

Abstract
(-)-Epigallocatechin-3-gallate (EGCG), a major component of green tea, protects against certain types of cancers, although the mechanism has not yet been determined. It was previously demonstrated that EGCG blocks aryl hydrocarbon receptor (AhR)-mediated transcription induced by the potent carcinogen 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Unlike other AhR antagonists that directly bind to the AhR, EGCG inhibits AhR-mediated transcription by binding to hsp90. We hypothesize that EGCG exerts anti-AhR and anti-cancer effects by acting as an hsp90 inhibitor. Using proteolytic footprinting, immunoprecipitation, and an ATP-agarose pull-down assay, EGCG was found to directly modulate the conformation of hsp90 and bind at or near to a C-terminal ATP binding site. Hsp90 chaperone function, as assessed by its ability to mediate refolding of denatured luciferase, was inhibited by EGCG treatment. Hsp90 dimerization, which occurs at the C-terminal end, was also inhibited by EGCG treatment. Co-immunoprecipitation studies showed that EGCG stabilizes an AhR complex that includes hsp90 and XAP2 (hepatitis B virus X-associated protein 2), and decreases the association of aryl hydrocarbon nuclear translocator (Arnt) with ligand-activated AhR. Thus, EGCG, through its ability to bind to hsp90, blocks AhR response element (AhRE) recognition. These studies indicate a novel mechanism whereby EGCG inhibits ligand-induced AhRE binding and AhR-mediated transcriptional activity. In EGCG-treated human ovarian carcinoma SKOV3 cells, decreased levels of several cancer-related hsp90 client proteins, such as ErbB2, Raf-1 and phospho-AKT were observed. EGCG also modified the association of hsp90 with several cochaperones. Overall, these data indicate that EGCG is a novel hsp90 inhibitor. Further studies are needed to determine if this has a role in the anti-tumor actions of EGCG.
Numerous animal studies have shown that green tea and several of its components protect against a variety of cancers, both spontaneous and chemically induced (1, 2). Epigallocatechin-3-gallate (EGCG), a major catechin in green tea, has been suggested to target several biomedically relevant molecules and disease-related cellular processes. These include pathways involved in induction of apoptosis and cell cycle arrest, modulation of cell signaling, and interference with angiogenesis and metastasis (3, 4, 5, 6). However, the precise molecular and cellular mechanisms by which EGCG acts to modulate tumorigenesis have yet to be determined.
We were interested to examine how EGCG may interact with the bHLH-PAS transcription factor aryl hydrocarbon receptor (AhR) to exert anti-cancer effects. Our laboratory has shown that EGCG blocks the AhR-mediated transcription induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), a potent toxicant and carcinogen (7, 8). In the unliganded state, the AhR is present in the cytoplasm associated with two molecules of the chaperone hsp90, an immunophilin-like protein XAP2, and the hsp90-interacting acidic protein p23 (9, 10, 11). Ligand binding initiates a cascade of events involving AhR translocation to the nucleus, release of hsp90, and dimerization with AhR nuclear translocator (Arnt). The ligand-bound AhR-Arnt complex recognizes and binds AhR-response elements (AhREs) located in the promoter region of responsive genes thereby modulating transcription (12). Unlike many other AhR antagonists that interact directly with the AhR, EGCG appears to block AhR-mediated transcription by binding to hsp90 (7).
Hsp90 is a chaperone protein that is highly conserved among species (13, 14). In mammalian cells, there are two genes encoding cytosolic hsp90 homologues, with the human hsp90α showing 85% sequence identity to hsp90β (13, 14). Hsp90 is present in high amounts in normal cells, whereas it is constitutively expressed at 2- to 10-fold higher levels in many cancer cells suggesting its importance for cell growth and survival (13, 14). Hsp90 dimerizes through its C-terminal end. Upon ATP binding, the two N-termini of the dimer associate to form a molecular “clamp” with a client protein to exert its chaperone function (13, 14). Several studies have found that hsp90 derived from many tumor cell types is in a high-affinity multi-chaperone complex with associated ATPase activity that is much greater than that found in normal primary cells (15, 16). This may be the main reason why tumor cells are more sensitive to the growth suppressive effects of hsp90 inhibitors compared to normal cells (16, 17). Many hsp90 client proteins are conformationally labile signal transducers that play a critical role in growth control, cell survival, and, under conditions of dysregulation, tumor development (13, 18, 19). Furthermore, hsp90 inhibitors, such as 17-allylamino-geldanamycin (17-AAG), are being intensively studied for anti-cancer therapy in clinical trials (20). As such, it is attractive to hypothesize that the anti-cancer activity of EGCG is due, at least in part, to its ability to interfere with hsp90 functions. To further test this hypothesis, we reasoned that it was essential to localize the binding site for EGCG on hsp90, and define the mechanism whereby EGCG interferes with hsp90 chaperone function. These investigations indicate that EGCG acts by binding at or near a C-terminal ATP binding site to inhibit dimerization and promote an hsp90 conformation that interferes with its chaperone activity for a client protein, the AhR. These studies also indicate that within intact cells, EGCG affects the stability of other hsp90 client proteins and cochaperones.
EXPRIMENTAL PROCEDURES
Protein expression and purification
Mouse AhR, Arnt and hsp90 proteins were expressed in the presence of 35S-Methionine by coupled transcription/ translation (TnT) in rabbit reticulocyte lysate (RRL) according to the manufacturer’s protocol (Promega, Madison, WI). A chicken hsp90 cDNA plasmid was a kind gift from Dr. Toft (Mayo Medical School, Rochester, MN). A glutathione-S-transferase (GST) tagged human hsp90β truncation (residues 520-724) fusion protein (GST-hHsp90-N520), a gift from Dr. Ratajczak (University of Western Australia, Australia), was expressed in DH5α competent cells (Invitrogen, Carlsbad, CA), and purified using MagneGST protein purification kit (Promega).
