiluminescence detection kit was from Amersham (Piscataway, NJ).). Lipofectamine 2000 transfection reagent was obtained from Invitrogen (Carlsbad, CA).
Cell Culture
H69, A549, H358 and MCF-7 cells were maintained in RPMI 1640 supplemented with 2 mM glutamine and 10% (v/v) heat-inactivated fetal bovine serum at 37°C. HeLa cells were maintained in DMEM supplemented with 2 mM glutamine and 7% (v/v) heat-inactivated fetal bovine serum at 37°C. Cells were kept in a humidified incubator at 37°C with 95% air and 5% CO2.
Transfection
Cells were transfected with 1 μg of pcDNA3 or vector containing WT p70S6K or mutant p70S6K using Lipofectamine 2000 (Invitrogen). Briefly, cells were seeded 1 day before transfection. 48 h following transfection, cells were treated as indicated in the text and processed for Western blot analysis. For flow cytometric analysis, cells transfected with an empty vector, wild-type (WT) p70S6K, the caspase cleavage mutant (D393A) or the N-terminal domain were grown in the presence of G418 for 4 days to select for a pool of stable cells. To knockdown caspases, control siRNA or targeted siRNA were introduced into A549 cells using Lipofectamine 2000 (Invitrogen) and manufacturer's protocol (39). Briefly, cells were seeded 1 day before transfection. 48 h following siRNA transfection, cells were treated as indicated in the text and processed for Western blot analysis.
Immunoblot Analysis
Cells were lysed in M-PER mammalian extraction buffer (Pierce, Rockford, IL) containing 1 mM DTT and protease inhibitors. Equal amounts of total protein were electrophoresed by SDS-PAGE and transferred electrophoretically to a poly(vinylidene difluoride) membrane. Western blot analyses were performed as described before (38). The blots were probed with antibody to either GAPDH or actin to control for loading differences.
Site-directed mutagenesis, in vitro translation and caspase cleavage assay
Full-length EE-tagged p70S6K obtained from Dr. Dennis Templeton (40) was cloned into pcDNA3 and synthesized with the T7 Quick TNT kit (Promega) in the presence of [35S]Met (38). Labeled proteins were incubated with human recombinant caspase-3 in 50 mM HEPES, pH 7.5, 0.1% CHAPS, 5 mM dithiothreitol, 10% glycerol, and 0.1 mM EDTA at 37 °C for 1 h. Proteins were separated by SDS-PAGE, and autoradiography was performed with the dried gel. Caspase cleavage site was mutated using the QuikChange site-directed mutagenesis kit (Stratagene) and the manufacturer's protocol. Sequences were confirmed by DNA sequencing.
Assessment of Apoptosis by Flow Cytometry
Cells were treated with varying concentrations of cisplatin for 72 h, harvested, washed with phosphate-buffered saline and fixed in 70% ethanol overnight. Nuclei were then stained with propidium iodide and DNA content was analyzed using the Beckman Coulter Cytomics FC 500 flow cytometer.
RESULTS
Cisplatin induces downregulation of p70S6K in lung cancer cells
Since p70S6K has been associated with cisplatin resistance, we determined the involvement of p70S6K in cisplatin-induced apoptosis. We treated human small cell lung cancer H69 and non-small cell lung cancer A549 cells with different concentrations of cisplatin. Figure 1 shows that cisplatin treatment caused a concentration-dependent decrease in the level of total p70S6K with a concomitant increase in a fragment with approximate molecular mass of 45-kDa in both H69 (Figure 1a) and A549 (Figure 1b) cells. These results suggest that downregulation of p70S6K by cisplatin may be due to proteolytic cleavage of the full-length protein.
Figure 1.
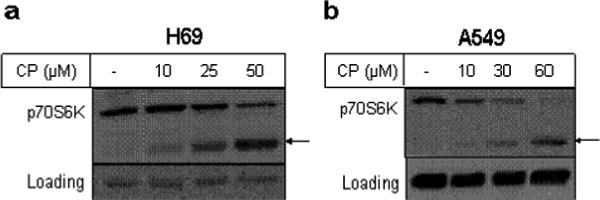
Cisplatin induced downregulation of p70S6K in lung cancer cells. H69 (a) and A549 (b) cells were treated with indicated concentrations of cisplatin for 48 h and 24 h, respectively. Western blot analysis was performed with total cellular extracts using indicated antibodies. GAPDH was used as a loading control. Results are representative of three independent experiments. Arrows indicate cleaved p70S6K band.
Downregulation of p70S6K is mediated by caspases
To understand the mechanism of p70S6K downregulation, we examined the effect of cell-permeable inhibitors of proteasomes (MG132), calpains (calpepetin) and caspases (zVAD) on cisplatin-induced p70S6K downregulation. As shown in Figure 2a, MG132 and calpeptin had little effect on the processing of p70S6K. However, the broad specificity caspase inhibitor z-VAD completely prevented the downregulation of full-length p70S6K as well as the generation of the 45-kDa fragment. These results suggest that cisplatin-induced activation of caspases was responsible for the proteolytic cleavage of p70S6K.
