

Abstract
The sequential organization of genomes, i.e. the relations between distant base pairs and regions within sequences, and its connection to the three-dimensional organization of genomes is still a largely unresolved problem. Long-range power-law correlations were found using correlation analysis on almost the entire observable scale of 132 completely sequenced chromosomes of 0.5 × 106 to 3.0 × 107 bp from Archaea, Bacteria, Arabidopsis thaliana, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster, and Homo sapiens. The local correlation coefficients show a species-specific multi-scaling behaviour: close to random correlations on the scale of a few base pairs, a first maximum from 40 to 3,400 bp (for Arabidopsis thaliana and Drosophila melanogaster divided in two submaxima), and often a region of one or more second maxima from 105 to 3 × 105 bp. Within this multi-scaling behaviour, an additional fine-structure is present and attributable to codon usage in all except the human sequences, where it is related to nucleosomal binding. Computer-generated random sequences assuming a block organization of genomes, the codon usage, and nucleosomal binding explain these results. Mutation by sequence reshuffling destroyed all correlations. Thus, the stability of correlations seems to be evolutionarily tightly controlled and connected to the spatial genome organization, especially on large scales. In summary, genomes show a complex sequential organization related closely to their three-dimensional organization.
Keywords: Genome organization, Nuclear architecture, Long-range correlations, Scaling analysis, DNA sequence classification
Introduction
While several genomes have been sequenced completely, their complex sequential and three-dimensional organization is largely unknown, despite the interwoven co-evolution of molecular structure, genetic information, and function: e.g. the regulation of genes, their transcription and replication, as well as the differentiation and function of cells are closely connected to this complex sequential and three-dimensional genome organization (Bernardi 1989, 1995; Lamond and Earnshaw 1998; Cremer and Cremer 2001; Dundr and Misteli 2001; Knoch 2002; Knoch 2003). The sequential and three-dimensional genome organization is characterized by its functional, sequential or structural elements. Sequentially, there are e.g. genes coding for proteins and RNAs, regulatory elements and binding sequences that cluster, respectively, in coding and noncoding locally or globally controlled regions. Furthermore, single nucleotide polymorphisms (SNP) and physically unstable breakpoint regions, repeat and duplication regions, and regions classified by their relatively homogenous base pair composition, i.e. isochores, or the abundance of genetic syndromes, i.e. dysfunctional regions related to illnesses, appear (Bernardi 1989, 1995). Structurally, these information elements are encoded in several architectural levels: the DNA double helix, the nucleosome, the chromatin fiber, chromatin fiber folding into a higher-order organization e.g. a further fiber level, chromatin loops and aggregation of these loops in e.g. rosettes, chromosomal interphase and metaphase bands, and whole spatial interphase territories and metaphase chromosomes orchestrated within the nucleus (Lamond and Earnshaw 1998; Cremer and Cremer 2001; Dundr and Misteli 2001; Knoch 2002, 2003).
The general sequential organization of genomes and their evolution has been of major interest since the discovery of DNA, its double-helical structure, and its role as the primary carrier of information and inheritance. The sequential organization covers the relative positioning of sequential and structural elements and their relations on a global, regional, and local (fine-structural) level, as well as the presence and functional effect of these elements and relations on other global, regional, or local levels. In practical terms: what relation has a base pair at position x to a base pair at position y being 102 or even 107 base pairs away and where does the relation originate from? The first investigations determining the chemical properties, sequential order, and self-reproduction of transfer ribonucleic acids (tRNA) showed both an organization into codons of 3 bp and a maximum stability of self-replicated tRNA at ~75 bp (Eigen and Winkler-Oswatitsch 1981a, b; Eigen et al. 1981). This pattern was also found by periodicity analysis in DNA sequences (Shephard 1981a, b), adding to the discussion about the previous hypothesis about a comma-less genetic code (Crick et al. 1957). Thus, the information on the sequence level of genomes evolved in a very defined and delicate interaction with its underlying material carrier—the DNA and other molecular agents involved. Until the development of high-throughput sequencing techniques (i.e. those giving continuous sequences >103 bp) and theoretic advances in correlation analyses (e.g. for texts, time series, languages, and music), further sequences could not be analysed (Mandelbrot 1983; Hsü and Hsü 1990, 1991; Rabinovich et al. 1992).
Long-range correlations at least up to 800 bp were found in the mostly noncoding (76% introns) gene of the human-blood coagulation factor VII by fitting the power spectrum P(f) of the mutual information function to a power law 1/fβ (Li 1991, 1997; Li and Kaneko 1992; Li et al. 1994). Despite limited statistics, the correlation coefficient β appeared to be different between intron- and exon-containing regions. This was explained by repetitive subsequences whose generation should be comparable to the copy-and-error mechanism of modern music composition. Mapping of several sequences to a two-state random walk extended long-range correlations to 103 bp in intron-rich genes (Peng et al. 1992). In genes lacking introns only random correlations were found. These observations were interpreted as non-equilibrium and equilibrium states, being of general fractal nature. Simultaneously, long-range correlations with similar extent and a “1/fβ-noise” character were found (Voss 1992) in 25,000 sequences (the total GenBank Release 68) in ten different organism groups (primate, rodent, mammal, vertebrate, invertebrate, plant, virus, organelle, bacterium, and phage). The use of the (equal-symbol) spectral density function (Reif 1965; Robinson 1974) also revealed a periodicity of 3 bp caused by the codon usage and a periodicity of 9 bp of unknown origin, but characteristic for primates, other vertebrates, and invertebrates.
Besides the widespread astonishment on how such correlations could have persisted and evolved over thousands of base pairs (Amato 1992; Maddox 1992), the reports induced a broad discussion about the validity of the results: On the one hand, the origin of correlations was questioned and attributed to the mere presence of regions with biased base pair composition (Nee 1992; Li et al. 1994; Li 1997). Computer generation of such patchy sequences seemed to support these results. Random mutation and reshuffling of such sequences as well as the bacteriophage lambda destroyed any correlation (Karlin and Brendel 1993). On the other hand, the existence of long-range correlations was totally rejected, since the results by Peng did not show an exactly linear power-law behaviour (Prabhu and Claverie 1992; Chatzidimitriou-Dreismann and Larhammar 1993). A Levy-Walk model for the sequences solved these inconsistencies (Buldyrev et al. 1993). Possibly, it also accounted better for the evolution of long-range correlations than their interpretation as stationary fractional Brownian Motion (Allegrini et al. 1998). Long-range correlations were finally regarded as established by Peng et al. (1994) through the development of detrended fluctuation analysis (DFA), which is an alternative method differentiating local patchiness from long-range correlations and believed to be even more insensitive to local random fluctuations, and by Li (1997). The existence of different correlation behaviours between sequences with and without introns, respectively, was also proven by DFA (Buldyrev et al. 1995). Concerning evolution and persistence, copy-and-deletion models were discussed (Li and Kaneko 1992; Li et al. 1994; Li 1997), and related to the earlier observation of isochores (Bernardi 1989, 1995; Li 2001, 2002), i.e. sequence regions with a relatively homogenous base pair distribution as well as close connections to the globular three-dimensional genome organization (Takahashi 1989; Grossberg et al. 1993; Stanley et al. 1994; Borovik et al. 1994; Mira et al. 2001).
Additionally, methods and results were further validated by comparing different methods (Borovik et al. 1994; Luo et al. 1998) and extended to fractal Cantor pattern recognition (Provata and Almirantis 2000), factorial moments analysis (Mohanty and Narayana-Rao 2000), rescaled range transition matrix analysis (Yu and Chen 2000), as well as two-dimensional visualizations (Yu et al. 2000; Hao et al. 2000a, b). Mechanisms of sequence evolution inspired by language evolution were also proposed (Hao et al. 2000b; de Oliveira 1993; Mackiewicz et al. 1999). Regarding periodicities or correlations connected to codon usage (Voss 1992) or nucleosomal binding sequences (Ambrose et al. 1990), only sequences known to contain these features were analysed and a variety of periodicities were found (Blank and Becker 1996; Liu and Stein 1997; Lowary and Widom 1998; Bailey et al. 2000).
Nevertheless, the complex sequential genome organization and its connection to its three-dimensional organization have remained largely unresolved. Therefore, we analysed the appearance of long-range correlations including its dependence on the scale of analysis (multi-scaling) as well as the presence of fine-structural features by correlation analysis in completely sequenced Archaea, Bacteria, and Eukarya genomes as a virtual microscope for genome organization. The origin of the fine-structured multi-scaling long-range correlations and their relations to the higher-order genome structure is investigated by comparison with artificial sequence designs, destruction of correlations by random sequence reshuffling, and predictions for the three-dimensional genome organization. The species-specificity of the correlations is investigated qualitatively by cluster analysis. In summary, a framework of the complex sequential organization of genomes is established.
Theory
Correlation analysis of DNA sequences and genomes
The analysis of long-range power-law correlations in genetic sequences attempted here, is based on the concentration profile of single nucleotides along the DNA sequence: The square root of the mean-square deviation between the concentration of nucleotides cl in a window of length l and the concentration 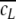 of nucleotides in the entire DNA sequence with length L was calculated
of nucleotides in the entire DNA sequence with length L was calculated
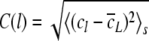 |
1 |
while averaging over all s = L − l + 1 possible window positions. Nucleotides used were adenine (A), thymine (T), guanine (G), and cytosine (C), as well as their grouping into purines (A + G) and pyrimidines (T + C). “Unknown” nucleotides were accounted for by using their general appearance probabilities. Since purines/pyrimidines are complementary, the results are equal and their analysis as base versus nonbase equals mapping the DNA sequence to the trajectory of a one-dimensional random walk. In the following, only the results for purines versus pyrimidines are considered.
For a fractal self-similar sequence such as a random walk the concentration fluctuation function C(l) shows power-law behaviour:
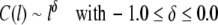 |
2 |
where −1.0 characterizes a negatively, −0.5 a randomly, and 0.0 a positively correlated sequence. The power-law behaviour of C(l) is connected to the power-law behaviour of the minimum and maximum deviation function F(l) ~ lα (Peng et al. 1992), the common autocorrelation function A(l) ~ lγ, and the power spectrum S(f) ~ (1/f)β with frequency f via
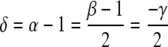 |
3 |
(Prabhu and Claverie 1992; Chatzidimitriou-Dreismann and Larhammar 1993; Borovik et al. 1994; Stanley et al. 1994). C(l) is related to the common autocorrelation function by double summation
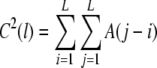 |
4 |
Thus, local random fluctuations are substantially reduced and the analysis leads to a more reliable characterization of the DNA sequence compared with, e.g. A(l) (Peng et al. 1992; Li et al. 1994; Li 1997). Numerical calculation of C(l) by using Eq. 1 in this sequence of operations
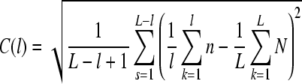 |
5 |
by means of the probabilities for a nucleotide at a certain position n = P(s + k), N = P(k), and e.g. P = 1 for purines and P = 0 elsewhere, leads to extreme numerical instabilities (Fig. 2a). These instabilities were avoided by expansion of Eq. 5 to
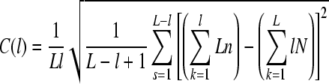 |
6 |
and by exact calculation provided by the GNU multiple precision package GMP. The greater stability is due to the start of deviations from the exact result (Fig. 1b) and becomes especially important for sequences longer than 105 base pairs. To save computing power, the program automatically adjusted the precision (guaranteeing >8 digits) depending on the sequence length.