Proteolytic footprinting of hsp90
Purified human hsp90α (Stressgen, Victoria, BC) or TnT-expressed hsp90 in HEDG buffer (25mM HEPES, 1.5mM Na2EDTA, 1mM dithiothreitol (DTT), 10% glycerol, pH 7.6) were incubated at room temperature (RT) in the presence of EGCG or vehicle (DMSO) for 1h. The reaction mixtures were digested as described previously (21, 22) with trypsin at the concentrations indicated in the figure. The reaction was stopped by the addition of leupeptin (10.7μg/ml) and chilling on ice. The resulting hsp90 fragments were separated by SDS-PAGE and detected by immunoblotting. For TnT samples, the gel was dried and 35S-labeled bands were visualized by phosphorImager (Molecular Dynamics, Sunnyvale CA).
Immunoblotting
Following SDS-PAGE, the resolved bands were transferred to PVDF membrane (Millipore, Billerica, MA) and blotted with antibodies against AhR (RPT-1), XAP2, p23, Cyp40, N-terminal hsp90 (PA3-013) (Affinity BioReagents, Golden, CO), C-terminal hsp90 (AC88), hsp70 (Stressgen), AKT, phospho-AKT (Cell Signaling Technology, Danvers, MA), ErbB2 or Raf-1 (Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were blocked for 1h with 3% nonfat dry milk in TBST (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.2% Tween-20), incubated with primary antibodies at 4°C for 2 h, then for 1 h with appropriate secondary antibodies conjugated to horseradish peroxidase (Jackson Immuno-research, West Grove, PA). Detection was by chemiluminescence, using reagents purchased from KPL (Gaithersburg, MD).
Immunoprecipitation
Protein A/G agarose (Santa Cruz) was pre-incubated with hsp90 antibodies AC88 or PA3-013 (2μg antibody /100uL agarose slurry) for 1h at 4°C. Purified human hsp90 was incubated with DMSO, 200μM EGCG, 10μg/ml GA, or 10μg/ml GA plus 200μM EGCG at RT for 1h. For every treatment, 0.05μg of hsp90 was added into 100uL of antibody-saturated Protein A/G Agarose, incubated with rocking for 2h or overnight at 4°C, and the samples were briefly centrifuged to remove the supernate. The pellets were washed twice with HEDGMo (HEDG + 20mM NaMoO4) plus 150mM NaCl buffer. Immunoprecipitated proteins were then separated by SDS-PAGE, and blotted with AC88 antibody.
ATP-agarose pull-down assay
Purified human hsp90 or hsp90N520 were incubated with 2-200μM EGCG or 1-10μg/ml geldanamycin (GA) at RT for 1h, and then 100μL of γ-phosphate-linked ATP agarose (Innova Biosciences, Cambridge, UK) or CN-Br activated control agarose (Sigma) in incubation buffer [10mM Tris (pH7.5), 5mM MgCl2, 50mM KCl, 2mM DTT, 0.01% NP40, 20mM NaMoO4, 2μg/ml bovine serum albumin] were added and incubated at 30°C for 20 min with frequent mixing. The beads were washed three times with washing buffer [10mM Tris (pH7.5), 5mM MgCl2, 50mM KCl, 2mM DTT, 0.01% NP40, 20mM NaMoO4] and finally eluted with 5mM ATP in washing buffer. The eluted proteins were separated by SDS-PAGE and blotted with AC88 antibody.
Dimerization assays
The ability of full-length and C-terminal hsp90β proteins to dimerize was assessed by chemical cross-linking using bis-sulfosuccinimidyl suberate (BS3) (Pierce chemical, Rockford, IL), an amine-reactive cross-linker. Purified human hsp90 or hHsp90N520 protein was diluted in DPBS to a final concentration of 2 μM. Stock solutions of 3mM BS3 were freshly prepared before diluting to the final assay concentration. Purified human hsp90 or GST-hHsp90N520 was treated with EGCG dissolved in DMSO and incubated at RT for 1 h, prior to chemical cross-linking. Cross-linking with 15 μM BS3, was carried out at RT for 1 h, and reactions were stopped by incubating with 50 mM Tris-HCl, pH 7.5, for an additional 15 min at RT. Each reaction mixture was then boiled in the presence of 2X sample buffer, and was subjected to SDS-PAGE. Proteins were then transferred and probed with AC88 antibody.
Chaperone function assay
Chaperone function was measured using a luciferase refolding assay (23). RRL, used as the source of hsp90, was supplemented with an ATP generating system (10 mM phosphocreatine, 1 mM ATP, and 100μg/ml creatine phosphokinase). EGCG ( 25-200μM) or an equal volume of DMSO vehicle were added to RRL and allowed to equilibrate for 30 min at RT. Firefly luciferase (Promega, Madison, WI) (100 nM) in Stability Buffer (SB; 25 mM Tris-HCl, pH 7.8, 8 mM MgSO4, 0.1 mM EDTA, 10μg/mL bovine serum albumin, 10% glycerol, and 0.25% Triton-X100) was heat-denatured for 8 min at 40°C and placed on ice for 10 min. Another portion of firefly luciferase in SB was maintained in its native state on ice to serve as a control. The above heat-denatured firefly luciferase was diluted in RRL containing EGCG or vehicle (DMSO) to a final concentration of 10nM in SB and incubated at 28°C for 1h. Aliquots of 5μL were transferred from each reaction to a cooled white-walled 96-well plate (Corning, Corning NY) and assayed for luciferase refolding, as determined by luciferase enzyme activity using a Packard LumiCount (Packard BioScience, Meriden, CT), following the addition of 100 μL assay buffer (25 mM Tris-HCl, pH 7.8; 8 mM MgSO4, 0.1 mM EDTA, 12 mM DTT, 100 μM luciferin, 240 μM CoA, and 0.5 mM ATP). As a control, the native luciferase was utilized in the assay instead of the denatured luciferase and the activity was measured to ensure that EGCG did not directly inhibit luciferase activity. Relative light units were determined as a measure of luciferase activity, and were normalized to control values. Values are expressed as mean ± SD. Six parallel samples were tested for each treatment. For some samples, exogenous human hsp90 (200 μg/ml) was added to diluted RRL prior to EGCG or DMSO treatment.