Figure 2.
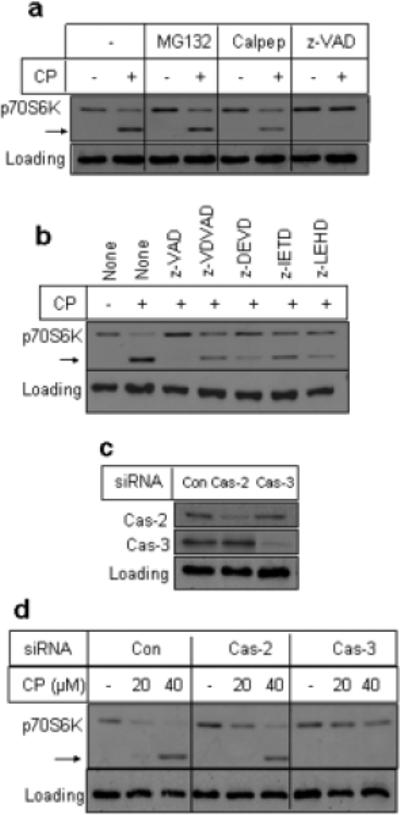
Cisplatin-induced p70S6K downregulation is mediated by caspase-3. (a), A549 cells were treated with 10 µM Mg132, 20 µM calpeptin and 20 µM z-VAD for 30 min prior to incubation with 30 μM cisplatin. (b), A549 cells were treated with 20 μM caspase inhibitors for 30 min prior to incubation with 30 μM cisplatin. z-VAD, broad specificity caspase inhibitor; z-VDVAD, caspase-2 inhibitor; z-DEVD, caspase-3 inhibitor; z-IETD, caspase-8 inhibitor; z-LEHD, caspase-9 inhibitor. Cells were processed following treatment with cisplatin for 24 h. Western blot analysis was performed with total cellular extracts using indicated antibodies. GAPDH was used as a loading control. The arrow indicates cleaved p70S6K band. Results are representative of two independent experiments. (c), A549 cells were transfected with control non-targeting siRNA or siRNA SMARTpool against caspase-2 or caspase-3 as described in “Experimental Procedures.” Western blot analysis was performed with total cellular extracts using indicated antibodies. (d), A549 cells transfected with control and caspase-2 siRNA were treated with or without cisplatin for 24 h and Western blot analysis was performed with total cell extracts. GAPDH was used as a loading control. The arrow indicates cleaved p70S6K band. Results are indicative of two independent experiments.
p70S6K is cleaved by caspase-3
To determine which caspase is responsible for the cleavage of p70S6K, we used cell-permeable peptide caspase inhibitors against caspase-2, −3, −8 and −9. As seen in Figure 2b, the caspase-3 inhibitor z-DEVD and the caspase-9 inhibitor z-LEHD effectively blocked the cleavage of p70S6K triggered by cisplatin whereas caspase-2 inhibitor z-VDVAD and caspase-8 inhibitor z-IETD had modest effects.
Since the peptide caspase inhibitors lack absolute specificity, we used small interfering RNA (siRNA) to deplete a specific caspase to determine which caspase cleaves p70S6K in intact cells. Figure 2c shows that siRNA against caspase-2 and −3 effectively depleted respective caspases as compared to cells transfected with control non-targeting siRNA. Depletion of caspase-3 but not caspase-2 prevented downregulation of p70S6K (Figure 2d). Knockdown of caspase-3 also attenuated cisplatin-induced cleavage of PARP in A549 cells (data not shown). We have also found that knockdown of caspase-8 or −9 did not prevent proteolytic cleavage of p70S6K (data not shown).
To directly demonstrate p70S6K as a substrate for caspase-3, we examined the effect of human recombinant caspase-3 on the cleavage of [35S]-labeled in vitro-translated p70S6K. As seen in Figure 3, in vitro-translated p70S6K appeared at a higher molecular mass (∼75-kDa) because it had an EE epitope tag. Treatment of p70S6K with human recombinant caspase-3 resulted in its cleavage to generate a major fragment above 50-kDa and a smaller fragment of approximately 20-kDa. Since the N-terminal domain of p70S6K contains the EE-tag, the larger fragment above 50-kDa is similar to the 45-kDa cleavage product seen when endogenous p70S6K is cleaved. We also detected a minor band above the larger cleaved fragment. This could be due to an additional minor caspase-3 cleavage site in p70S6K.
Figure 3.