Fig. 2.

Correlations in Homo sapiens and their fine-structural features: the correlation coefficient δ(l) shows strong positive correlations for human chromosomes (a, b). In general, δ increases from a starting value until a plateaued maximum, before a decrease and a second statistically significant maximum for chromosomes XX, XXI, and XXII. Finally, δ decreases to values characteristic for random sequences and enters the region of fluctuation. Within this general behaviour, a distinct fine-structure is visible in all chromosomes (c, f), which survives averaging (d, e; Figs. 6, 9). The very pronounced local maximum at 11 bp might be related to the double-helical pitch, whereas the local minima and maxima are related to the nucleosome, which is obvious for 146 bp, but less obvious for 172, 205, 228, and 248 bp (d, e). The second maximum around 105 might be related to chromatin loops of the three-dimensional genome organization
Fig. 1.

Introduction to the correlation function C(l) and the correlation coefficient δ(l): a The correlation function C(l) of random sequences shows power-law behaviour as expected for a fractal self-similar sequence (legend in c). The error caused by inexact numerics is shown for C(l) of Homo sapiens chromosome XXI (red line) and the absolute numerical error (b). The slope is the correlation coefficient δ, whose value in the linear region is −0.5 (yellow line), indicating random correlations. The finite sequence length generates a cut-off after which the power-law behaviour breaks down, thus concatenation of two sequences creates a double cut-off. Sequences of Homo sapiens exhibit not only a positively correlated power-law behaviour due to a δ bigger than −0.5, but also four regions (numbers 1–4) with different degrees of correlation. The detailed correlation behaviour is given by the local correlation coefficient δ(l) (c), which fluctuates around −0.5 for random sequences. The fluctuations become larger as the window size approaches the cut-off. Homo sapiens reveals a distinct positively correlated pattern with less fluctuations. To distinguish real from statistical correlations, the standard deviation was computed from 20 random sequences with similar base pair distribution as in Homo sapiens for C(l) (c, thick) and δ(l) (d, thin). The standard deviation of δ(l) shifts only to higher window sizes depending on the sequence length (colors as in c)
To determine the local correlation coefficient δ(l) for the analysis of the general behaviour and fine-structural features of long-range correlations as a function of window size l, the following asymmetric finite difference quotient of second order was applied to 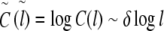 with
with 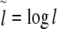 :
:
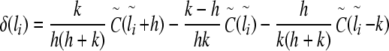 |
7 |
with
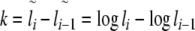 |
8 |
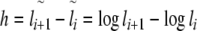 |
9 |
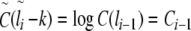 |
10 |
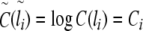 |
11 |
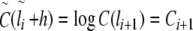 |
12 |
To reduce the enormous computing power needed to calculate C(l) and δ(l) for every possible l, every l from 1 to 104 bp and only 250 logarithmically distributed l for every order of magnitude thereafter were chosen. Calculations were performed on PCs and IBM SP2s, using ~5,000 h of central processing unit (CPU) time. On the latter the analyses were split into jobs of a few minutes, computing a small number of windows each, thus being an extremely efficient “gap-filler” in batch mode of parallel machines. These computations are also ideal for grid computing, e.g. screensaver applications.
Design of artificial random DNA sequences/genomes
To investigate the error behaviour and to determine the origin of various correlation properties, artificial sequences based on different assumptions about their composition were constructed:
Random sequences were constructed from a uniform distribution of base pairs using a R250 random number generator based on 16 parallel copies of a linear shift register with a period of 2250 − 1 (Kirkpatrick and Stoll 1981). This is a far greater period compared with the linear congruent generator used normally and thus produces series with no structure resulting from the random number generator. The R250 generator is computationally faster as well (Maier 1991). The base pair composition was either equal (A, C, G, T, each 25%) or biased by the human base pair distribution (A: 30%, C: 20%, G: 20%, T: 30%). Other biases were not chosen here, since a simple base pair bias does not result in different general, multi-scaling or fine-structure correlation behaviours.
Random block sequences were assembled from blocks of random length with a base pair composition that was biased randomly. The block length B was chosen uniformly from the interval [0, B] or [B − 10%, B + 10%] with B of 5 × 101, 1 × 102, 5 × 102, 1 × 103, 1 × 104, 1 × 105, or 1 × 106. The degree of bias in the base pair composition defining the difference magnitude between blocks, was chosen independently for each block. The concentration of purines per block varied uniformly in [0.5 − D, 0.5 + D] with D of 0.050, 0.075, 0.100, 0.150, 0.200, 0.250, 0.300, 0.350, 0.400, 0.450, or 0.500. One block was appended to the other to compose the random block sequence.
Random codon sequences were composed by random arrangement of codons biased in their frequency of appearance by the codon usage tables provided by the Kazusa DNA Research Institute, Kisarazu, Japan (http://www.kazusa.jp/, downloaded on 13th October 2001). Random arrangement of codons using a uniform distribution, i.e. without an appearance bias of each codon, equals the construction of totally random sequences.
Random gene sequences were designed as hybrids between totally unbiased random sequences and random codon sequences: Codons with a distribution biased by codon usage tables were distributed randomly within connected blocks. These blocks of 999 bp long simulated genes were placed equally, i.e. at a fixed interval, in a totally unbiased random sequence. Therefore, variation of the fraction of blocks in the sequence led to a change not only in their number but also in the length of the random sequence separating them. Thus, random gene sequences resemble some aspects of random block sequences.
Random nucleosome sequences were either based on a 230 bp consensus sequence or two special sequence motifs of nucleosomal binding sites. These were arranged in 2,750 bp long genes/blocks as described for random gene sequences. For the consensus sequence, the three nucleosomal binding sequences 602nvp_rev, 605nvp, and 618nvp_rev found by SELEX experiments were compared (Bailey et al. 2000). Base pairs present in at least two of the sequences were kept constant, while the other base pairs were chosen in an unbiased random manner: nnnGnnTGnT TCnnTnAnACC GAnnnnATCn nTTnnGnnAT GGACTACGnn GnGnCCnnGA GnnnnCnGGT GCCnnnnnCG CnCAATnnnG TnnAGACnnT CTAGnnCCGC TTAAACGCnn nTACnnCTnT CCCCCnCnTA nCGCCAAGGGG nnTnCnnnCT AGTCnCnAnn CACnTGTnnGn AnnCnTAAnC TGCAnnnnnT nACAnnGnCC TTGCC. Blocks, consequently, are not a mere concatenation of the same consensus sequence, and thus irrelevant correlations are reduced. The special sequence motifs GCTCTAGAGC GCTCTAGAGC GCTCTAGAGC and CGTTTAAGCG TATCTAGAGC were suggested (Lowary and Widom 1998) to be the underlying motifs for nucleosomal binding. Blocks contained a random mixture of both sequences with a ratio of 60%:40% according to their length.
Results
The concentration fluctuation function C(l) (Eq. 1) and its exponent the local correlation coefficient δ(l) (Eq. 7) were calculated for 6 high-quality chromosome sequences of Homo sapiens, 3 chromosome sequences of the fruitfly Drosophila melanogaster, all 16 chromosome sequences of the yeast Saccharomyces cerevisiae, 3 preliminary chromosome sequences of the yeast Schizosaccharomyces pombe, 4 chromosome sequences of the plant Arabidopsis thaliana (Table 1), as well as for the completely sequenced genomes of 16 Archaea (Table 2) and 84 sequences of 80 Bacteria, four of which are bi-chromosomal (Table 2). The sequence length varied from 3 × 105 bp for the yeast chromosome III to 2.8 × 107 bp for a fragment of the human chromosome XIV. Longer stretches of undefined base pairs were not present, except for a few nucleotides (especially in the human sequences). Since most Archaea and Bacteria sequences are circular (with the single exception here of Agrobacterium tumefaciens), the linear data base sequences were overlap-free concatenated to cover the entire range of possible sequence correlations.
Table 1.
Attributes and correlation properties of analysed Eukarya genomes
| Eukarya | Accession number | Category | Length [bp] | Correlation properties | ||||
|---|---|---|---|---|---|---|---|---|
| Start [N,R,P] [bp] | 1st max. [bp] | Transition [bp] | 2nd max. [F,R] [bp] | Fine-struc. [C,F] | ||||
| Arabidopsis thaliana Chr. I top + bottom | AE00517 & AE005173 | P | 28,890,626 | P | 60/600 | 171 | 580 | C |
| Arabidopsis thaliana Chr. I top | AE00517 | P | 14,221,746 | P | 60/550 | 160 | 550 | C |
| Arabidopsis thaliana Chr. I bottom | AE005173 | P | 14,668,880 | P | 60/650 | 185 | 620 | C |
| Arabidopsis thaliana Chr. II | AE002093 | P | 19,646,744 | P | 60/680 | 180 | 660 | C |
| Arabidopsis thaliana Chr. IV | NC001268 | P | 17,549,956 | P | 60/650 | 160 | 680 | C |
| Saccharomyces cerevisiae Chr. I | NC001133(1) | Y | 230,203 | P | 500 | – | – | C |
| Saccharomyces cerevisiae Chr. II | NC001133(1) | Y | 813,139 | P | 435 | – | – | C |
| Saccharomyces cerevisiae Chr. III | NC001133(2) | Y | 316,613 | P | 450 | – | – | C |
| Saccharomyces cerevisiae Chr. IV | NC001133(2) | Y | 1,531,929 | P | 410 | – | – | C |
| Saccharomyces cerevisiae Chr. V | NC001133(2) | Y | 576,870 | P | 640 | – | – | C |
| Saccharomyces cerevisiae Chr. VI | NC001133(1) | Y | 270,148 | P | 640 | – | – | C |
| Saccharomyces cerevisiae Chr. VII | NC001133(1) | Y | 1,090,936 | P | 540 | – | – | C |
| Saccharomyces cerevisiae Chr. VIII | NC001133(2) | Y | 562,638 | P | 620 | – | – | C |
| Saccharomyces cerevisiae Chr. IX | NC001133(1) | Y | 439,885 | P | 460 | – | – | C |
| Saccharomyces cerevisiae Chr. X | NC001133(1) | Y | 745,440 | P | 460 | – | – | C |
| Saccharomyces cerevisiae Chr. XI | NC001133(1) | Y | 666,445 | P | 580 | – | – | C |
| Saccharomyces cerevisiae Chr. XII | NC001133(1) | Y | 1,078,173 | P | 560 | – | – | C |
| Saccharomyces cerevisiae Chr. XIII | NC001133(1) | Y | 924,430 | P | 560 | – | – | C |
| Saccharomyces cerevisiae Chr. XIV | NC001133(1) | Y | 784,330 | P | 450 | – | – | C |
| Saccharomyces cerevisiae Chr. XV | NC001133(1) | Y | 1,091,284 | P | 550 | – | – | C |
| Saccharomyces cerevisiae Chr. XVI | NC001133(1) | Y | 875,709 | P | 420 | – | – | C |
| Schizosaccharomyces pombe Chr. I | V-011213 | Y | 5,602,103 | P | 900 | 1.2*^4 | R1.0*^5 | C |
| Schizosaccharomyces pombe Chr. II | V-011213 | Y | 4,430,733 | P | 850 | 1.4*^4 | R1.0*^5 | C |
| Schizosaccharomyces pombe Chr. III | V-011213 | Y | 2,467,649 | P | 610 | 2.0*^4 | R1.0*^5 | C |
| Drosophila melanogaster Chr. 2L | 2L-1011210 | I | 2,265,1956 | P | 40/3,100 | – | – | C |
| Drosophila melanogaster Chr. 2R | 2R-2-011210 | I | 14,631,223 | P | 40/3,800 | – | – | C |
| Drosophila melanogaster Chr. 3R | 3R-1-011210 | I | 28,460,979 | P | 40/3,400 | – | – | C |
| Homo sapiens sapiens Chr. XI | NT009151 | Pr | 19,322,668 | P | 200 | 1.0*^5 | R3.5*^5 | N |
| Homo sapiens sapiens Chr. XIV | NT026437 | Pr | 28,334,988 | P | 200 | 1.7*^4 | F1.4*^5 | N |
| Homo sapiens sapiens Chr. XV | NT010321 | Pr | 9,197,381 | P | 200 | 2.0*^4 | 1.0*^5 | N |
| Homo sapiens sapiens Chr. XX | NT011362 | Pr | 24,982,240 | P | 200 | 1.2*^4 | 1.3*^5 | N |
| Homo sapiens sapiens Chr. XXI | Hattori51 | Pr | 33820172 | P | 200 | 2.0*^4 | 1.3*^5 | N |
| Homo sapiens sapiens Chr. XXII | TIGR, WLC010213 | Pr | 33,705,278 | P | 200 | 1.2*^4 | 2.0*^4 | N |
To simulate the whole chromosome I of Arabidopsis thaliana the sequences of the top and bottom arm that are separated by an unsequenced centromeric region were concatenated. Accession numbers of Saccharomyces cerevisiae are annotated with the version in brackets. The sequences of Schizosaccharomyces pombe are the preliminary from 10/12/2001. The sequences of Drosophila melanogaster are the three largest available sequences with “Gold Standard Quality” downloaded on 10/12/2001 from http://www.fruitfly.org/. A human sequence of chromosome XXI was used (Hattori et al. 2000) with no apparent accession number. The sequence of chromosome XXII was downloaded from The Institute for Genome Research (TIGR) website at http://www.tigr.org/. Taxonomic categories are plant (P), yeast (Y), insect (I), and primate (Pr). Properties of correlation are classified with N for negative (crossing the random regime with a value of 0.5 in bp), R for random, and P for positive correlation coefficients at window sizes of a few base pairs. The transition to the second maximum can be a minimum (M). Second maxima are dividable into those with a fine-structure not attributable to statistics (F) and those not clearly separable from fluctuations based on the cut-off length of sequences (R). The fine-structure is categorized by codon usage (C) and nucleosomal binding (N)
Table 2.