Cell culture and preparation of cytosol and whole cell lysate
Mouse hepatoma cells, Hepa 1c1c7, were grown at 5% CO2 in Minimum Essential Medium (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum, sodium pyruvate, L-glutamine, sodium bicarbonate, and Gentamycin. Human ovarian carcinoma SKOV3 cells [a kind gift from Dr L. Li (University of Rochester Medical Center, Rochester, NY)] were grown at 5% CO2 in McCoy 5A modified medium (Invitrogen, Carlsbad, CA). Preparation of cytosols and whole cell lysate of Hepa cells was as described previously (7) with some modification. When reaching 90% confluence, cells were harvested by scraping and centrifugation, and then homogenized in HEDG buffer containing protease inhibitors (Complete mini cocktail tablet, Roche Applied Science, Indianapolis, IN). Cytosols were prepared by centrifugation of the homogenate at 100,000g for 40 min. For whole cell lysate, the homogenates were further incubated in the presence of 0.42M NaCl at 4°C for 30min and centrifuged at 100,000g for 40 min. Total protein concentrations were determined by the Coomassie Plus assay (Pierce Chemical). Whole cell lysate of SKOV3 cells was collected by incubating the cells with passive lysis buffer (Promega).
Co-immunoprecipitation
Protein A/G agarose was pre-incubated with specific or non-specific antibodies (2μg antibody /50uL agarose slurry) for 1h at 4°C. The antibodies used were anti-AhR [rabbit polyclonal, against N-terminal peptide, Multiple Peptides System, San Diego, CA (Figure 4) and mouse monoclonal, RPT-9, Affinity BioReagents (Figure 5)], and anti-hsp90 (PA3-013). Cytosol was adjusted to 2.5mg/ml of total protein concentration by adding HEDGMo buffer, treated with DMSO, 1nM TCDD, 200μM EGCG, or T+E for 2h at RT and diluted to 1mg/ml with HEDGMo buffer. The in vitro expression mix in TnT system was diluted 1:4 by adding HEDGMo buffer before the 2h compound treatment as for the cytosol, and then diluted 1:2.5 further with HEDGMo before co-immunoprecipitation. Whole cell lysate was diluted with HEDGMo buffer to a final NaCl concentration of 0.12-0.15M before co-immunoprecipitation. For every treatment, up to 500μg of total protein was added into 50uL of antibody-saturated Protein A/G agarose, incubated with rocking for 2h or overnight at 4°C, and the samples were briefly centrifuged to remove the supernatant. The pellets were washed twice with HEDGMo plus 150mM NaCl buffer. Immunoprecipitated proteins were then separated by SDS-PAGE.
Fig 4.

EGCG alters the interaction of hsp90 with the AhR, an hsp90 client protein, and stabilizes an AhR complex that includes hsp90 and XAP2. A, B. Hepa cytosol was incubated with DMSO, 1nM TCDD (T), 100μM EGCG (E) or T plus E for 2h at RT and immunoprecipitated with anti-hsp90 (PA3-013) (A), anti-AhR (B) or non-specific antibody. C. Hepa cells were incubated with DMSO, 1nM TCDD (T), 100μM EGCG (E) or T plus E for 1h. Whole cell lysates were collected and then immunoprecipitated with anti-AhR or non-specific antibody. Precipitated proteins as well as untreated cytosol or lysate (Input) were resolved by SDS-PAGE and blotted with anti-AhR, anti-hsp90, anti-XAP2, and anti-p23 antibody respectively.
Fig 5.
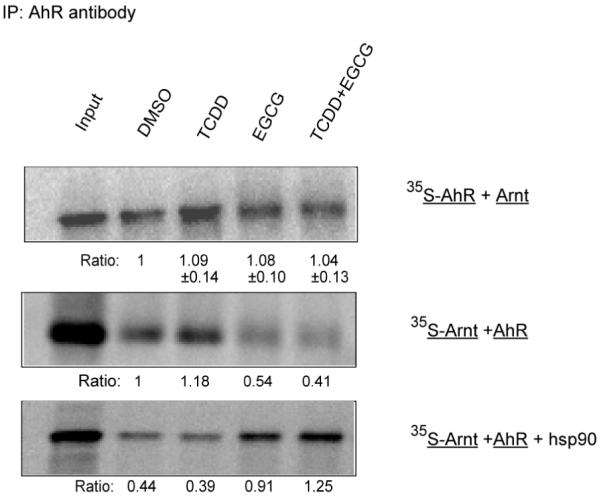
EGCG decreases the association of Arnt with TCDD-activated AhR. Mouse AhR and Arnt were separately in vitro translated in rabbit reticulocyte lysate. Only one of them was expressed each time in the presence of [35S]-Methionine. Equal volumes of diluted AhR and Arnt translation were mixed, incubated with DMSO, 1nM TCDD (T), 200μM EGCG (E) or T plus E for 2h at RT and immunoprecipitated with anti-AhR antibody. For some samples, exogenous human hsp90 (200μg/ml) was added before treatments. All samples were separated by 7.5% SDS-PAGE and dried gels were visualized and quantified by PhosphorImager. Input lysate (Input) was loaded as positive control. Numbers below the 35S-AhR bands (Ratio) are the relative amounts of immunoprecipitated AhR from different treatments normalized to DMSO treatment (mean± SD, n=4). Numbers below the 35S-Arnt bands (Ratio) are the calculated fold induction of Arnt associated with AhR by different treatments normalized to DMSO treatment in the absence of additional hsp90. No 35S-AhR or -Arnt were detectable when nonspecific IgG was used for immunoprecipitation. Representative of at least three experiments.
MTT assay
Upon confluence, cultured cells were washed by PBS without phenol red. MTT in PBS (5mg/ml) were added into wells in a 24-well plate and incubated at 37°C for 3 h. At the end of the incubation period, the medium was moved. The converted dye was then solubilized with acidic isopropanol (0.04 M HCl in isopropanol). Absorbance of the converted dye is measured at a wavelength of 570nm with background subtraction at 650nm.