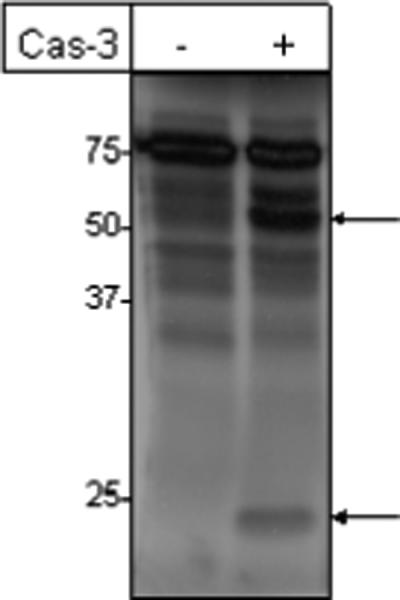
In vitro-translated p70S6K is cleaved by caspase-3. EE-tagged p70S6K cloned into pcDNA3 was synthesized with the T7 Quick TNT kit (Promega) in the presence of [35S]Met and incubated with or without human recombinant caspase-3 as described in the “Experimental Procedures.” The reaction products were separated by SDS-PAGE and analyzed by autoradiography. Arrows indicate cleaved p70S6K bands. The results are representative of three separate experiments.
To further confirm the involvement of caspase-3 on cisplatin-induced proteolytic cleavage of p70S6K in intact cells, we used MCF-7 breast cancer cells that lack functional caspase-3. As shown in Figure 4a, cisplatin failed to downregulate p70S6K even when MCF-7 cells were treated with 50 μM cisplatin. However, ectopic expression of caspase-3 in these cells resulted in downregulation of p70S6K with the emergence of a cleaved band (Figure 4b). These results unambiguously demonstrate that p70S6K is indeed a substrate for caspase-3.
Figure 4.
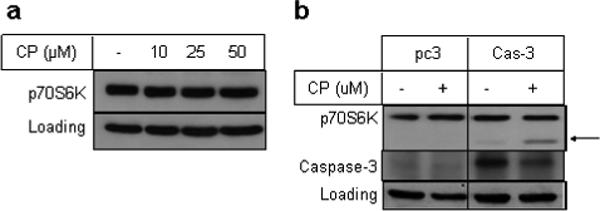
Cisplatin fails to cleave p70S6K in MCF-7 cells. (a), MCF-7 cells were treated with indicated concentrations of cisplatin. (b), MCF-7 cells were transfected with pcDNA3 vector alone or vector containing caspase-3 construct and then treated with 25 μM cisplatin for 24 h. Cells were then processed and Western blot analysis was performed with total cellular extracts using indicated antibodies. GAPDH was used as a loading control. The arrow indicates cleaved p70S6K band.
Caspase-3 cleaves p70S6K after the aspartic acid residue at 393
To determine the functional significance of caspase-3-mediated cleavage of p70S6K, we first wanted to determine the site at which p70S6K is cleaved. p70S6K did not contain any consensus caspase-3 recognition motif Asp-Glu-Xaa-Asp. Since Asp-Ser-Pro-Asp396 resembled weak caspase-3 cleavage recognition motif (Asp-Xaa-Xaa-Asp) in the human p70S6K protein sequence, we first mutated Asp residue at 396 to Ala and determined the ability of human recombinant caspase-3 to cleave mutant D396A p70S6K. As shown in Figure 5a, mutation of p70S6K at D396 to Ala did not prevent its cleavage by human recombinant caspase-3 in the in vitro TNT assay. Based on the molecular size of the cleaved fragment, we mutated several aspartic acid residues to alanine to identify the potential caspase cleavage site and examined the ability of cisplatin to induce cleavage of wild-type and mutant p70S6K in intact cells (Figure 5b). We used EE epitope-tagged p70S6K so that we could differentiate overexpressed p70S6K from the endogenous p70S6K. We were unable to express p70S6K in A549 cells; therefore, we introduced wild-type and mutant p70S6K in HeLa cells. As shown in Figure 5b, antibody against EE epitope detected p70S6K in cells expressing wild-type and mutant constructs. Cisplatin caused cleavage of EE-p70S6K to generate an N-terminal fragment that was recognized by the EE antibody. Mutation of Asp residue at amino acid 322, 342, 378, 383 and 397 to Ala did not prevent cisplatin-induced cleavage of p70S6K. However, mutation of Asp at 393 to Ala (D393A) inhibited the cleavage of p70S6K. These results suggest that p70S6K is primarily cleaved at the Thr-Pro-Val-Asp↓-Ser site in intact cells.
Figure 5.