Attributes and correlation properties of the Archaea (A) and Bacteria (B) genomes analysed
| Archaea and Bacteria | Accession number | Category | Length [bp] | Correlation properties | |||||
|---|---|---|---|---|---|---|---|---|---|
| Start [N,R,P] [bp] | 1st max. [bp] | Transition [L,PL,M] [bp] | 2nd max. [K,F,R,T] [bp] | Fine-struc. [C,F] | Class [A,A′] [A′′,B] | ||||
| Aeropyrum pernix K1 | BA000002 | A | 1,669,695 | P | 460 | M7.0*^4 | F6.0*^4 | C | A |
| Archaeoglobus fulgidus DSM4304 | AE000782 | A | 2,178,400 | P | 420 | M1.8*^4 | R | C | A |
| Halobacterium NRC-1 | AE004437 | A | 2,014,239 | N < 21 | 900 | L | F1.0*^5 | C | B |
| Methanocaldococcus jannaschii L77117 | L77117 | A | 1,664,957 | P | 500 | M2.0*^4 | R2.3*^5 | C | A |
| Methanopyrus kandleri AV19 | AE094390 | A | 1,694,969 | P | 630 | M1.0*^4 | F1.0*^5 | C | A |
| Methanosarcina acetivorans C2A | AE010299 | A | 5,751,492 | P | 540 | L | R3.0*^5 | C | A′′ |
| Methanosarcina mazei Goe1 | AE008384 | A | 4,096,345 | P | 825 | M2.5*^4 | F1.5*^5 | C | A′′ |
| Methanobacterium thermoautotrophicum delta-H | AE000666 | A | 1,751,377 | P | 1,100 | M3.7*^4 | R2.0*^5 | C | A′′ |
| Pyrobaculum aerophilum | AE009441 | A | 2,222,430 | P | 385 | Ms | R | C | A |
| Pyrococcus abyssi | AL096836 | A | 1,765,118 | P | 400 | M2.0*^4 | R | C | A |
| Pyrococcus furiosus DSM3638 | AE009950 | A | 1,908,256 | P | 400 | M1.5*^4 | R3.0*^5 | C | A |
| Pyrococcus horikoshii | BA000001 | A | 1,738,505 | P | 400 | M2.0*^4 | R | C | A |
| Sulfolobus solfataricus | AL596259 | A | 2,992,245 | P | 370 | M8.6*^4 | R | C | A |
| Sulfolobus tokodaii | BA000023 | A | 2,694,756 | P | 385 | M6.6*^4 | R | C | A |
| Thermoplasma acidophilum | AL139299 | A | 1,564,906 | P | 440 | M3.0*^4 | R | C | A |
| Thermoplasma volcanium | BA000011 | A | 1,584,854 | P | 440 | M6.0*^4 | R | C | A |
| Agrobacterium tumefaciens C58 circular chromosome (Cereon) | AE007869 | B | 2,841,581 | N < 4 | 800 | M | T3.2*^5 | C | A′ |
| Agrobacterium tumefaciens C58 linear chromosome (Cereon) | AE007870 | B | 2,074,782 | N < 4 | 700 | M | T3.2*^5 | C | A′ |
| Agrobacterium tumefaciens C58 circular chromosome (University of Washington) | AE007870 | B | 2,074,782 | N < 4 | 700 | M | T3.2*^5 | C | A′ |
| Agrobacterium tumefaciens C58 linear chromosome (University of Washington) | AE007870 | B | 2,074,782 | N < 4 | 700 | M | T3.2*^5 | C | A′ |
| Aquifex aeolicus VF5 | AE000657 | B | 1,551,335 | P | 370 | M1,6*^4 | – | C | A |
| Bacillus halodurans | BA000004 | B | 4,202,353 | P | 1,000 | T5.0*^3 | K1.0*^5 | C | B |
| Bacillus subtilis subsp. subtilis 168 | AL009126 | B | 4,214,814 | P | 850 | T3.5*^3 | F1.0*^5 | C | B |
| Borrelia burgdorferi B31 | AE000783 | B | 910,681 | P | 600 | M5.0*^4 | R1.5*^5 | C | A |
| Brucella melitensis 16 M Chr. I | AE008917 | B | 2,117,144 | N | 720 | M1.0*^4 | T2.5*^6 | C | A′ |
| Brucella melitensis 16 M Chr. II | AE008918 | B | 1,177,787 | N | 830 | M2.0*^4 | T1.0*^5 | C | A′ |
| Buchnera aphidicola APS | BA000003 | B | 640,681 | P | 850 | M6.6*^4 | R1.5*^5 | C | A |
| Campylobacter jejuni subsp. jejuni NCTC 11168 | AL111168 | B | 1,641,481 | P | 660 | M2.0*^4 | T1.2*^5 | C | A′ |
| Caulobacter crescentus CB15 | AE005673 | B | 4,016,947 | N < 65 | 790 | M7.0*^3 | T4.0*^5 | C | A′ |
| Chlamydia muridarum | AE002160 | B | 1,069,393 | P | 650 | L | K6.0*^4 | C | B |
| Chlamydia trachomatis | AE001273 | B | 1,042,519 | P | 650 | L | F6.0*^4 | C | B |
| Chlamydophila pneumoniae AR39 | AE002161 | B | 1,229,784 | P | 500 | PL | F1.0^5 | C | B |
| Chlamydophila pneumoniae CWL029 | AE001363 | B | 1,230,230 | P | 500 | PL | F1.0^5 | C | B |
| Chlamydophila pneumoniae J138 | BA000008 | B | 1,228,266 | P | 500 | PL | F1.0^5 | C | B |
| Clostridium acetobutylicum ATCC824 | AE001437 | B | 3,940,880 | P | 630 | PL | K1.0*^5 | C | B |
| Clostridium perfringens 13 | BA000016 | B | 3,031,430 | P | 615 | PL | K.10*^5 | C | B |
| Corynebacterium glutamicum | AX114121 | B | 330,400 | N < 12 | 1,000 | PL | K.4*^5 | C | B |
| Deinococcus radiodurans Chr. I | AE001825 | B | 2,648,577 | N < 5 | – | – | – | C | A′ |
| Deinococcus radiodurans Chr. II | AE001825 | B | 412,344 | N < 5 | – | – | – | C | n.d. |
| Escherichia coli K12 | U00096 | B | 4,639,221 | N < 5 | 860 | M8*^3 | F2.2*^4 | C | B |
| Escherichia coli O157:H7-EDL933 | AE005174 | B | 5,468,733 | N < 5 | 1,000 | PL | F2.2*^4 | C | B |
| Escherichia coli O157:H7-RIMD0509952 | BA000007 | B | 5,498,450 | N < 5 | 1,000 | PL | F2.2*^5 | C | B |
| Fusobacterium nucleatum subsp. nucleatum ATCC 25586 | AL731704 | B | 2,174,500 | P | 1,400 | PL | R3.0*^5 | C | B |
| Haemophilus influenzae Rd KW20 | L42023 | B | 1,830,023 | P | 720 | M7.5^4 | R1.8*^5 | C | A |
| Helicobacter pylori 26695 | AE000511 | B | 1,667,825 | P | 860 | M6.0*^4 | T2.0*^5 | P | A |
| Helicobacter pylori J99 | AE001439 | B | 1,643,831 | P | 860 | M1.8^4 | T2.0*^5 | C | A |
| Lactococcus lactis subsp. lactis IL1403 | AE005176 | B | 2,365,589 | P | 950 | L | K1.0*^5 | C | B |
| Listeria innocua Clip 11262 | AL592020 | B | 3,011,208 | P | 600 | L | K1.0*^5 | C | B |
| Listeria monocytogenes EGD | AL591824 | B | 2,944,528 | P | 600 | L | K1.0*^5 | C | B |
| Mesorhizobium loti MAFF303099 | BA000012 | B | 7,036,071 | N < 10 | 660 | M5.0^4 | T8*^5 | C | A′ |
| Mycobacterium leprae TN | AL450380 | B | 3,268,203 | N < 25 | 700 | L | F1.7*^5 | C | B |
| Mycobacterium tuberculosis CDC1551 | AE000516 | B | 4,403,661 | N < 22 | 1,000 | L | F3.6*^5 | C | B |
| Mycobacterium tuberculosis H37Rv | AL123456 | B | 4,411,529 | N < 22 | 1,000 | L | F3.6*^5 | C | B |
| Mycoplasma genitalium G37 | L43967 | B | 580,074 | P | 900 | PL | 6.0*^4 | C | B |
| Mycoplasma pneumoniae M129 | U00089 | B | 816,394 | P | 1,000 | PL | F8*^4 | C | B |
| Mycoplasma pulmonis UAB-CTIP | AL445566 | B | 963,879 | P | 630 | M8.3*^4 | R1.3*^5 R2.6*^5 | C | A |
| Neisseria meningitidis Sero Group A, Strain Z2491 | AL157959 | B | 2,184,406 | R | 1,100 | PL | F3.5*^5 | C | B |
| Neisseria meningitidis Sero Group B, Strain MC58 | AE002098 | B | 2,272,351 | R | 1,300 | PL | F3.5*^5 | C | B |
| Nostoc PCC7120 | BA000019 | B | 6,413,771 | R | 690 | M3.5*^4 | F1.6*^5 | C | B |
| Pasteurella multocida PM70 | AE004439 | B | 2,257,487 | R | 650 | PL | F1.0*^5 | C | B |
| Pseudomonas aeruginosa PA01 F(13) | AE004091 | B | 6,264,403 | N < 11 | 950 | L | F3.8*^5 | C | B |
| Ralstonia solancearum GMI1000 | AL646052 | B | 3,716,413 | N | 1,900 | PL | K2.0*^5 | C | B |
| Rickettsia conorii Malish 7 | AE006914 | B | 1,268,755 | P | 690 | ML | R2.5*^5 | C | B |
| Rickettsia prowazekii Madrid-E | AJ235269 | B | 1,111,523 | P | 690 | M1.2*^5 | R2.5*^5 | C | A |
| Salmonella enterica subsp. enterica serovar Typhi CT18 | AL513382 | B | 4,809,037 | N < 4 | 950 | L | T2.5*^5 | C | B |
| Salmonella typhimurium LT2 | AE006468 | B | 4,857,432 | N < 4 | 950 | L | T2.5*^5 | C | B |
| Sinorhizobium meliloti 1021 | AL591688 | B | 2,160,837 | P | 750 | M1.0*^4 | F3.0*^5 | C | B |
| Staphylococcus aureus subsp. aureus MW2 | BA000033 | B | 2,820,462 | R | 1,000 | L | K8.3*^4 | C | B |
| Staphylococcus aureus subsp. aureus Mu50 | BA000017 | B | 2,878,134 | R | 1,000 | L | K8.6*^4 | C | B |
| Staphylococcus aureus subsp. aureus N315 | BA000018 | B | 2,813,641 | R | 1,000 | L | K1.2*^5 | C | B |
| Streptococcus pneumoniae R36 | AE007317 | B | 2,038,615 | P | 860 | L | F8.3*^5 | C | B |
| Streptococcus pneumoniae TIGR4 | AE005672 | B | 2,160,837 | P | 860 | L | F8.3*^5 | C | B |
| Streptococcus pyogenes M1 GAS | AE004092 | B | 1,852,441 | P | 860 | L | F8.3*^4 | C | B |
| Streptococcus pyogenes MGAS8232 | AE014074 | B | 1,900,521 | P | 850 | L | F8.3*^4 | C | B |
| Streptomyces coelicolor A3(2) | AL644882 | B | 8,667,507 | N < 5 | 720 | M2.0*^4 | F1.0*^5 | C | B |
| Synechocystis sp. PCC6803 | AB001339 | B | 3,573,470 | P | 500 | M8.0*^4 | R1.0*^5 | C | A |
| Thermoanaerobacter tengcongensis | AE008691 | B | 2,689,445 | P | 580 | M2.5*^3 | K1.0*^5 | C | B |
| Thermotoga maritima | AE000512 | B | 1,860,725 | P | 690 | M6.0*^5 | R1.8*^5 | C | A |
| Treponema pallidum subsp. pallidum Nicholas | 2275888 | B | 1,137,944 | P | 1,000 | L | K | C | B |
| Ureaplasma urealyticum | 1503438 | B | 751,719 | R | 900 | M2.5*^4 | F1.0*^5 | C | A |
| Vibrio cholerae O1 biovar eltor N16961 Chr. I | AE003852 | B | 2,961,116 | N < 5 | 630 | L | F2.0*^5 | C | B |
| Vibrio cholerae O1 biovar eltor N16961 Chr. II | AE003853 | B | 1,072,311 | N < 5 | 630 | L | F1.0*^5 | C | B |