RESULTS
EGCG elicits a characteristic hsp90 proteolytic footprint, indicating its binding to the C-terminal region of hsp90
The hsp90 inhibitors, geldanamycin and novobiocin, bind to different domains in hsp90, N-terminal and C-terminal, respectively, and therefore lead to different fragmentation patterns following proteolytic footprinting (22, 24). Here, footprinting of hsp90 was performed to determine whether the binding of EGCG induces a conformational change that is similar to or different from other hsp90-binding agents. 35S-labeled chicken hsp90 was expressed in vitro in rabbit reticulocyte lysate (RRL) and then incubated with EGCG or DMSO. The footprint of hsp90 was obtained by treatment with different concentrations of trypsin. There was a clear EGCG-dependent stabilization of a radiolabeled band at approximately 85 kDa representing the full-length hsp90 (Fig. 1B). In addition, a radiolabeled band at approximately 50 kDa appeared with increasing concentrations of trypsin. However, from these results we could not determine, based on the major trypsin cleavage sites in hsp90 (Fig. 1A), whether the 50 kDa band represented an hsp90 fragment at the N-terminal or C-terminal end. Furthermore, these data could not conclusively distinguish whether EGCG binds the C-terminal fragment directly or through another protein in the TnT lysate that may be associated with the C-terminal fragment. To determine this, we utilized antibodies specific to the N-terminal and C-terminal regions of hsp90 to define a trypsinolytic footprint of purified unlabeled hsp90. These data (Fig. 1C) strongly suggest that the ∼50 kDa band is a C-terminal fragment. This same pattern of proteolytic footprint was found at EGCG concentrations as low as ∼5μM (not shown). Together, these studies indicate that EGCG changes the conformation of full-length hsp90 such that its susceptibility to trypsin-mediated degradation is altered. Furthermore, along with our previous finding that EGCG-conjugated Sepharose binds to a C-terminal fragment (residues 538-738) of chicken hsp90 (7), we interpret these data to indicate that EGCG binds directly to a site on the C-terminal region of hsp90. Notably, novobiocin-treated hsp90 also presented a similar footprint compared to that of the EGCG treated hsp90 (not shown), while the footprint of geldanamycin (GA)-bound hsp90 resembled the DMSO treated hsp90 footprint (not shown) (22, 24).
Fig 1.

EGCG modulates the conformation of hsp90 and binds to the C-terminal region of hsp90. A. Major trypsin cleavage sites of Hsp90 in its native conformation and proposed major cleavage sites of Hsp90 in its EGCG bound conformation are shown on the top. Origins of N- and C-terminal Hsp90 fragments are shown in the lower part of the panel. B. Chicken hsp90 was translated in vitro in the presence of [35S]-Methionine, diluted, incubated with DMSO or 200μM EGCG and then treated with trypsin at indicated concentrations. All samples were separated by 10% SDS-PAGE, and visualized by phosphorImager. C. Purified human hsp90 protein was incubated with DMSO or 200μM EGCG and then treated with trypsin at indicated concentrations. All samples were separated by 10% SDS-PAGE, transferred, and blotted with antibody recognizing the N-terminal (PA3-013) or C-terminal (AC88) of hsp90. D. Purified human hsp90 was incubated with DMSO (D), 200μM EGCG (E), 10μg/ml GA, or 10μg/ml GA plus 200μM EGCG (E) and immunoprecipitated with AC88 or PA3-013. Immunoprecipitated hsp90 was subjected to SDS-PAGE and blotted with AC88. E. Purified hsp90 or hsp90N520 were treated with 2-200μM EGCG or 1-10μg/ml geldanamycin (GA) at RT for 1h, and incubated with ATP-agarose or control agarose. The eluted proteins were separated by SDS-PAGE and blotted with AC88 antibody. Representative of at least three experiments.
EGCG affects the ability of a C-terminal hsp90 antibody to immunoprecipitate hsp90
To further investigate the effect of EGCG on hsp90 conformation, we examined the ability of the AC88 anti-hsp90 antibody to immunoprecipitate purified hsp90. AC88, for which the epitope maps to residues 661-677 (25), does not immunoprecipitate hsp90 present in the complex with client proteins (26), and molybdate or novobiocin decreases the ability of AC88 to immunoadsorb hsp90 from reticulocyte lysate (22). As shown in Figure 1D, AC88 effectively immunoprecipitated DMSO-treated hsp90. By comparison, EGCG treatment impaired the ability of AC88 to immunoprecipitate hsp90, while GA enhanced this immunoprecipitation. Co-treatment with EGCG and GA decreased the immunoprecipitation compared to GA treatment alone. In contrast, EGCG treatment did not impair, but slightly enhanced the ability of PA3-013 antibody (against N-terminus of hsp90) to immunoprecipitate hsp90. EGCG had no effect on the recognition of hsp90 by the AC88 antibody by Western blotting following SDS-PAGE (not shown). These observations provide additional evidence that EGCG modifies the conformation of the C-terminal portion of hsp90.
EGCG inhibits the binding of hsp90 to ATP-agarose
In addition to an N-terminal ATP binding site, a second ATP binding site has been identified in the C-terminal region of hsp90 (residues 663-676) to which novobiocin also binds (27, 28). Given the above findings, it is reasonable to speculate that EGCG binds at or near to this ATP binding domain. To further test this hypothesis, an ATP-agarose pull-down assay was performed to determine whether EGCG could inhibit the binding of ATP to purified hsp90 or its C-terminal fragment that contains the C-terminal ATP binding domain. As shown in Figure 1E, EGCG inhibited the binding of hsp90 or hsp90N520 to ATP-agarose in a concentration-dependent manner. As expected, GA inhibited the binding of hsp90 but not that of hsp90N520 to ATP-agarose. Control agarose did not bind Hsp90 or hsp90N520 with either treatment. These data suggest that EGCG binds at or near an ATP binding domain in the C-terminal region.