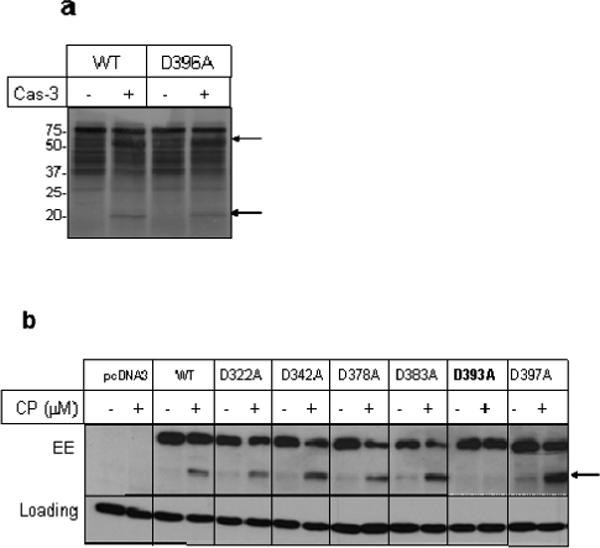
Caspase-3 cleaves p70S6K after the aspartic acid residue at 393. (a), In vitro translated wild-type and D396A mutant p70S6K was treated with human recombinant caspase-3 as described under legend to Figure 3. (b), HeLa cells were transfected with empty vector (pcDNA3) or vector containing wild-type or mutant p70S6K. 24 h after transfection, cells were treated with cisplatin for 18 h. Western blot analysis was performed with total cellular extracts using indicated antibodies. GAPDH was used as a loading control. The arrow indicates cleaved p70S6K band. The results are representative of two separate experiments.
Cleavage of p70S6K by caspase-3 results in augmentation of cell death
To determine if proteolytic cleavage of p70S6K is important for cisplatin-induced cell death, we deleted amino acids 394 to 525 of p70S6K [Δ(394−525)] to create the large N-terminal fragment of the protein (Figure 6a). We introduced pcDNA3 or vector containing wild-type p70S6K (WT-p70S6K), non-cleavable mutant (D393A) or the N-terminal fragment Δ(394−525) in HeLa cells (Figure 6b) and treated cells with cisplatin. We have consistently found that the level of transfected N-terminal fragment was less compared to WT or D393A mutant p70S6K. This may be because cells expressing the N-terminal fragment underwent apoptosis or the truncated p70S6K was less stable inside cells. We quantified apoptosis by the appearance of sub-G1 peak in a flow cytometer (Figure 6c). Based on the results from three independent experiments, 3±2% cells appeared in the sub-G1 phase of HeLa cells transfected with an empty vector, and cisplatin treatment increased apoptotic peak to 20.6±5%. Transfection of wild-type p70S6K alone had little effect on apoptosis and fraction of cells in the sub-G1 peak increased from 4.6±2% to 24.9±4% by cisplatin treatment. Overexpression of D393A mutant alone had little effect on apoptosis and the fraction of cells in the sub-G1 peak was 3.4±1% but it attenutated cisplatin-induced apoptosis to 15.4±1%. In contrast, ectopic expression of the N-terminal fragment alone increased apoptotic peak to 11.3±1% and cisplatin treatment further enhanced the apoptotic peak to 42.7±4%. Thus, cleavage of p70S6K by caspase-3 enhanced cisplatin-mediated apoptosis.
Figure 6.
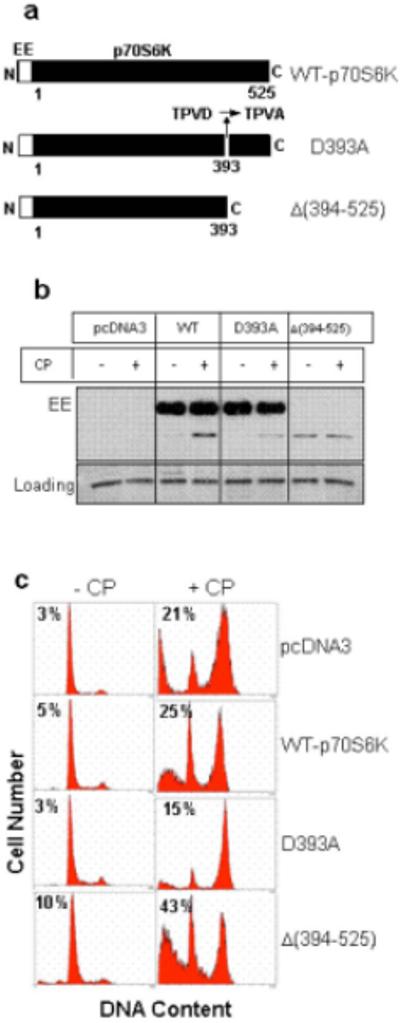
The proteolytic cleavage of p70S6K by caspase-3 enhanced cisplatin-induced cell death. (a), A representative cartoon showing the p70S6K mutants that were generated. (b), HeLa cells were transfected with an empty vector or EE-tagged WT or mutant p70S6K as described under “Experimental Procedures.” Cells were treated with 2 μM cisplatin for 18 h and Western blot analysis was performed with antibody against EE epitope. GAPDH was used to control for loading differences. (c), Nuclei were stained with propidium iodide and analyzed using a flow cytometer. Results are representative of three separate experiments.