| Wigglesworthia glossinidia endosymbiont of Glossina brevipalpis | BA000021 | B | 697,721 | P | 562 | M2.0*^4 | F | C | A |
| Xanthomonas axonopodis pv. citri 306 | AE008923 | B | 5,175,554 | N < 13 | 3.7*^3 | L | F3.5*^5 | C | B |
| Xanthomonas campestris pv. campestris ATCC 33913 | AE008922 | B | 5,076,188 | N < 13 | 3.7*^3 | L | F2.9*^5 | C | B |
| Xylella fastidiosa 9a5c | AE003849 | B | 2,679,306 | N < 10 | 2,200 | M4.0*^4 | R | C | A′′ |
| Yersinia pestis CO92 | AL590842 | B | 4,653,728 | N < 5 | 900 | L | F1.7*^5 | C | B |
Properties of correlation are classified with N for negative (crossing the random regime is given in bp), R for random, and P for positive correlation coefficients for window sizes of a few base pairs. The transition between maxima is characterized by a steadily linear increase (L), a plateau with a more or less fast increase to the second maximum (P) or by a distinct minimum (M). Second maxima are dividable into those in the shape close to a cap without much structure (K), those with a plateau including a fine-structure not attributable to statistics (F), those not clearly separable from fluctuations based on the cut-off length of sequences (R), and those being a mixture of F and R (T). The general fine-structure is categorized into codon usage (C) or another distinct fine-structure (F). The more general classification into the classes A, A′, A′′ and B (Fig. 10) is based on qualitative visual inspection as well as Pearson correlation and unweighted pair group method with arithmetic mean (UPGMA) cluster analysis of the whole curves in Fig. 10, i.e. the classification in the previous columns might lead to slightly different groupings
The exact calculation of C(l), in principal being only a simple counting problem, required the use of a numerically stable algorithm (Eq. 6) and the multiple precision package GMP for the longest sequences. This prevented fast-growing numerical errors and function breakdowns for large l (Fig. 1a, b). The calculation of δ(l) was also exact, considering the chosen resolution of l to save computer power: from l to 104 bp every l, and for >104 bp 250 logarithmically distributed l were selected. Thus, for l > 104 bp local variances in C(l) resulting in correlations δ(l) with high frequencies are in general smoothed out, although they could also increase the fluctuation depending on the local non-static behaviour of C(l) for a given triplet of l used to calculate δ(l).
Appearance of long-range correlations
In all sequences analysed, the concentration fluctuation function C(l) shows power-law behaviour with varying slopes, indicating a nontrivial degree of correlation (Figs. 1a, 10b). This is corroborated by the local correlation coefficient δ(l) with varying values significantly >−0.5, the characteristic value for random sequences (Fig. 1c). Thus, positive long-range correlations of nonrandom origin were found across almost the entire sequence scale, i.e. 107, but certainly up to 105 to 106 bp respective to the sequence length, in all analysed sequences (Figs. 2a, b; 3a–c; 4a–d; 5a–c; 6a–l; 10a).
Fig. 10.

Comparison of averages of correlation coefficients δ(l) for all genomes analysed: a Shown are the averages taken over δ(l) for each of the Eukarya genomes, the Archaea and the classes A (without the Archaea), A′, A′′, and B. Comparison reveals that only Homo sapiens does not show the zigzag pattern due to the codon usage, although it shows a fine-structure not present in any other genome or class. All genomes show a maximum between window sizes of 100–1,000 bp of which only the maxima present in Homo sapiens seem to be connected to the nucleosome. The classes A′ and B show a second maximum after a decrease of δ with very high correlations for window lengths of ~105 bp in contrast to the other genomes. Only Homo sapiens shows also a second maximum, although in the mean it is washed out and is not statistical significantly in contrast to analysis of some of the single human chromosomes analysed. b For comparison purposes, the means of the concentration fluctuation function C(l) for the same averages are shown
Fig. 3.

Correlations in Drosophila melanogaster: the sequences of Drosophila melanogaster analysed show positive correlations (a–c). The averaged δ (b) has two main maxima (40 and 3,400 bp), with several local maxima in-between (108, 146, 251, 850, 2,033, and 2,370 bp), and two major minima (302 and 1,100 bp). These features appear in all chromosomes (c), similar to those of Arabidopsis thaliana (Fig. 5)
Fig. 4.

Correlations in Saccharomyces cerevisiae and Schizosaccharomyces pombe: correlations appear up to 104 to 105 bp for Saccharomyces cerevisiae and up to 105.5 bp for Schizosaccharomyces pombe. The general behaviour of δ(l) is characterized by an increase of δ to maxima around 500 and 900 bp, respectively. Thereafter, δ decreases until random correlations are reached for Saccharomyces cerevisiae, or in case of Schizosaccharomyces pombe a minimum between 1.2 to 2.0 × 104 bp followed by a second maximum around 105 bp
Fig. 5.

Correlations in chromosomes of Arabidopsis thaliana: Arabidopsis thaliana reveals positive correlations (a–d). The averaged δ (c) increases to two main maxima (60 and 600 bp), two small local maxima in-between (112 and 270 bp), and one major minimum (178 bp). These features appear in all chromosomes (d) and are similar to those of Drosophila melanogaster (Fig. 3). The zigzag visible fine-structure is due to correlations based on the codon usage (b) and is still present for large window sizes
Fig. 6.

Correlations in Archaea and Bacteria genomes and their classification: the analysis of the correlation coefficient δ(l) of Archaea (a, b) and Bacteria (c–l) reveals behaviours separable into four major classes, referred to as A, A′, A′′, and B, as revealed by cluster analysis. Members of each class were averaged, respectively (Fig. 10). In general, Archaea and Bacteria are characterized by a first maximum below 103 bp with decreased height and increased position, influenced by a second maximum. The transition exhibits a minimum or a saddle point also connected to the growing presence of the second maximum. The often extreme degree of correlation is unlike that found in any of the Eukarya. A prime example for Archaea is Archeoglobus fulgidus, for class A Aquifex aeolicus, and for class A′ Campylobacter jejuni. Class A′′ is a mixture of class A and A′ consisting, e.g. of Methanobacterium thermoautotrophicum delta-H and Xylella fastidiosa. Class B consists e.g. of Bacillus halodurans or Clostridium acetobutylicum and is characterized by an extreme degree of correlation and a sharp descent without fluctuations. Sequences from the same Archaea or Bacteria species but different strains show almost identical behaviour
Naturally, the finite length of the sequences generates a cut-off for the local concentration cl approaching the mean concentration 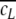 for large l (Fig. 1a), resulting in the breakdown of the power-law behaviour. The concatenation of sequences leads to a double cut-off. Since for cut-off approaching l the number of sequence windows s = L − l in general, and the number of truly independent windows s = L/l over which the average is taken (Eq. 1) decreases rapidly, random deviations do not average out anymore and fluctuations with increasing frequency and amplitude appear in C(l) and more apparently in δ(l). The sampling for l > 104 bp has, of course, an influence here (see above), but neither masks the exact correlation behaviour considering every l nor changes the relative comparison between different sequences (see below).
for large l (Fig. 1a), resulting in the breakdown of the power-law behaviour. The concatenation of sequences leads to a double cut-off. Since for cut-off approaching l the number of sequence windows s = L − l in general, and the number of truly independent windows s = L/l over which the average is taken (Eq. 1) decreases rapidly, random deviations do not average out anymore and fluctuations with increasing frequency and amplitude appear in C(l) and more apparently in δ(l). The sampling for l > 104 bp has, of course, an influence here (see above), but neither masks the exact correlation behaviour considering every l nor changes the relative comparison between different sequences (see below).