EGCG inhibits hsp90 dimerization at the C-terminal end
The ability of hsp90 to dimerize mediates one of its important functions. By dimerizing through its C-terminal domain, hsp90 exhibits molecular chaperone activity by cycling between an open and closed conformation, like a molecular clamp, that has differential ability to interact with client proteins (29). Since we demonstrated that EGCG alters the conformation of hsp90 through apparent binding at its C-terminal region, we further hypothesized that EGCG would affect hsp90 dimerization. Full-length and C-terminal hsp90 dimerization were assessed by chemical cross-linking following EGCG treatment. As shown in Figure 2A, EGCG inhibited chemically induced dimerization of purified full-length hsp90 in a concentration-dependent manner. Under the experimental conditions used and in the presence of the cross-linking agent, the expressed C-terminal proteins tended to also form tetramers (Fig. 2B). Oligomerization of hsp90 and a C-terminal hsp90 fragment has also been detected by others (30). Nevertheless, EGCG similarly inhibits the oligomerization of these C-terminal hsp90 fragments. The ability of EGCG to affect hsp90 dimerization is consistent with the binding of EGCG at the C-terminal region of hsp90.
Fig 2.

Dose-dependent inhibition of hsp90 dimerization by EGCG. A. Full length human hsp90 was treated with DMSO or 25-200μM EGCG at RT for 1h, prior to chemical cross-linking using BS3 at RT for 1h. Reactions were subsequently stopped by incubating with 50 mM Tris-HCl, pH 7.5, for a further 15 min. Each reaction mixture was subjected to 6% SDS-PAGE. Resolved protein bands were probed with AC88 to detect Hsp90. B. A similar experiment was performed with a C-terminal fragment hsp90N520 of human hsp90 and using 12% SDS-PAGE. Representative of at least three experiments.
EGCG inhibits hsp90 chaperone function
A large number of proteins are known to interact with hsp90 at different domains, such as the C-terminal tetratricopeptide (TPR) acceptor site or the N-terminal p50cdc37 acceptor site (28, 31). If EGCG significantly modifies hsp90 conformation, then the ability of client proteins to bind, in a quantitative or qualitative manner, to these sites would be affected and the subsequent chaperone function would be modified. An important end result of hsp90 chaperone activity is the maintenance of a client protein in a functional conformation. One way to determine if hsp90 chaperone activity is altered by EGCG is to examine its effect on hsp90-facilitated refolding of denatured firefly luciferase (32). This was performed using RRL that is rich in hsp90 as well as other chaperones, such as hsp70 and hsp40, that are necessary for the proper refolding of luciferase to occur (32, 33). As shown in Figure 3, EGCG dose-dependently inhibited luciferase activity in the presence of RRL. In the absence of RRL, EGCG alone did not affect luciferase activity (not shown). The addition of exogenous hsp90 (200μg/ml) prior to EGCG treatment partially restored luciferase activity in all EGCG-treated groups. The addition of bovine serum albumin (200μg/ml), instead of hsp90, did not show any ability to restore the refolding (not shown).
Fig 3.

EGCG inhibits the refolding of denatured luciferase in a dose dependent manner. Diluted rabbit reticulocyte lysate (RRL) was incubated with indicated concentrations of EGCG or vehicle for 30min at RT. Heat-denatured luciferase was diluted in the treated lysate to a final concentration of 10nM and incubated for 40min at 28°C. For each sample, 5uL of the mixture was transferred to a 96-well plate reader and mixed with 100uL of assay buffer. Relative light units were determined as a measure of the luciferase activity. Six parallel samples were tested for each treatment. For some samples, exogenous human hsp90 (200μg/ml) was added to diluted RRL before EGCG treatment. Relative light units were normalized to control values. Values are the mean ± SD.
EGCG alters the interaction of hsp90 with the AhR, an hsp90 client protein, and stabilizes an AhR complex that includes hsp90 and XAP2
The investigations above indicate that EGCG modifies the conformation of hsp90 and alters its ability to serve as a chaperone for client proteins. Additional studies were performed to show that EGCG directly alters the ability of hsp90 to interact with these client proteins. Previous studies in our lab demonstrated that EGCG affects the function of the AhR, an hsp90 client protein, by binding directly to hsp90 but not the AhR (7). To further investigate how EGCG modifies the AhR complex and its ability to regulate gene transcription, as well as to determine if this occurs through an EGCG-elicited alteration in the ability of hsp90 to directly associate with the AhR, immunoprecipitation with hsp90 antibody was performed. Hepa cell cytosol was treated with DMSO, 1nM TCDD, 100μM EGCG, or 1nM TCDD plus 100μM EGCG. Hsp90 was immuno-precipitated with hsp90 antibody and associated proteins were determined by immunoblotting. As shown in Figure 4A, EGCG treatment increased the association of AhR and XAP2 with hsp90, with or without the presence of TCDD. There was little or no effect on the association of another cochaperone, p23, with the complex.
In order to more thoroughly examine the components of the EGCG-induced AhR complex, immunoprecipitation with AhR antibody was performed. Hepa cell cytosol was treated with DMSO, 1nM TCDD, 100μM EGCG, or 1nM TCDD plus 100μM EGCG. AhR was immunoprecipitated with AhR antibody and associated proteins were determined by immunoblotting. As shown in Figure 4B, EGCG treatment increased the association of hsp90, XAP2 and p23 with the AhR and blocked their TCDD-induced dissociation from AhR. To validate the EGCG effect on AhR complex in a cell system, whole cell lysate was collected after Hepa cells were treated with DMSO, 1nM TCDD, 100μM EGCG, or 1nM TCDD plus 100μM EGCG for 1h. The whole cell lysate was then utilized in the co-immunoprecipitation with AhR antibody. Similar to Figure 4B, Figure 4C shows that EGCG treatment stabilized the association of hsp90 and XAP2 with AhR. There was little or no effect on the association of p23 with the complex. The total levels of the above proteins were not affected by EGCG treatment (not shown). Together, these data indicate that, both in vitro and in intact cells, a consequence of an EGCG-elicited change in hsp90 conformation is the stabilization of an AhR complex that includes at least hsp90 and XAP2, and possibly p23. This is consistent with our previous data using a sucrose gradient sedimentation assay (7).