Since p70S6K is not cleaved in MCF-7 cells which lack caspase-3, we transfected the N-terminal fragment Δ(394−525) in MCF-7 cells. As shown in Figure 7a, the cells rounded up and detached from the tissue culture dish when transfected with a vector containing the N-terminal fragment but not when transfected with the empty vector pcDNA3. Since MCF-7 cells lack caspase-3 they do not undergo DNA fragmentation. PARP is a substrate for both caspase-3 and −7, and the cleavage of full-length 116-kDa PARP to the 85-kDa cleaved form is a convenient way to monitor cell death by apoptosis. Figure 7b shows that the level of full-length PARP decreased substantially with concominant increase in the level of cleaved 85-kDa PARP when cells were transfected with the N-terminal fragment of p70S6K in MCF-7 cells. Since MCF-7 cells overexpress p70S6K, we also introduced pcDNA3 or vector containing WT-p70S6K, non-cleavable mutant D393A or the N-terminal fragment in lung cancer H358 cells. As shown in Figure 7c, the cleaved fragment of p70S6K but not the caspase cleavage mutant induced cell death. These results suggest that the cleavage of p70S6K may contribute to cell death.
Figure 7.
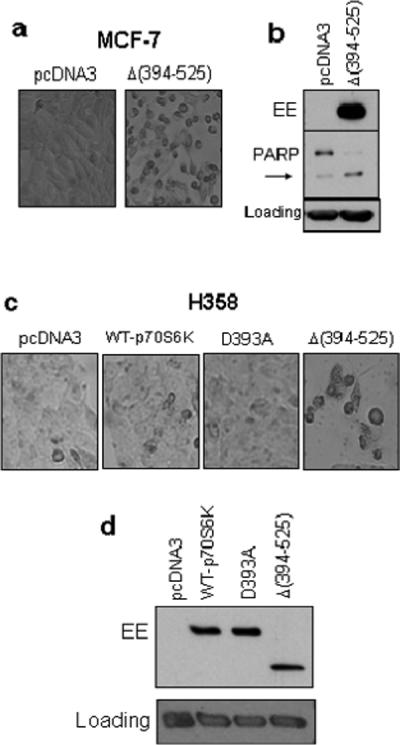
The cleavage product of p70S6K enhanced cell death in MCF-7 and H358 cells. MCF-7 (a, b) or H358 (c, d) cells were transfected with empty vector or vector containing p70S6K constructs. (a and c), 48 h after transfection cellular morphology was examined under a microscope. (b and d), Western blot analysis was performed with total cellular extracts using indicated antibodies. The arrow indicates cleaved PARP. (c) H358 Actin was used as a loading control.
DISCUSSION
In the present study, we made a novel observation that p70S6K is a substrate for caspase-3. While, the best recognized caspase substrates are caspases themselves (13), other caspase substrates include proteins involved in metabolism, cell cycle, DNA repair, signaling pathways and human diseases (23-26). The importance of proteolytic processing of various proteins during apoptosis is well established. Cleavage of certain proteins, such as PARP, DNA-dependent protein kinase, and lamin B has been causally linked to apoptosis (7, 13, 14) whereas cleavage of anti-apoptotic proteins, such as Akt relieves their negative regulation on apoptosis (27). In addition, cleavage of antiapoptotic proteins, such as Bcl-2 converts them to proapoptotic proteins (28). We have shown that proteolytic cleavage of p70S6K was important for apoptosis induced by the DNA damaging agent cisplatin.
The results of our present study demonstrated p70S6K as a substrate for caspase-3. We have shown that the general caspase inhibitor but not calpain or proteasome inhibitor prevented cisplatin-induced downregulation of p70S6K. Although peptide caspase inhibitor of caspase-3 effectively inhibited cisplatin-induced processing of p70S6K, inhibitors of other caspases, especially caspase-9 also attenuated cisplatin-induced cleavage of p70S6K. This may be because these inhibitors lack absolute specificity. However, knockdown of caspase-3 by siRNA also inhibited proteolytic cleavage of p70S6K, demonstrating the requirement for caspase-3 in the cleavage of p70S6K. There is a report suggesting that p70S6K is downregulated in the presence of Fas ligand in Jurkat cells (29). Since p70S6K downregulation was attenuated in Jurkat cells containing mutant caspase-8 compared to cells expressing wild-type caspase-8, the authors suggested that p70S6K downregulation is mediated by caspase-8 (29). We have found that cisplatin induced cleavage of p70S6K in H69 cells which lack caspase-8, suggesting that caspase-8 is not directly involved in the cleavage of p70S6K. Since caspase-8 is an apical caspase and acts upstream of caspase-3, it is possible that activation of caspase-8 results in the activation of caspase-3, causing cleavage of p70S6K. Our in vitro cleavage assay with human recombinant caspase-3 demonstrated that caspase-3 is capable of cleaving p70S6K directly. Furthermore, treatment with as high as 50 μM cisplatin had no effect on the cleavage of p70S6K in MCF-7 cells that lack functional caspase-3 but overexpression of caspase-3 in MCF-7 cells resulted in proteolytic cleavage of p70S6K in response to cisplatin. Cleavage of p70S6K was observed in several different cell lines, including A549, H69, H358 and HeLa cells and in response to several different apoptotic stimuli, including cisplatin, doxorubicin, TNF and TRAIL (data not shown), suggesting that proteolytic cleavage of p70S6K during apoptosis is a general phenomenon.