To distinguish real from these statistical correlations, random sequences with an initial length of 2, 4, or 34 Mbp as well as their concatenation were created, using either equal or biased human base pair distributions. These random sequences show the same behaviour, since C(l) is based on the concentration deviation from the mean concentration. Only the onsets of fluctuations and cut-offs differ according to the length of the sequence. Therefore, the standard deviation calculated from 20 such sequences for each length could be fitted with the same (but shifted) exponential function (Fig. 1d). The standard deviations for C(l) and δ(l) remain small, e.g. SDδ(l) is <0.1 up to ~1.3 and <0.05 up to ~1.6 orders of magnitude below the maximum sequence length. Consequently, positive long-range correlations are indeed present almost up to the entire scale of the sequences analysed, when the standard deviation as a function of the sequence length is taken into account.
Multi-scaling of long-range correlations
Beyond the appearance of simple long-range correlations with a single slope covering the whole length scale, the concentration fluctuation function C(l) has a far more complex behaviour. In all sequences analysed, the slopes vary considerably between different scaling regions, i.e. the sequences show multi-scaling behaviour (Figs. 1, 10). The local coefficient of correlation is the more sensitive measure to investigate these general patterns within the limit of the chosen resolution of l. On scales with minor fluctuations and small standard deviation (Fig. 1c, d), δ(l) generally shows a global maximum between 40 and 3,400 bp. This maximum can be followed by a region of one or several significant maxima around 6 × 104 to 3 × 105 bp (Figs. 2a, b; 3a–c; 4a–d; 5a–d; 6a–l; 10a–d). Both regions are connected either directly or via a transition zone characterized by one or several minima. Consequently, in all the analysed sequences positive multi-scaling long-range correlations up to almost the entire length were found beyond the simple power-law behaviours also known from literature. The specific characteristics of these multi-scaling properties allow the clustering of genomes into different morphologic classes concerning the behaviour of δ(l) (Tables 1, 2). These as well as their possible origin and interpretation are discussed in the following sections:
General behaviour of the multi-scaling in Eukarya
Homo sapiens: Six sequences from chromosomes XI, XIV, XV, XX, XXI, and XXII with lengths from 9 × 106 to 3.8 × 107 bp were analysed (Table 1). Sequences of chromosomes XX, XXI, and XXII cover huge chromosomal regions with many ideogram bands, in contrast to those of chromosomes XI, XIV, and XV. In all human sequences δ(l) increases from an initial value around −0.42 to a maximum between −0.26 and −0.22, located at ~200 bp (Fig. 2a, b). Despite the very similar ascent, the descent to the minimum between −0.40 and −0.35 at 2 × 104 to 3 × 104 bp diverges: a transition from a slower to a faster descent is characteristic for chromosome XI, XIV, XV, and XXI, relative to an initially steeper descent for chromosome XX and XXII. The transition is located between 2,000 and 4,000 bp in all six sequences. Thereafter, a second maximum was found for chromosome XXII at ~4 × 104 bp and for chromosomes XX and XXI at 1.3 × 105 bp. The significance of these maxima is not only highlighted with respect to the standard deviation (Fig. 1d) but also in their steadiness compared with the spiked fluctuations of random sequences (Fig. 1c). Chromosomes XI, XIV, and XV also exhibit significant peaks in the region between 105 and 5 × 105 bp, although their appearance is accompanied by a high degree of fluctuation. Whether these fluctuations or the substructure of the well-defined maxima of chromosomes XX, XXI, and XXII feature real regularity, might remain unclear until the truly complete (i.e. gap-free) sequence of all 24 human chromosomes can be analysed.
Drosophila melanogaster: The three Drosophila sequences analysed (Table 1), contain in contrast to human, yeast, Archaea, and Bacteria two flat maxima below 104 bp (c.f. Arabidopsis thaliana) with −0.347 and −0.345 at 40 and 3,400 bp, separated by a major minimum of −0.37 at ~304 bp (Fig. 3a, c). Several smaller local maxima at 108, 146, 251, 850, 2,033, and 2,370 bp and one local minimum at 1,100 bp are present in-between, and survive averaging (Fig. 3b, c). Above scales of 3,400 bp, δ decreases to values characteristic of random correlations.
Saccharomyces cerevisiae: In the 16 completely sequenced yeast chromosomes of 3 × 105 to 1.5 × 106 bp (Table 1), δ increases linearly from −0.45 to a maximum around −0.25 between 400 and 650 bp, and thereafter decreases until the random correlation and fluctuation region is reached (Fig. 4a–d). The significance of the peaks and fluctuations on scales >104 bp is unclear. Below 104 bp, however, the behaviour of δ is astonishingly similar in every yeast chromosome.
Schizosaccharomyces pombe: In the case of the three preliminarily sequenced chromosomes of 2.4 × 106 to 5.6 × 106 bp length (Table 1), δ increases from −0.45 linearly to a maximum around −0.23 between 600 and 900 bp, thereafter decreases to a minimum between 1.2 × 104 and 2.0 × 104 bp, before reaching a second significant maximum region around 105 bp that contains many fluctuations (Fig. 4d). Despite the much longer sequences, the behaviour is remarkably similar to that of Saccharomyces cerevisiae below the first maximum.
Arabidopsis thaliana: Here the two sequences of chromosome II and IV and the top and bottom arm of chromosome I as well as their concatenation to test changes from single arms to a complete chromosome were analysed (Table 1). While the genomes of human, yeast, Archaea, and Bacteria possess one maximum below 104 bp, Arabidopsis thaliana, like Drosophila melanogaster, shows two flat maxima of −0.342 and −0.345 at 60 and 600 bp, separated by a major minimum of −0.36 at ~178 bp (Fig. 5a, d). In-between, two smaller local maxima are present at 112 and 270 bp. Averaging all sequences leaves these structures unchanged (Fig. 5c, d). Above 600 bp, δ decreases to values characteristic of random correlations. The growing fluctuations are statistically insignificant, despite the length of the sequences between 1.5 × 107 and 2.8 × 107 bp. Concatenation of the top and bottom arm did not lead to changes below 104 bp, but structures present in the separated arms discussed above were averaged out.
General behaviour of the multi-scaling in Archaea and Bacteria
Archaea and Bacteria (Table 2) revealed a more diverse behaviour than expected from the similarity between the chromosomes of the respective Eukarya under study. This suggested that the classification of this variety into groups based on the distinct curve shapes is possible. After extensive qualitative visual comparisons, as a first quantitative attempt for clarification, an unweighted pair group method with arithmetic mean (UPGMA) clustering approach based on pair-wise distances derived from Pearson correlation coefficients led to an appropriate representation of the appearance of the fractal behaviours (Knoch et al. 2000; Knoch 2002, 2003; Lefkovith 1993): let sij be the coefficient of correlation between the values measured at a certain window size for genomes i and j, respectively. The distance dij between both genomes may then be defined as dij := ln (0.5 + 0.5sij). Such a simple approach is not based on any model of genome evolution but is intended to be purely descriptive and is seen here as a matter for further investigation. Nevertheless, this simple clustering already revealed four major classes with distinct multi-scaling behaviour, which in the following will be referred to as A, A′, A′′, and B, respectively, and which agree very well with the visual inspection.
In class A, consisting of some Bacteria (e.g. Aquifex aeolicus) and most of the Archaea (e.g. Aeropyrum pernix and except Halobacterium sp. NRC1), δ increases up to a general maximum around −0.14 at ~550 bp and decreases afterwards with growing fluctuations (Fig. 6a, b). Separate analyses of Archaea and Bacteria within class A reveal a shift of the maximum position with −0.15 at ~450 bp and −0.13 at 650 bp, respectively. The region of second local maxima at around 105 bp within the present fluctuations seems, due to the limited number of available sequences, statistically insignificant, although the second maxima become more significant between 5 × 104 and 105 bp for Bacteria. Class A′ (e.g. Campylobacter jejuni), possesses a lower first maximum around −0.27 at ~850 bp, followed by a minimum of around −0.35 between 5,000 and 2.5 × 104 bp. Continuing with a linear increase, a statistically significant plateaued maximum between 6 × 104 and 3 × 105 bp, in which small fluctuations are present, is reached. Finally, the plateau decreases sharply without much fluctuation. Class A′′ consists of, e.g. Methanobacterium thermoautotrophicum delta-H and Xylella fastidiosa, and seems to be a mixture of classes A and A′. Yet another behaviour is shown by the largest class B (e.g. Bacillus halodurans and Clostridium acetobutylicum). Here, the first maximum is only hinted at after the usual increase and reaches plateaued saddle points at ~2,000 bp. Thereafter, δ rises towards a second maximum at ~105 bp with an extreme degree of correlation sometimes even above −0.1. For window sizes > 105 bp δ decreases sharply with hardly any fluctuation, supporting again the statement that commonly high correlation degrees suppress fluctuations.
In summary, the general correlation behaviour of Archaea and Bacteria is characterized by a first maximum below 103 bp with decreased height and increased position, before a second maximum appears. The transition between these maxima exhibits a minimum or a saddle point, depending on the influence of the second maximum. The sometimes extreme degree of correlation is unlike that found in any Eukarya. Notably, the different strains from the same Archaea or Bacteria species behave very similarly, suggesting evolutionary constancy of correlations below the species level. Higher-order groups in the cluster analysis are barely consistent with monophyletic groups. For instance, the four main classes each contain a mixture of Archaea and Bacteria. On the other hand, some of the clusters may point to convergent adaptations to environmental conditions, e.g. extremophiles seem to behave very similar.
Origin and interpretation of multi-scaling
The distinct morphologic classes found within the general correlation behaviour by visual inspection and a simple quantitative approach, imply a higher degree of sequential organization than being caused by a merely statistical multi-scaling behaviour, since the correlation degree is distinctly varying with the scale. To determine quantitatively a possible origin of these multi-scaling behaviours, random sequences were designed assuming a block organization of genomes. For Eukarya, such a block organization has already been proposed by structures such as isochores of ideogram bands in metaphase chromosomes (Bernardi 1989, 1995; Li 2001, 2002), differing e.g. in their AT/GC content (Francke 1994), or as part of the three-dimensional organization of genomes (Knoch et al. 2000; Knoch 2002, 2003). These results might also point to a more sophisticated organization, e.g. blocks within blocks or periodicities. However, due to the lack of an irreversible unique determination after a superposition, this might not be traceable back in every case, i.e. it could be a block, a periodicity or both, which leads to the same behaviour.
Random block sequences with a total length of 10 Mbp were composed from blocks with a random length B chosen either from [0, B] or [B − 10%, B + 10%]. This avoids artificial correlations due to a fixed block length (see below). While [0, B] approximates a primitive fractal block pattern with a certain degree of self-similarity due to the broadly distributed block length, [B − 10%, B + 10%] models a softened periodicity. The differences between blocks were created by changing the uniform purine/pyrimidin compositions to concentrations chosen uniformly from [0.5 − D, 0.5 + D] with D varying from 0.00 to 0.50. The overall composition remained therefore unchanged, since the local differences are averaged out on larger scales.
All created block sequences have one global maximum, whose position, width, and descent are proportional to the block length. The ascent and initial values are proportional and the maximum height is inversely proportional to the concentration deviation D (Fig. 7a, b). This agrees with the measurement process leading to C(l) and δ(l). Both block length distributions used, yielded similar results with slightly smaller values for the block length distribution from [0, B] (Fig. 7a). Remarkably, fluctuations common in random sequences with uniform or biased base pair composition become apparent only after the descent (Fig. 1c). Consequently, these fluctuations are suppressed by correlations induced by the blocks, the suppression being proportional to the block length. In detail, the maximum height changes from −0.42 to nearly −0.005 and its position shifts from 35 to 1.5 × 104 bp for blocks from 50 to 106 bp and a deviation D of 0.100 (Fig. 7a). For D from 0.050 to 0.500, the maximum height changes from −0.27 to −0.03 and from −0.04 to −0.005 for blocks of 103 and 105 bp. Thus, blocks of large length and/or large concentration deviations create correlations of extremely high degree. The correlation degree for δ(l = 3) as a function of the deviation D, follows δ(l = 3, D) = −0.5 + 0.113D + 0.855D2, a quadratic fit with R = 0.99, in contrast to the linear dependence found in the simulation of the fine-structural pattern due to codon usage (see below).