EGCG decreases the association of Arnt with TCDD-activated AhR
A critical function of hsp90 in regulating the signaling actions of many client proteins is the shuttling of these proteins to the nucleus in a conformation that would become transcriptionally active. For the AhR, hsp90 appears to be an important participant in the translocation of the ligand-bound AhR complex containing at least hsp90 and XAP2 (12, 34). Within the nucleus, processes that are not fully understood mediate the dissociation of AhR from hsp90 and the formation of a heterodimer with Arnt (34, 35). The above data indicate that EGCG stabilizes a complex composed of the AhR, hsp90 and XAP2. EGCG treatment of intact cells also appears to result in enhanced nuclear uptake of the AhR, with, however, loss of AhRE recognition and transcriptional activity (7). This suggests that either the EGCG-modified AhR complex fails to mediate the formation of the AhR-Arnt heterodimer, or that this dimer lacks AhRE binding ability and transcriptional activity. To address this question, we examined the ability of EGCG to affect the association between AhR and Arnt. Mouse AhR and Arnt were separately expressed in RRL, with only one being labeled with [35S] in each experiment. Equal amounts of AhR and Arnt were combined, incubated with DMSO, 1nM TCDD, 200μM EGCG or TCDD plus EGCG, then immunoprecipitated with anti-AhR antibody (RPT-9). As shown in Figure 5, the amounts of 35S-AhR precipitated by the anti-AhR antibody were equivalent across all treatments, and the presence of EGCG substantially decreased the heterodimerization of AhR and Arnt activated by TCDD. This effect of EGCG could be blocked by addition of purified human hsp90 (200μg/ml) prior to EGCG treatment. No 35S-AhR or -Arnt were detectable when nonspecific IgG was used for immunoprecipitation (not shown).
EGCG enhances the association of hsp90 with hsp70 and Cyp40
In addition to its direct influence on client folding, hsp90 locally concentrates co-chaperone activity within the client complex, and dynamic exchange of co-chaperones on hsp90 facilitates sampling of co-chaperone activities that can act on the client protein (36). Hsp90 inhibitors, such as GA and novobiocin, have been shown to alter the association of hsp90 with cochaperones such as hsp70, HOP, Cdc37 and p23 (22, 37). To investigate whether EGCG modifies the interaction of hsp90 with the cochaperones and whether this would provide any mechanistic insight into how EGCG affects hsp90 chaperone activity, co-immunoprecipitation was done in Hepa cell cytosol. Hepa cell cytosols were treated with DMSO or 200μM EGCG, immunoprecipitated with hsp90 antibody and associated proteins were determined by immunoblotting. As shown in Figure 6A, EGCG enhanced the association of hsp90 with hsp70 and Cyp40. These experiments were also done in reticulocyte lysate with a similar result (not shown).
Fig.6.

EGCG modifies the interaction of hsp90 with cochaperones, and leads to the degradation of the hsp90 client proteins ErbB2, Raf-1 and phospho-AKT. A. Hepa cytosol was incubated with DMSO or 200μM EGCG (E) for 1h at RT and immunoprecipitated with anti-hsp90 (PA3-013) or non-specific antibody. Precipitated proteins as well as untreated cytosol (Input) were resolved by SDS-PAGE and blotted with anti-hsp70, anti-hsp90 and anti-Cyp40 antibody. B. SKOV-3 cells were treated with 2-50μM EGCG for up to 20h (8h for pAKT and AKT). Cell lysate was collected by incubating with passive lysis buffer, resolved by SDS-PAGE and blotted with anti-hsp70, anti-ErbB2, anti-Raf-1, anti-AKT, anti-pAKT and anti-GAPDH antibody.
EGCG alters cellular levels of cancer-related hsp90 client proteins
The significance of some hsp90 inhibitors, such as 17-AAG, for cancer therapy is that they cause proteasome-mediated degradation of cancer-related hsp90 client proteins, such as ErbB2 and Raf-1 (38). To test whether EGCG affects the levels of these hsp90 client proteins, we treated the SKOV3 cells with 0-50μM EGCG. SKOV3 cells are among the most sensitive cancer cells lines to EGCG’s growth suppressive effect (39). As shown in Figure 6B, EGCG elicits a dose-dependent decrease of the level of ErbB2, Raf-1 and phospho-AKT (pAKT), with a slight increase of the level of hsp70. Cell viability, as determined by MTT assay, was not affected up to 50μM EGCG at this time point (not shown).
DISCUSSION
Catechins are the major constituents in green tea, accounting for 30-42% of the dry weight, and more than 50% of the total catechins are EGCG (2). Abundant studies have suggested that tea consumption has protective effects against cancer and many other diseases in animal models (1, 2). Epidemiological studies, though inconclusive, have also suggested an association between tea consumption and a lower risk of cancer in humans. One study suggested a significantly reduced risk of breast cancer among green tea drinkers (40). Phase II clinical trials have indicated effectiveness of green tea extract against UV-induced skin injuries, oxidative DNA damage, and prostate cancer (41). Most studies of EGCG in isolated cells have demonstrated effective concentrations ranging from 10 to 100μM for inhibition of tumor cell growth (3, 4, 5, 6, 39). In contrast, EGCG concentrations found in human plasma through tea consumption are only in the high nanomolar range (42). Thus, it may be difficult to achieve, through tea consumption, effective concentrations of EGCG that have anti-cancer activity; this may contribute to the inconclusive results of some epidemiological investigations. However, better understanding of the possible mechanisms might allow design of more bioavailable EGCG derivatives which could be developed for chemotherapeutic purposes.
All of the data we presented here and elsewhere (7) are consistent with the notion that EGCG is an inhibitor of hsp90 function through its interaction with the C-terminal end of this protein, in particular at or near an ATP binding site. ATP plays an important role in hsp90 chaperone function in terms of stabilizing the hsp90 structure as well as completion of the chaperone cycle (29). There are two ATP binding sites, one on the N-terminus and the other on the C-terminus (27). The binding of ATP to the N-terminal site induces the association of the N-terminal domains forming a closed conformation and resulting in stabilization of client protein binding (27). Binding of GA to the N-terminal ATP site prevents this closed conformation, and permits destabilization of hsp90-client protein complexes. The precise function of the C-terminal ATP binding site is less understood. Previous studies have indicated that N-terminal ATP binding is required for the C-terminal site to become available for nucleotide binding (27, 28, 43). Our finding that GA enhances the ability of AC88 to immunoprecipitate hsp90 is consistent with the N-terminal ATP binding site being involved in the unmasking of the C-terminal site. ATP was found to stabilize a C-terminal hsp90 fragment in a trypsinolytic footprinting (22), suggesting that ATP binding modifies the conformation of the C-terminal region. Novobiocin, which binds to the C-terminus of hsp90, is believed to bind to this ATP binding site residing within residues 663-676 (28), and this binding has been shown also to lead to the destabilization of hsp90-client protein interactions. Thus, it has been suggested that hsp90 function is regulated by the cyclic binding, hydrolysis and/or exchange of ATP at these two sites resulting in hsp90 conformations available for and stabilizing the interactions with client proteins and cochaperones.