Cleavage of substrates by caspases may result in their activation or inactivation but there are also proteins that are cleaved with the cleavage having no effect on their functions (23, 30-32). These caspase substrates have been described as ‘innocent bystanders’ (24, 30). Thus, to examine the functional significance of caspase-3-mediated p70S6K cleavage, we first wanted to determine the site at which p70S6K is cleaved. Active caspases cleave key proteins by recognizing a set of four neighboring amino acids in their substrate termed P4-P3-P2-P1 and have a stringent requirement for aspartic acid at the P1 position (13, 25, 33, 34). Although p70S6K contained Asp-Ser-Pro-Asp, which had weak resemblance to the caspase-3 cleavage recognition motif Asp-Glu-Xaa-Asp, the mutation of Asp at 396 to Ala had no effect on caspase-3-mediated cleavage of p70S6K. Using an antibody that recognizes the N-terminal domain of p70S6K, we have demonstrated that the cleavage of p70S6K generates a fragment of an approximate molecular mass of 45-kDa. We have shown that treatment of in vitro translated p70S6K with human recombinant caspase-3 generated two fragments of approximate molecular mass 45-kDa and 20-kDa, representing the cleavage of full-length protein into two fragments. Therefore, we mutated several Asp residues that may serve as recognition sites for caspase-3, and identified Thr-Pro-Val-Asp↓-Ser as the cleavage site for caspase-3. The discovery of substrate cleavage by caspase at non-canonical sites is now becoming increasingly common (35-38). It has been reported that caspase-3 is more tolerant to variations of the cleavage site and the presence of Asp at the P-4 position is not absolutely necessary (36). We have, however, detected a minor cleavage fragment above the major N-terminal cleavage product when in vitro translated EE-p70S6K was incubated with human recombinant caspase-3 (Figure 3). In addition, we could detect a faint band corresponding to the cleavage fragment of p70S6K when HeLa cells expressing D393A mutant p70S6K were treated with cisplatin (Figure 6b). It is conceivable that cleavage of p70S6K at D393 or mutation of Asp-393 to Ala exposes other caspase cleavage sites as we have seen previously during caspase-7-mediated cleavage of PKCε (38).
Since p70S6K is cleaved by caspase-3, the cleavage of p70S6K is a part of the apoptotic process. Our results suggest that the proteolytic cleavage of p70S6K can also contribute to cisplatin-induced apoptosis in several cell lines. However, it remains to be seen if this is a general phenomenon or if it is cell type-dependent. The mutation of Asp 393 residue at the caspase cleavage site to Ala attenuated cisplatin-induced apoptosis. Since the N-terminal fragment of p70S6K was the major cleavage product, we also generated the N-terminal domain by deleting the amino acid residues after caspase-3 cleavage site at 393 to directly demonstrate the importance of proteolytic cleavage of p70S6K on apoptosis. Introduction of the N-terminal domain alone was sufficient to induce cell death and it further enhanced cisplatin-induced cell death. p70S6K regulates multiple cellular functions, including cell proliferation, protein translation and autophagy. Future studies should determine if the ability of the cleaved p70S6K to enhance apoptosis is due to its ability to influence autophagy. Since proteolytic cleavage of p70S6K was associated with cell death, a mutation in the caspase cleavage site may result in resistance to chemotherapeutic drugs. Alternatively, the signaling pathways that lead to caspase-3 activation and cleavage of p70S6K may be defective in drug resistant cells.
In summary, we have made several important and novel observations in the present study. Although the involvement of p70S6K in cell proliferation and protein translation has been extensively studied, we have provided evidence for its new role in apoptosis. We have identified p70S6K as a novel substrate for caspase-3. In addition, we have identified a novel non-canonical cleavage site for caspase-3 in p70S6K. Finally, we have demonstrated that the proteolytic cleavage of p70S6K can contribute to cisplatin-induced apoptosis.
ACKNOWLEDGEMENT
The authors gratefully acknowledge Dr. Dennis Templeton for providing us EE-p70S6K construct, Dr. Swarajit Biswas for cloning caspase-3 and Savitha Sridharan for critical reading of the manuscript.
† This work was supported by the grant [CA071727] to Alakananda Basu from the NIH/NCI. Rohini Dhar was supported by the NSF Score grant [0440334].