Fig. 7.

Appearance and simulation of the block structure of genomes: simulation of random sequences using blocks of random length B either from the intervals B ± 10% or 0 to B, and with deviations from the uniform purine/pyrimidine concentration, leads to a global maximum in the correlation coefficient (a, b). Its position, height, and descent are proportional to the block length (a; B ± 10%: solid line, 0−B: dotted line, B: see legend, for a deviation of 0.100) and the ascent to the maximum and its height are proportional to whereas its position is inversely proportional to the concentration deviation (b; B ± 10% with B = 103, solid line, B = 105: dotted line, deviation see legend). The descent is remarkably smooth, although fluctuations increase exponentially as a function of the window size l (Fig. 1). The degree of correlation follows a quadratic dependence δ(l = 3, D) = −0.5 + 0.113D + 0.855D2 with R = 0.99 (c), in contrast to the linear dependence found for simulations of the codon usage
To understand the obvious evolutionary persistence of the multi-scaling long-range behaviour, simple random rearrangements of blocks with the same properties as those used to create the random block sequences were applied to these sequences: The multi-scaling properties were highly reduced after 104 and completely disappeared after 105 rearrangements. Consequently, evolutionary persistence seems only guaranteed by defined and not totally random rearrangements in real genomes. At least for correlations on scales >103 bp this requires most likely the involvement of the three-dimensional organization of genomes and vice versa, i.e. the involvement of the local nucleosomal as well as the higher-order 30 nm chromatin fiber conformation in the form of chromatin loops and aggregates thereof, because these are the mutational units on this scale. This seems obvious with respect to the fact that most of the larger genomic rearrangements are lethal and take place in a defined manner allowing e.g. the determination of breakpoint regions (Bernardi 1989, 1995; Lamond and Earnshaw 1998; Cremer and Cremer 2001; Dundr and Misteli 2001; Knoch 2002, 2003). Thus, the general sequential and the three-dimensional organization seem indeed to be closely interwoven, as already hypothesized (Takahashi 1989; Grossberg et al. 1993; Stanley et al. 1994; Borovik et al. 1994; Mira et al. 2001).
Consequently, the general morphology of the multi-scaling correlation behaviour in all analysed sequences is at least partly explained by a relatively simple block organization with evolutionary persistence. In reality, of course, the mixture of block length and deviations is more complex than assumed here. Especially integration of blocks within blocks could fine-tune the general behaviour as already proposed above. Nevertheless, the detailed description of the general morphology can already be quantified reasonably well:
In the case of Homo sapiens the first maximum could be due to blocks of ~500 bp and concentration deviations of 0.050–0.075. The second maximum present in the sequences of chromosomes XX, XXI, and XXII cannot be explained by a simple block structure on the order of 105 bp, although its smooth and fluctuation-less appearance is similar to those of large blocks, i.e. this second maximum cannot be generated from the behaviour of the random block sequences (Fig. 7a, b). This holds also for the superposition of a small and large block organization, considering the relatively small difference between the two methods of block length simulation [0, B] and [B − 10%, B + 10%] and concerning the concentration deviation. However, a more pronounced periodicity, consisting of evenly spaced blocks with a deviation in base pair composition and a length of around 105 bp, could be the origin of these second maxima. Such periodicities were found in the simulation of the codon usage and nucleosomal binding sites (see below; Figs. 8e, f; 9a, c).
Fig. 8.

Appearance and simulation of the codon fine-structure of genomes: in all but the human sequences a fine-structure with a periodicity of 3 bp is present up to window length of several hundred base pairs, which is related to the codon usage (a, b). Already a uniform distribution of the 20 amino acids in artificial random sequences causes this feature. Species-specific codon usage is responsible for the starting behaviour δ(3) < −0.5 or δ(3) > −0.5. Pseudomonas aeruginosa PA01 has an additional dominating periodicity of 12 bp which cannot be explained simply by codon usage (b). The appearance and visibility of the codon usage as well as the degree of correlation at δ(3) is proportional to the concentration ccodon,gene of codons distributed as in the human genome codons within a random sequence and is more apparent for codons organized in genes/blocks (c, for 100% see a). The degree of correlation follows a linear dependence with δ(l = 3, ccodon,gene) = −0.5 + 0.046ccodon,gene and R = 0.99 (d). Organization of codons in genes/blocks leads to a δ(l) maximum and oscillations due to the gene/block length and separation (c, e–g; Fig. 7)
Fig. 9.

Appearance and simulation of the nucleosomal fine-structure of genomes: the fine-structure present in all human sequences (Fig. 1) is in agreement with the pattern found in simulations using a consensus nucleosomal binding sequence (a, b, d) organized in a block/gene fashion (Fig. 7). The positions of the local maxima are mostly the same as in the human genome (dark numbers/arrows are in agreement within ±1 bp), whereas the similarity of the position of the local minima is difficult to compare as they smear out in the human sequence due to the block structure of genomes (Fig. 1). Use of a mixture of two special sequence motifs results in highly ordered periodicities of 10 bp, attributable to the helical pitch and the base pairs bound to the nucleosomal core (c). The appearance, visibility, as well as the degree of correlation is again proportional to the concentration of the blocks/genes in the random sequence (see legend in b), leading also to a general maximum and oscillations of δ(l) (a, embedding hull in c)
The behaviour of chromosomes from Saccharomyces cerevisiae and Schizosaccharomyces pombe are best described by a block length of 5,000 bp and deviations of 0.05. Sequences of Arabidopsis thaliana can be regarded as a mixture of two block sizes of 50 to 100 bp and 5,000 bp, with deviations below 0.05.
Concerning Archaea and Bacteria, the first maximum in the morphologic classes of Archaea, A, A′, and A′′ is described best by 5,000 to 104 bp blocks with deviations from 0.30 to 0.075. The second maxima increasing from A′ to A′′ can be explained by increasing presence of large blocks or by more pronounced periodicities as e.g. for Homo sapiens. In class B this interpretation is more obvious by merging blocks of 5,000 bp and 105 to 106 bp with deviations in the base pair concentration >0.075. These block arrangements agree very well with the suggested topology of the genomic higher-order structure due to clustering of DNA loops in Archaea and Bacteria or chromatin loops and their clustering in Eukarya (Lamond and Earnshaw 1998; Cremer and Cremer 2001; Dundr and Misteli 2001; Knoch 2002, 2003) considering their spatial scaling behaviour (Knoch 2002, 2003). The latter is based on simulation of the chromatin fiber topology (Knoch 2002, 2003) assuming the so-called Multi-Loop-Subcompartment (MLS) topology (Lamond and Earnshaw 1998; Cremer and Cremer 2001; Dundr and Misteli 2001; Knoch 2002, 2003) proposing chromatin loops from 60 to 256 kbp organized in rosettes resembling interphase ideogram bands and connected by a similarly sized linker as well as assuming the so-called Random-Walk/Giant-Loop (RWGL) topology (Lamond and Earnshaw 1998; Cremer and Cremer 2001; Dundr and Misteli 2001; Knoch 2002, 2003), where large 1 to 5 Mbp loops are connected to a backbone. Only for the MLS topology there is good agreement between spatial and sequential scaling behaviour (actually the similarity is very striking) according to the already proposed theme “what is near in sequence space should be near in real space” (Nee 1992; Karlin and Brendel 1993; Prabhu and Claverie 1992; Chatzidimitriou-Dreismann and Larhammar 1993; Buldyrev et al. 1993), i.e. that the sequential and three-dimensional organization seem really to be tightly interrelated. Although this seems obvious, the general multi-scaling behaviour and its persistence strengthens this connection (see also fine-structure behaviour).
Fine-structuring of multi-scaling long-range correlations and its origin
Within the multi-scaling long-range correlations further fine-structures were found which are attributable to codon usage and nucleosome-associated sequences according to the results of a detailed comparison of real with artificially designed random sequences. This leads clearly to the conclusion that the sequential organization of genomes is in many aspects related to its three-dimensional spatial arrangement, as will be explained in detail in the following sections.
Codon-usage-associated fine-structure
A fine-structure with a periodicity of 3 bp is well known (Eigen and Winkler-Oswatitsch 1981a, b; Eigen et al. 1981; Shephard 1981a, b; Crick et al. 1957). Here, it is demonstrated up to window lengths of several hundred base pairs (Fig. 5g) or even a few thousand base pairs in all but the human sequences (Figs. 2a, b; 8a). In the bacterium Pseudomonas aeruginosa PA01, the 3 bp periodicity is dominated by another periodicity of 12 bp (Figs. 6i; 8b). The sequences of Homo sapiens show yet another fine-structure (Fig. 2c–f). To relate this fine-structure to codon usage and to distinguish it from those found in human and Pseudomonas aeruginosa PA01, 10 Mbp long random sequences were generated, consisting completely of codons with a distribution based on codon usage tables. As expected, uniformly distributed codons, the simplest codon usage table, totally lack a fine-structure (Fig. 8c), since this resembles a completely random organization of single base pairs. However, a random distribution of amino acids based on the human codon usage distribution, with an imbalance towards the frequency of each single codon, already introduces enough imbalance to create the 3 bp fine-structure. Random codon sequences based on the respective codon usage table displayed the fine-structure for all analysed sequences. Thus, neither the fine-structure present in Homo sapiens nor the 12 bp periodicity in Pseudomonas aeruginosa PA01 are based on the codon usage. The latter possibly is due to an uncommon but distinct succession of codons. The simulations also correctly reproduce the correlation degree at δ(l = 3) and whether this starting value is greater than or less than −0.5. The fine-structure also rapidly approximates -0.5, thereafter fluctuating around it. Thus, no increase of δ is created as in the real sequences, i.e. this general increase is finally attributed to the block structure of genomes.
To investigate the codon concentration ccodon,gene needed to produce the fine-structure, codons from a variety of usage tables were either randomly mixed into a random sequence (random codon sequence) or organized in blocks of 333 or 999 bp codons. The blocks were distributed equally in the sequence (random gene sequence). Whereas the former approach simulates mutated, distorted or free for deletion genes, the latter comes close to functional genes. The fine-structure appearance is proportional to the codon concentration and starts at concentrations of ~10% for gene and >50% for codon sequences (Fig. 8c). Thus, the earlier onset for gene sequences is caused by the uninterrupted succession of codons within a gene. This proximity enhancement is not present in random codon sequences. The degree of correlation for the human codon distribution at δ(l = 3) follows a linear dependency with δ(l = 3, ccodon,gene) = −0.5 + 0.046ccodon,gene and R = 0.99 for random codon as well as gene sequences (Fig. 9d). For Drosophila melanogaster, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Arabidopsis thaliana, Chlamydia muridarum, Mycobacterium tuberculosis, and Pseudomonas aeruginosa PA01, similar linear laws were found with slopes of 0.047, 0.043, 0.043, 0.045, 0.044, −0.055, and −0.056, respectively. Consequently, the dependence is based on the degree of correlation within the codon usage.