In the present study, EGCG was found to protect a C-terminal hsp90 fragment from lysis by trypsin, in similar fashion as both ATP and novobiocin (22, 28). EGCG also impaired the ability of AC88, whose epitope was mapped to residues 661-676 (25), to immunoprecipitate hsp90, and competed with hsp90 or its C-terminal fragment to bind to ATP-agarose. These data indicate that EGCG binds at or near the C-terminal ATP binding site on hsp90. Consistent with this, we further demonstrated that EGCG effectively blocks hsp90 dimerization as well as chaperone function determined by the ability to mediate refolding of a denatured client, luciferase. Together, these data indicate that the mechanism of EGCG acting as an hsp90 inhibitor is through its ability to bind at or near to the C-terminal ATP binding site of hsp90, disrupting ATP binding, inducing an inappropriate conformational change, and inhibiting hsp90 chaperone function. A recent study found that EGCG decreases the K+-ATP channel’s sensitivity to ATP, and suggested that the 5’OH group in the B-ring in EGCG is critical for binding to the ATP binding pocket (44). EGCG was also found to compete with ATP for binding to a glucose-regulated protein 78 (GRP78), an hsp70 family member (5). In addition, EGCG was found to bind to an ATP binding site on insulin-like growth factor-1 receptor (IGF-1R) (6). It remains to be determined whether the ability of EGCG to affect ATP binding sites in a variety of proteins may be a general feature of EGCG and other EGCG-like compounds.
To show more effectively the ability of EGCG to modify chaperone activity, we examined in more detail the mechanism whereby EGCG inhibits the transcriptional activity of the AhR, an hsp90 client protein. Based on current knowledge, our previous data (7), and the data we present here, we propose a model in which the AhR exists in multiple forms within the cytoplasm coupled with hsp90, but also determined by the relative absence or presence of XAP2 and/or p23 (Figure 7). TCDD binding initiates transformation of the cytosolic AhR to a nuclear AhR-Arnt heterodimer which then binds to AhRE. The binding of EGCG to hsp90 stabilizes an AhR complex that includes at least hsp90 and XAP2, and possibly p23. However, the interaction of EGCG with the hsp90-AhR complex also apparently modifies AhR conformation to expose the NLS sequence resulting in translocation of the AhR complex into the nucleus (7). The stabilization of the AhR-hsp90-XAP2 complex prevents TCDD-mediated AhR-Arnt association and blocks AhR-mediated transcriptional activity.
Fig 7.

A proposed model for the AhR complex response to TCDD and its modification by EGCG. In the cytoplasm, the AhR forms a complex with hsp90, XAP2, and p23. TCDD binding initiates the translocation of AhR to the nucleus, its dissociation from XAP2, p23, and hsp90, its dimerization with Arnt and the binding of the heterodimer to DNA. Binding of EGCG to hsp90 stablizes an AhR complex that includes hsp90 and XAP2, prevents TCDD-mediated AhR-Arnt association, and blocks AhR-mediated transcriptional activity. This AhR complex is capable of nuclear translocation through exposure of the Nuclear Localization Sequence (NLS).
GA, which binds to the N-terminal ATP binding site of hsp90, has also been found to block TCDD-induced transcription by affecting the AhR complex. In contrast to EGCG, GA destabilizes the hsp90-AhR interaction, induces AhR nuclear translocation and release of p23, does not prevent the TCDD-induced AhR-Arnt heterodimerization, but leads to increased proteosomal degradation of the AhR (25). Molybdate, which binds the C-terminus of hsp90, enhances the association of hsp90-AhR complex, prevents AhR nuclear translocation, and prevents TCDD-induced Arnt hetero-dimerization and AhR degradation (45). Our studies, presented here and elsewhere (7), indicate that EGCG stabilizes the AhR complex, induces AhR nuclear translocation, but prevents the TCDD-induced Arnt heterodimerization and degradation of the AhR. Notably, EGCG was also shown to increase the nuclear accumulation of Nrf-2 (NF-E2-related factor). However, contrary to that of AhR, the DNA binding and transcriptional activity of Nrf-2 were enhanced by EGCG treatment in human breast epithelial (MCF10A) cells (46). Therefore, EGCG appears to act uniquely among the hsp90 inhibitors. The uniqueness of EGCG as an hsp90 inhibitor is also examplified by its modification on the hsp90-cochaperone complex. In these investigations, we illustrated that EGCG alters the interaction of hsp90 with the cochaperones. Differently from any other hsp90 inhibitors, EGCG stabilizes the association of hsp70, Cyp40 and XAP2 to hsp90. Notably, another C-terminal hsp90 inhibitor, novobiocin significantly reduces the interaction of hsc70, FKBP52 and p23 with hsp90 (22, 47). Generally, it is thought that hsp70 binds to hsp90 in an “intermediate” complex through HOP, and dissociates with HOP from the late complex that contains TPR co-chaperones, such as CyP40 (26). Our observations suggest that EGCG could stabilize a novel hsp90-co-chaperone complex which may contribute to the different effects of EGCG compared with other hsp90 inhibitors.