1 Abbreviation
- CP
cisplatin
- mTOR
mammalian target of rapamycin
- p70S6K
p70 ribosomal S6 kinase
- PARP
poly(ADP-ribose) polymerase
- PI
propidium iodide
- PKC
protein kinase C
- TNF
tumor necrosis factor-α
- TRAIL
TNF-related apoptosis-inducing ligand
- WT
wild-type
- z-DEVD-fmk
benzyloxycarbonyl-Asp-Glu-Val-Asp-7-amino-4-fluoromethylketone
- z-IETD-fmk
benzyloxycarbonyl-Ileu-Glu-Thr-Asp-fluoromethylketone
- z-LEHD-fmk
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone
- z-VAD-fmk
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
- z-VDVAD-fmk
benzyloxycarbonyl-Val-Asp-Val-Ala-Asp-fluoromethylketone
REFERENCES
- 1.Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5'TOP mRNA translation through inhibition of p70s6k. The EMBO journal. 1997 Jun 16;16(12):3693–3704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barlund M, Forozan F, Kononen J, Bubendorf L, Chen Y, Bittner ML, et al. Detecting activation of ribosomal protein S6 kinase by complementary DNA and tissue microarray analysis. Journal of the National Cancer Institute. 2000 Aug 2;92(15):1252–1259. doi: 10.1093/jnci/92.15.1252. [DOI] [PubMed] [Google Scholar]
- 3.Dhar R, Basu A. Constitutive activation of p70 S6 kinase is associated with intrinsic resistance to cisplatin. Int J Oncol. 2008 May;32(5):1133–1137. [PubMed] [Google Scholar]
- 4.Klos KS, Wyszomierski SL, Sun M, Tan M, Zhou X, Li P, et al. ErbB2 increases vascular endothelial growth factor protein synthesis via activation of mammalian target of rapamycin/p70S6K leading to increased angiogenesis and spontaneous metastasis of human breast cancer cells. Cancer research. 2006 Feb 15;66(4):2028–2037. doi: 10.1158/0008-5472.CAN-04-4559. [DOI] [PubMed] [Google Scholar]
- 5.Liu LZ, Zhou XD, Qian G, Shi X, Fang J, Jiang BH. AKT1 amplification regulates cisplatin resistance in human lung cancer cells through the mammalian target of rapamycin/p70S6K1 pathway. Cancer research. 2007 Jul 1;67(13):6325–6332. doi: 10.1158/0008-5472.CAN-06-4261. [DOI] [PubMed] [Google Scholar]
- 6.Seufferlein T, Rozengurt E. Rapamycin inhibits constitutive p70s6k phosphorylation, cell proliferation, and colony formation in small cell lung cancer cells. Cancer research. 1996 Sep 1;56(17):3895–3897. [PubMed] [Google Scholar]
- 7.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell death and differentiation. 1999 Nov;6(11):1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 9.Salvesen GS, Dixit VM. Caspase activation: The induced-proximity model. Proc Natl Acd Sci USA. 1999;96:10964–10967. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baliga BC, Read SH, Kumar S. The biochemical mechanism of caspase-2 activation. Cell death and differentiation. 2004 Nov;11(11):1234–1241. doi: 10.1038/sj.cdd.4401492. [DOI] [PubMed] [Google Scholar]
- 11.Duan H, Dixit VM. RAIDD is a new ‘death’ adaptor molecule. Nature. 1997 Jan 2;385(6611):86–89. doi: 10.1038/385086a0. [DOI] [PubMed] [Google Scholar]
- 12.Tinel A, Tschopp J. Science. 5672. Vol. 304. New York, NY: May 7, 2004. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. pp. 843–846. [DOI] [PubMed] [Google Scholar]
- 13.Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J. 2004 Dec 1;384(Pt 2):201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997 Nov 14;91(4):443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 15.Beuvink I, Boulay A, Fumagalli S, Zilbermann F, Ruetz S, O'Reilly T, et al. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell. 2005 Mar 25;120(6):747–759. doi: 10.1016/j.cell.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 16.Grunwald V, DeGraffenried L, Russel D, Friedrichs WE, Ray RB, Hidalgo M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer research. 2002 Nov 1;62(21):6141–6145. [PubMed] [Google Scholar]
- 17.Mabuchi S, Altomare DA, Cheung M, Zhang L, Poulikakos PI, Hensley HH, et al. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clin Cancer Res. 2007 Jul 15;13(14):4261–4270. doi: 10.1158/1078-0432.CCR-06-2770. [DOI] [PubMed] [Google Scholar]
- 18.Shi Y, Frankel A, Radvanyi LG, Penn LZ, Miller RG, Mills GB. Rapamycin enhances apoptosis and increases sensitivity to cisplatin in vitro. Cancer research. 1995 May 1;55(9):1982–1988. [PubMed] [Google Scholar]