Beyond the fine-structure, the random gene results, in obvious contrast to the codon sequence results, also demonstrate a general multi-scaling behaviour as for random block sequences: a first maximum before 103 bp is followed by periodicities proportional to the different separations between genes for different ccodon,gene (Fig. 8d–g). The height and position of the first maximum is more pronounced the greater the deviations between the genes and the rest of the sequence are, and thus is the greatest for ccodon,gene = 60% with a δ of −0.44 at 480 bp. Consequently, the multi-scaling created by genes has a smaller influence on δ in comparison with the block organization discussed above, since compared with blocks, much higher deviations in base pair compositions (~30%) are necessary to achieve high peak values in agreement with the argument about correlation strength at δ(l = 3). Nevertheless, small sequence regions with a strongly deviating base pair concentration in connection with a periodic spacing could explain the second maxima found around 105 bp in the human sequences, which are not interpretable with the simple block approach (see above). A straightforward calculation, based on the total length of the haploid human genome of ~3.5 × 109 bp and the ~35,000 genes so far found, also results in an average gene spacing of 105 bp. Thus, the second maxima found there might originate from gene spacing or density within these sequences.
Nucleosomal binding-associated fine-structure
The fine-structure is practically identical even in detail in all human sequences (Fig. 2c–f). It is far more complex than could result from the codon usage effects alone: The very pronounced local maximum at 11 bp might be associated to the double-helical pitch, whereas the local minima and maxima thereafter seem related to the nucleosome. The obvious maximum at 146 bp (exactly the DNA length wrapped around the octamer histone protein core of the nucleosome) is supplemented by less pronounced maxima at 172, 205, 228, and 248 bp.
No codon-like fine-structure is visible within these peaks. To confirm this relation to the nucleosome, i.e. nucleosomal associated sequences, again 10 Mbp long random nucleosome sequences were created in which nucleosome “binding” sequences were organized in blocks. The blocks were equally distributed, i.e. with a fixed distance in-between, within a totally random sequence. The gene size of 2,750 bp was either designed from a consensus sequence of 230 bp or a mixture of two special sequence motifs of 30 and 20 bp. All three motifs were based on nucleosomal binding studies. The consensus sequence, which contains constant as well as variable sites, is somewhat more resistant against periodicities than the exact mixture of the motifs. The fine-structure of the consensus sequence exhibits a very similar pattern, with 75% of maxima found within ±1 bp of the position of the real human sequences, e.g. at 146 bp (Fig. 9b, d). The low similarity of ~33% for local minima is, however, difficult to compare due to the smearing out caused by the general multi-scaling behaviour of the human sequences. As in the real human sequence, no codon-associated fine-structure is present. Furthermore, a correlation between 2,000 and 4,000 bp, attributable to the transition of the multi-scaling behaviour, was not found. It could, however, be associated to short-range correlations between entire nucleosomes and thus to the conformation of nucleosomes within the chromatin fiber. The general two-peaked multi-scaling behaviour as found in Arabidopsis thaliana also remains unsupported. The appearance, the visibility, as well as the degree of correlation are once again proportional to the concentration of the nucleosomal gene blocks within the random sequence. Accordingly a concentration of nucleosomal binding sequences of at least 5–10% but including more sequence motifs perhaps even 50–70% in human sequences may cautiously be predicted. The use of the mixture of two sequence motifs results in a first maximum at 13 bp as for the consensus sequence and in a highly ordered periodicity of 10 bp (Fig. 9c), being strongly proportional to the concentration. This periodicity is attributable to the double-helical pitch and not to the short motif length.
Both kinds of random nucleosome sequences again produce the multi-scaling behaviour suggested by the block/gene organization as in the investigation of the general block organization or of the codon usage. The fine-structure is embedded within (Fig. 9a, c). Especially for the mixture of the sequence motifs, these fine-structured periodicities propose an embedding hull defining the block/gene-based periodicity (Fig. 9c). Thus, the general multi-scaling behaviour is basically associated with a general block organization, which here might indeed be composed of nucleosomal associated blocks. In contrast, the opposite causality—that the mere multi-scaling behaviour would be associated to the nucleosome—remains speculative without the existence of a fine-structure.
Thus, on the nucleosomal level the interaction as well as the co-evolution between sequence and structure is now more clearly demonstrated by the difference between genomes with a relatively high density of genes/coding regions in relation to the total sequence size. For Archaea, Bacteria, Saccharomyces cerevisiae, Schizosaccharomyces pombe, and less for Arabidopsis thaliana, as well as Drosophila melanogaster this ratio is high in contrast to Homo sapiens where a large part of the genome (>90%) is believed to be gene-free or noncoding (which does not imply that this majority is biologically unimportant). In these regions, the structural integrity of the chromatin fiber as well as the importance of the epigenetic histone code seem to have been dominant in evolution and to have influenced the fine-structural correlation behaviour, quite in contrast to the apparently underrepresented codon feature. This does, of course, not imply that there is no codon-associated fine-structure within genes or coding regions. Rather, due to its underrepresentation with respect to total sequence length, it could be expected to not significantly affect the correlation properties. This is in agreement with the concentration dependence of the codon-associated fine-structure demonstrated above. Thus, the link between sequence and structure already predicted from the general multi-scaling behaviour, especially on scales >103, is further supported. Correspondingly, our results point with seemingly unprecedented clarity to the tight co-evolutionary connection between the sequential and three-dimensional organization, as hypothesized earlier (Nee 1992; Karlin and Brendel 1993; Prabhu and Claverie 1992; Chatzidimitriou-Dreismann and Larhammar 1993; Buldyrev et al. 1993).
Conclusion
The complex sequential and three-dimensional genome organization as well as its evolutionary persistence is still little understood, despite the fundamental importance of the interwoven co-evolution of molecular structure and genetic information for organismic function and regulation. Only recently has it become feasible to address this organization in detail due to huge research efforts and advances such as e.g. the human genome project. Here, we investigated the sequential large-scale genome organization with respect to the appearance, features, origins, persistence, specificity, classification, and, finally, its relation to its three-dimensional organization of the genome:
The concentration fluctuation function C(l) and its exponent δ(l), the local correlation coefficient, were calculated using numerically exact algorithms for a total of 201 complete genome sequences 0.5 × 106 to 3.0 × 107 bp in length from Homo sapiens, Drosophila melanogaster, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Arabidopsis thaliana, Archaea, and Bacteria. The results revealed long-range correlations almost up to the entire length scale in all sequences, but at least up to 105 to 106 bp. This is an increase of 2–3 orders of magnitude compared with earlier studies. Beyond the appearance of a simple power-law behaviour, the long-range correlations presented a more complex behaviour: δ(l) showed a maximum between 50 and 2,000 bp and sometimes a region containing one or more second maxima at ~105 bp. Especially the human sequences display very pronounced second maxima. Likewise, many Bacteria show a remarkable degree of correlation at this scale, whose origin remains unknown. This so-called multi-scaling behaviour was species-specific and may point to convergent adaptations to environmental conditions. Since these classes seemed unconnected to any single parameter such as base pair composition or gene content, correlation analysis might lead to a new classification system, which integrates different properties of the general organization of whole genomes. Results of cluster analysis mostly were incongruent with the phylogeny of the taxa under study. Nevertheless, some clusters indicate convergent adaptive evolution, as several Archaea and Bacteria living under extreme environmental conditions were grouped together. Thus, such clustering approaches may be of use in future studies on the evolution of general genome architecture.
Analysis of computer-generated random sequences suggests that the multi-scaling might originate from a block-wise sequence organization. Investigation of the evolutionary persistence of multi-scaling by simulation of random sequence reshuffling resulted in total loss of (multi-scaling) correlations. Thus, persistence of multi-scaling in evolution can only be caused by nonrandom rearrangements in real genomes. This result points to a close connection with the three-dimensional genome structure. A nonrandom arrangement in blocks agrees very well with the suggested higher-order genome topology due to clustering of DNA loops in Archaea and Bacteria or chromatin loops and their clustering in Eukarya. Within the multi-scaling correlation behaviour, additional species-specific fine-structures were found which are attributable to codon usage. An exception is the human genome in which the fine-structure is connected to nucleosome association or “binding.” Both connections were also clarified by artificial random sequence design. Obviously, again a strong co-evolution and close relations within the sequence (especially the dominance of gene/coding regions) as well as between sequence and structure can be inferred.
Consequently, our analysis of the appearance, characteristics, origins, persistence, and specificity of the fine-structured multi-scaling long-range correlations observed in completely sequenced genomes proposes a complex sequential genome organization co-evolutionarily interwoven with the three-dimensional genome organization. We provide a consistent and unifying framework for this connection by using a “virtual microscopy” approach.
Acknowledgments
Part of the research presented here was conducted at the German Cancer Research Center (DKFZ), the Kirchhoff Institute for Physics, University of Heidelberg, both in Heidelberg, Germany, and the Erasmus MC, Rotterdam, The Netherlands. Dr. Agnes Hotz-Wagenblatt and Dr. Karl-Heinz Glatting (both DKFZ) and the European Bioinformatics Institute (EMBL-EBI) provided the completely sequenced genomes, which is gratefully acknowledged. Dr. Malte Wachsmuth, Dr. Katalin Fejes-Toth, Dr. Karsten Rippe, and Dr. Michael J. Moorhouse are thanked for critical reading of the manuscript. We are also grateful to the Supercomputing Center Karlsruhe (SCC; grant ChromDyn), the Computing Facility of the DKFZ for access to their IBM SP2s, as well as the German MediGrid, German D-Grid Initiative, and the Erasmus Computing Grid. This work was partly supported by the Bundesministerium für Bildung und Forschung (BMBF) under grant #01 KW 9602/2 (Heidelberg 3D Human Genome Study Group, German Human Genome Project) and #01 AK 803 A-H (German MediGRID) and #01 IG 07015 G (German Services@MediGRID). T. A. Knoch was kindly provided with a dissertation grant from the DKFZ at the beginning of this work.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
This article has been submitted as a contribution to the festschrift entitled “Uncovering cellular sub-structures by light microscopy” in honor of Professor Cremer’s 65th birthday.