- 19.Wan X, Helman LJ. Effect of insulin-like growth factor II on protecting myoblast cells against cisplatin-induced apoptosis through p70 S6 kinase pathway. Neoplasia. 2002 Sep-Oct;4(5):400–408. doi: 10.1038/sj.neo.7900242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu C, Wangpaichitr M, Feun L, Kuo MT, Robles C, Lampidis T, et al. Overcoming cisplatin resistance by mTOR inhibitor in lung cancer. Molecular cancer. 2005;4(1):25–34. doi: 10.1186/1476-4598-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calastretti A, Rancati F, Ceriani MC, Asnaghi L, Canti G, Nicolin A. Rapamycin increases the cellular concentration of the BCL-2 protein and exerts an anti-apoptotic effect. Eur J Cancer. 2001 Nov;37(16):2121–2128. doi: 10.1016/s0959-8049(01)00256-8. [DOI] [PubMed] [Google Scholar]
- 22.Law M, Forrester E, Chytil A, Corsino P, Green G, Davis B, et al. Rapamycin disrupts cyclin/cyclin-dependent kinase/p21/proliferating cell nuclear antigen complexes and cyclin D1 reverses rapamycin action by stabilizing these complexes. Cancer research. 2006 Jan 15;66(2):1070–1080. doi: 10.1158/0008-5472.CAN-05-1672. [DOI] [PubMed] [Google Scholar]
- 23.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 24.Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003 Jan;10(1):76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem. 1997 Jul 18;272(29):17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 26.Stennicke HR, Renatus M, Meldal M, Salvesen GS. Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem J. 2000 Sep 1;350(Pt 2):563–568. [PMC free article] [PubMed] [Google Scholar]
- 27.Jahani-Asl A, Basak A, Tsang BK. Caspase-3-mediated cleavage of Akt: involvement of non-consensus sites and influence of phosphorylation. FEBS letters. 2007 Jun 26;581(16):2883–2888. doi: 10.1016/j.febslet.2007.05.033. [DOI] [PubMed] [Google Scholar]
- 28.Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, et al. Science. 5345. Vol. 278. New York, NY: Dec 12, 1997. Conversion of Bcl-2 to a Bax-like death effector by caspases. pp. 1966–1968. [DOI] [PubMed] [Google Scholar]
- 29.Morley SJ, Jeffrey I, Bushell M, Pain VM, Clemens MJ. Differential requirements for caspase-8 activity in the mechanism of phosphorylation of eIF2alpha, cleavage of eIF4GI and signaling events associated with the inhibition of protein synthesis in apoptotic Jurkat T cells. FEBS Lett. 2000 Jul 21;477(3):229–236. doi: 10.1016/s0014-5793(00)01805-6. [DOI] [PubMed] [Google Scholar]
- 30.Timmer JC, Salvesen GS. Caspase substrates. Cell Death Differ. 2007 Jan;14(1):66–72. doi: 10.1038/sj.cdd.4402059. [DOI] [PubMed] [Google Scholar]
- 31.Luthi AU, Martin SJ. The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 2007 Apr;14(4):641–650. doi: 10.1038/sj.cdd.4402103. [DOI] [PubMed] [Google Scholar]
- 32.Utz PJ, Anderson P. Life and death decisions: regulation of apoptosis by proteolysis of signaling molecules. Cell Death Differ. 2000 Jul;7(7):589–602. doi: 10.1038/sj.cdd.4400696. [DOI] [PubMed] [Google Scholar]
- 33.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004 Nov;5(11):897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 34.Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, et al. Substrate specificities of caspase family proteases. J Biol Chem. 1997 Apr 11;272(15):9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- 35.Butkinaree C, Cheung WD, Park S, Park K, Barber M, Hart GW. Characterization of β-NAcetylglucosaminidase Cleavage by Caspase-3 during Apoptosis. The Journal of biological chemistry. 2008 Aug 29;283(35):23557–23566. doi: 10.1074/jbc.M804116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kipp M, Schwab BL, Przybylski M, Nicotera P, Fackelmayer FO. Apoptotic cleavage of scaffold attachment factor A (SAF-A) by caspase-3 occurs at a noncanonical cleavage site. The Journal of biological chemistry. 2000 Feb 18;275(7):5031–5036. doi: 10.1074/jbc.275.7.5031. [DOI] [PubMed] [Google Scholar]
- 37.Lee MW, Hirai I, Wang HG. Caspase-3-mediated cleavage of Rad9 during apoptosis. Oncogene. 2003 Sep 25;22(41):6340–6346. doi: 10.1038/sj.onc.1206729. [DOI] [PubMed] [Google Scholar]
- 38.Basu A, Lu D, Sun B, Moor AN, Akkaraju GR, Huang J. Proteolytic activation of protein kinase C-epsilon by caspase-mediated processing and transduction of antiapoptotic signals. The Journal of biological chemistry. 2002 Nov 1;277(44):41850–41856. doi: 10.1074/jbc.M205997200. [DOI] [PubMed] [Google Scholar]
- 39.Lu D, Huang J, Basu A. Protein kinase Cepsilon activates protein kinase B/Akt via DNA-PK to protect against tumor necrosis factor-alpha-induced cell death. The Journal of biological chemistry. 2006 Aug 11;281(32):22799–22807. doi: 10.1074/jbc.M603390200. [DOI] [PubMed] [Google Scholar]
- 40.Mahalingam M, Templeton DJ. Constitutive activation of S6 kinase by deletion of amino-terminal autoinhibitory and rapamycin sensitivity domains. Molecular and cellular biology. 1996 Jan;16(1):405–413. doi: 10.1128/mcb.16.1.405. [DOI] [PMC free article] [PubMed] [Google Scholar]