References
- Allegrini P, Buiatti M, Grigolini P, West BJ (1998) Fractional Brownian motion as a nonstationary process: an alternative paradigm for DNA sequences. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics 57(4):4558–4567. doi:10.1103/PhysRevE.57.4558
- Amato I (1992) DNA shows unexpected patterns writ large. Science 257:747. doi:10.1126/science.1496395 [DOI] [PubMed]
- Ambrose C, Lowman H, Rajadhyaksha A, Blasquez V, Bina M (1990) Location of nucleosomes in Simian Virus 40 chromatin. J Mol Biol 214:875–884. doi:10.1016/0022-2836(90)90342-J [DOI] [PubMed]
- Bailey KA, Pereira SL, Widom J, Reeve J (2000) Archael histone selection of nucleosome positioning sequences and the procaryotic origin of histone-dependent genome evolution. J Mol Biol 303:25–34. doi:10.1006/jmbi.2000.4128 [DOI] [PubMed]
- Bernardi G (1989) The isochore organization of the human genome. Annu Rev Genet 23:637–661. doi:10.1146/annurev.ge.23.120189.003225 [DOI] [PubMed]
- Bernardi G (1995) The human genome: organization and evolutionary history. Annu Rev Genet 29:445–476. doi:10.1146/annurev.ge.29.120195.002305 [DOI] [PubMed]
- Blank TA, Becker PB (1996) The effect of nucleosome phasing sequences and DNA topology on nucleosome spacing. J Mol Biol 260:1–8. doi:10.1006/jmbi.1996.0377 [DOI] [PubMed]
- Borovik AS, Grosberg AY, Frank-Kamenetskii MD (1994) Fractality of DNA texts. J Biomol Struct 12(3):655–669 [DOI] [PubMed]
- Buldyrev SV, Goldberger AL, Havlin S, Peng CK, Simons M, Stanley HE (1993) Generalized Lévy-walk model for DNA nucleotide sequences. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics 47(6):4514–4523. doi:10.1103/PhysRevE.47.4514 [DOI] [PubMed]
- Buldyrev SV, Goldberger AL, Havlin S, Mantegna RN, Matsa ME, Peng CK, Simons M, Stanley HE (1995) Long-range correlation properties of coding and noncoding DNA sequences: GenBank analysis. Phys Rev 51(5):5084–5091 [DOI] [PubMed]
- Chatzidimitriou-Dreismann CA, Larhammar D (1993) Long-range correlations in DNA. Nature 361:212–213. doi:10.1038/361212b0 [DOI] [PubMed]
- Cremer T, Cremer C (2001) Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet 2:292–301. doi:10.1038/35066075 [DOI] [PubMed]
- Crick FHC, Griffith JS, Orgel LE (1957) Codes without comma. Proc Natl Acad Sci USA 43:416–421. doi:10.1073/pnas.43.5.416 [DOI] [PMC free article] [PubMed]
- de Oliveira PMC (1993) Studying DNA evolution through successive file editions. Physica A 273:70–74. doi:10.1016/S0378-4371(99)00341-6 [DOI]
- Dundr M, Misteli T (2001) Functional architecture in the cell nucleus. Biochem J 356(Pt 2):297–310. doi:10.1042/0264-6021:3560297 [DOI] [PMC free article] [PubMed]
- Eigen M, Winkler-Oswatitsch R (1981a) Transfer-RNA, an early gene? Naturwissenschaften 68:282–292. doi:10.1007/BF01047470 [DOI] [PubMed]
- Eigen M, Winkler-Oswatitsch R (1981b) Transfer-RNA: the early adaptor. Naturwissenschaften 68:217–228. doi:10.1007/BF01047323 [DOI] [PubMed]
- Eigen M, Gardiner W, Schuster P, Winkler-Oswatitsch R (1981) Ursprung der genetischen Information. Spektrum Wiss 6:36–56
- Francke U (1994) Digitized and differentially shaded human chromosome ideograms for genomic applications. Cytogenet Cell Genet 65:206–219. doi:10.1159/000133633 [DOI] [PubMed]
- Grossberg A, Rabin Y, Havlin S, Neer A (1993) Crumpled globule model of the three-dimensional structure of DNA. Europhys Lett 23(5):373–378. doi:10.1209/0295-5075/23/5/012 [DOI]
- Hao B, Lee HC, Zhang S (2000a) Fractals related to long DNA sequences and complete genomes. Chaos Solitons Fractals 11:825–836. doi:10.1016/S0960-0779(98)00182-9 [DOI]
- Hao B, Xie H, Yu Z, Chen G (2000b) Factorizable language: from dynamics to bacterial complete genomes. Physica A 288:10–20. doi:10.1016/S0378-4371(00)00411-8 [DOI]
- Hattori M et al (2000) The DNA sequence of human chromosome 21. The chromosome 21 mapping and sequencing consortium. Nature 405:311–319. doi:10.1038/35012518 [DOI] [PubMed]
- Hsü KJ, Hsü AJ (1990) Fractal geometry of music. Proc Natl Acad Sci USA 87:938–941. doi:10.1073/pnas.87.3.938 [DOI] [PMC free article] [PubMed]
- Hsü KJ, Hsü AJ (1991) Self-similarity of the “1/f noise” called music. Proc Natl Acad Sci USA 88:3507–3509. doi:10.1073/pnas.88.8.3507 [DOI] [PMC free article] [PubMed]
- Karlin S, Brendel V (1993) Patchiness and correlations in DNA sequences. Science 259:677–680. doi:10.1126/science.8430316 [DOI] [PubMed]
- Kirkpatrick S, Stoll E (1981) Implementation of the R250 random number generator. J Comput Phys 40:517. doi:10.1016/0021-9991(81)90227-8 [DOI]
- Knoch TA (2002) Approaching the three-dimensional organization of the human cell nucleus: structural-, scaling- and dynamic-properties in the simulation of interphase chromosomes and cell nuclei, long-range correlations in complete genomes, in vivo quantification of the chromatin distribution, construct conversions in simultaneous co-transfections. PhD thesis, Ruperto-Carola University, Heidelberg, Germany, and TAK Press, Dr. Tobias A. Knoch, Mannheim, Germany, ISBN 3-00-009960-3. URN: urn:nbn:de:bsz:16-opus-31055 and URL: http://www.ub.uni-heidelberg.de/archiv/3105
- Knoch TA (2003) Towards a holistic understanding of the human genome by determination and integration of its sequential and three-dimensional organization. In: Krause E, Jäger W, Resch M (eds) High Performance Computing in Science and Engineering 2003. High-Performance Computing Center (HLRS) Stuttgart, University of Stuttgart, Springer, Berlin, ISBN 3-540-40850-9, pp 421–440
- Knoch TA, Münkel C, Langowski J (2000) Three-dimensional organization of chromosome territories in the human interphase nucleus. In: Krause E, Jäger W (eds) High Performance Computing in Science and Engineering 1999. High-Performance Computing Center (HLRS) Stuttgart, University of Stuttgart, Springer, Berlin, ISBN 3-540-66504-8, pp 229–238
- Lamond AI, Earnshaw WC (1998) Structure and function in the nucleus. Science 280:547–553. doi:10.1126/science.280.5363.547 [DOI] [PubMed]
- Lefkovith LP (1993) Optimal set covering for biological classification: theory of conditional clustering & its use in biological classification & identification. Accents Publications Service, Ottawa
- Li W (1991) Expansion-modification systems: a Model for spatial 1/f spectra. Phys Rev A 43:5240–5260. doi:10.1103/PhysRevA.43.5240 [DOI] [PubMed]
- Li W (1997) The study of correlation structures of DNA sequences: a critical review. Comput Chem 21(4):257–271. doi:10.1016/S0097-8485(97)00022-3 [DOI] [PubMed]
- Li W (2001) Delineating relative homogeneous G + C domains in DNA sequences. Gene 276:57–72. doi:10.1016/S0378-1119(01)00672-2 [DOI] [PubMed]
- Li W (2002) Are isochore sequences homogeneous? Gene 300:129–139. doi:10.1016/S0378-1119(02)00847-8 [DOI] [PubMed]
- Li W, Kaneko K (1992) Long-range correlation and partial 1/fα spectrum in a noncoding DNA Sequence. Europhys Lett 17(7):655–660. doi:10.1209/0295-5075/17/7/014 [DOI]
- Li W, Marr TG, Kaneko K (1994) Understanding long-range correlations in DNA sequences. Physica D 75:392–416. doi:10.1016/0167-2789(94)90294-1 [DOI]
- Liu K, Stein A (1997) DNA sequence encodes information for nucleosome array formation. J Mol Biol 270(4):559–573. doi:10.1006/jmbi.1997.1136 [DOI] [PubMed]
- Lowary PT, Widom J (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276:19–42. doi:10.1006/jmbi.1997.1494 [DOI] [PubMed]
- Luo L, Lee W, Jia L, Ji F, Tsai L (1998) Statistical correlation of nucleotides in a DNA sequence. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics 58(1):861–871. doi:10.1103/PhysRevE.58.861
- Mackiewicz P, Gierlik A, Kowalczuk M, Szczepanik D, Dudek MR, Cebrat S (1999) Mechanisms generating long-range correlation in nucleotide composition of the Borrelia burgdorferi genome. Physica A 273:103–115. doi:10.1016/S0378-4371(99)00345-3 [DOI]
- Maddox J (1992) Long-range correlations within DNA. Nature 358:103. doi:10.1038/358367a0 [DOI] [PubMed]
- Maier WL (1991) A fast pseudo random number generator. Dr. Dobb’s Journal 176
- Mandelbrot BB (1983) The fractal geometry of nature. W. H. Freeman and Company, New York, ISBN 0-7167-1186-9
- Mira A, Ochman H, Moran N (2001) Deletional bias and the evolution of bacterial genomes. Trends Genet 17(19):589–596. doi:10.1016/S0168-9525(01)02447-7 [DOI] [PubMed]
- Mohanty AK, Narayana-Rao AVSS (2000) Factorial moments analysis show characteristic length scale in DNA sequences. Phys Rev Lett 84(8):1832–1835. doi:10.1103/PhysRevLett.84.1832 [DOI] [PubMed]
- Nee S (1992) Uncorrelated DNA walks. Nature 357:450. doi:10.1038/357450a0 [DOI] [PubMed]
- Peng CK, Buldyrev SV, Goldberger AL, Havlin S, Sciortino F, Simons M, Stanley HE (1992) Long-range correlations in nucleotide sequences. Nature 356:168–170. doi:10.1038/356168a0 [DOI] [PubMed]
- Peng CK, Buldyrev SV, Havlin S, Simons M, Stanley HE, Goldberger AL (1994) Mosaic organization of DNA nucleotides. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics 49(2):1685–1689. doi:10.1103/PhysRevE.49.1685 [DOI] [PubMed]
- Prabhu VV, Claverie JM (1992) Correlations in intronless DNA. Nature 359:782. doi:10.1038/359782a0 [DOI] [PubMed]
- Provata A, Almirantis Y (2000) Fractal Cantor patterns in the sequence structure of DNA. Fractals 8(1):15–27. doi:10.1142/S0218348X00000044 [DOI]
- Rabinovich MI, Fabrikant AL, Tsmiring LS (1992) Finite-dimensional disorder. Sov Phys Usp 35(8):629–649. doi:10.1070/PU1992v035n08ABEH002253 [DOI]
- Reif F (1965) Fundamentals of statistical and thermal physics. McGraw-Hill, New York
- Robinson FNH (1974) Noise and fluctuations. Clarendon, Oxford
- Shephard JCW (1981a) Method to determine the reading frame of a protein from the purine/pyrimidine genome sequence and its possible evolutionary justification. Proc Natl Acad Sci USA 78(3):1596–1600. doi:10.1073/pnas.78.3.1596 [DOI] [PMC free article] [PubMed]
- Shephard JCW (1981b) Periodic correlations in DNA sequences and evidence suggesting their evolutionary origin in a comma-less genetic code. J Mol Evol 17:94–102. doi:10.1007/BF01732679 [DOI] [PubMed]
- Stanley HE, Buldyrev SV, Goldberger AL, Goldberger ZD, Havlin S, Mantegna RN, Ossadnik SM, Peng CK, Simons M (1994) Statistical mechanics in biology: how ubiquitous are long-range correlations? Physica A 205:214–253. doi:10.1016/0378-4371(94)90502-9 [DOI] [PubMed]
- Takahashi M (1989) A fractal model of chromosomes and chromosomal DNA replication. J Theor Biol 141:117–136. doi:10.1016/S0022-5193(89)80012-8 [DOI] [PubMed]
- Voss RF (1992) Evolution of long-range fractal correlations and 1/f noise in DNA base sequences. Phys Rev Lett 68(25):3805–3808. doi:10.1103/PhysRevLett.68.3805 [DOI] [PubMed]
- Yu ZG, Chen GY (2000) Rescaled range and transition matrix analysis of DNA sequences. Commun Theor Phys Beijing 33:673–678
- Yu Z, Anh VV, Wang B (2000) Correlation property of length sequences based on global structure of the complete genome. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics 63(1):673–678. doi:10.1103/PhysRevE.63.011903 [DOI] [PubMed]