Our data further indicate that EGCG binds not only to AhR-bound hsp90 but also purified hsp90. This suggests that EGCG is likely to affect other hsp90 client proteins through binding to hsp90 and inhibition of hsp90 functions such as dimerization and chaperone activity. Shown in the current investigation, as an example of hsp90 client proteins, AhR activity is affected by EGCG but not the level of AhR protein. However, the effects of EGCG on other hsp90 client proteins may not be the same as for the AhR, as shown in figure 6B and elsewhere (1, 2, 3, 39). In our investigations, we demonstrated that EGCG decreases the level of the hsp90 client proteins ErbB2, Raf-1 and pAKT in SKOV3 cells. Other data have shown that EGCG alters levels and/or activity of many hsp90 client proteins including ErbB2, AKT, CDK4, and Raf-1 both in vivo and in vitro in several different cell types (1, 2, 3, 39). Much evidence derived from the use of the GA-derived hsp90 inhibitors and novobiocin has shown these client proteins to be effective biomarkers of hsp90 inhibition (48). Clearly, further research is needed to define effects of EGCG on different client proteins and the mechanisms for these effects. We recognize that the ability of EGCG to affect hsp90 function may not be the only mechanism whereby EGCG exerts its anti-cancer effect, as many mechanisms have been postulated (3, 4, 5, 6). However, the ability to affect hsp90 may be a common mechanism for several of them that have been proposed. Interestingly, several proteins reported to bind to EGCG are either hsp90 client proteins (vimentin, IGF-1R, Grp78) (4, 5, 6) or closely related (laminin receptor) (49) to hsp90. Furthermore, inhibition of hsp90 disrupts their signaling pathways or leads to their degradation respectively (50). Additional studies are necessary to determine whether hsp90 plays a role in their binding to EGCG.
It is known that different clients have different binding sites on hsp90, and also likely require different interactions with cochaperone proteins other than or in addition to XAP2, hsp70 and Cyp40 (26). Moreover, different types of tumor cells are likely to depend on different, select and/or multiple hsp90 client protein-dependent signaling pathways for their growth and survival. As such, the ultimate client protein-dependent signaling pathway affected by EGCG that leads to inhibition of tumor growth may be tumor cell specific. Critical experiments providing conclusive evidence that inhibition of hsp90 is responsible for the anti-tumor effects of EGCG are likely to be of many types, cell specific, and reasonably complex.
That EGCG acts as an hsp90 inhibitor is a novel finding. Since hsp90 inhibitors such as 17-AAG, have been intensively studied for their anti-cancer effects, developing derivatives or other hsp90 inhibitors that are selective and more bioavailable becomes advantageous for identifying agents with increased therapeutic efficacy. Our identification here of the specific region on hsp90 with which EGCG interacts provides some momentum for research that may ultimately lead to the use of EGCG and/or of its derivatives for chemotherapeutic purpose.
ACKNOWLEDGEMENT
We thank Drs. D.O. Toft, T. Ratajczak and L. Li respectively for providing the hsp90 constructs and SKOV-3 cells utilized in these studies.
ABBREVIATIONS
- AhR
aryl hydrocarbon receptor
- Arnt
aryl hydrocarbon receptor nuclear translocator
- AhRE
AhR response element
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- EGCG
(-)-epigallocatechin-3-gallate
- GA
geldanamycin
- 17-AAG
17-allylamino-geldanamycin
- hsp90
90kDa heat shock protein
- XAP2
hepatitis B virus X-associated protein 2
- NLS
nuclear localization signal
- RT
room temperature
- BS3
bis-sulfosuccinimidyl suberate
Footnotes
The work was supported by NIH Grant ES014364, and Center Grant ES01247.
REFERENCE
- 1.Zaveri NT. Green Tea and its Polyphenolic Catechins: Medicinal Uses in Cancer and Noncancer Applications. Life Sci. 2006;78:2073–2080. doi: 10.1016/j.lfs.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 2.Yang CS, Chen L, Lee MJ, Landau JM. Effects of Tea on Carcinogenesis in Animal Models and Humans. Adv. Exp. Med. Biol. 1996;401:51–61. doi: 10.1007/978-1-4613-0399-2_5. [DOI] [PubMed] [Google Scholar]
- 3.Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Targeting Multiple Signaling Pathways by Green Tea Polyphenol (-)-Epigallocatechin-3-Gallate. Cancer Res. 2006;66:2500–2505. doi: 10.1158/0008-5472.CAN-05-3636. [DOI] [PubMed] [Google Scholar]
- 4.Ermakova S, Choi BY, Choi HS, Kang BS, Bode AM, Dong Z. The Intermediate Filament Protein Vimentin is a New Target for Epigallocatechin Gallate. J. Biol. Chem. 2005;280:16882–16890. doi: 10.1074/jbc.M414185200. [DOI] [PubMed] [Google Scholar]
- 5.Ermakova SP, Kang BS, Choi BY, Choi HS, Schuster TF, Ma WY, Bode AM, Dong Z. (-)-Epigallocatechin Gallate Overcomes Resistance to Etoposide-Induced Cell Death by Targeting the Molecular Chaperone Glucose-Regulated Protein 78. Cancer Res. 2006;66:9260–9269. doi: 10.1158/0008-5472.CAN-06-1586. [DOI] [PubMed] [Google Scholar]
- 6.Li M, He Z, Ermakova S, Zheng D, Tang F, Cho YY, Zhu F, Ma WY, Sham Y, Rogozin EA, Bode AM, Cao Y, Dong Z. Direct Inhibition of Insulin-Like Growth Factor-I Receptor Kinase Activity by (-)-Epigallocatechin-3-Gallate Regulates Cell Transformation. Cancer Epidemiol. Biomarkers Prev. 2007;16:598–605. doi: 10.1158/1055-9965.EPI-06-0892. [DOI] [PubMed] [Google Scholar]
- 7.Palermo CM, Westlake CA, Gasiewicz TA. Epigallocatechin Gallate Inhibits Aryl Hydrocarbon Receptor Gene Transcription through an Indirect Mechanism Involving Binding to a 90 kDa Heat Shock Protein. Biochemistry. 2005;44:5041–5052. doi: 10.1021/bi047433p. [DOI] [PubMed] [Google Scholar]
- 8.Palermo CM, Hernando JI, Dertinger SD, Kende AS, Gasiewicz TA. Identification of Potential Aryl Hydrocarbon Receptor Antagonists in Green Tea. Chem. Res. Toxicol. 2003;16:865–872. doi: 10.1021/tx025672c. [DOI] [PubMed] [Google Scholar]
- 9.Meyer BK, Perdew GH. Characterization of the AhR-hsp90-XAP2 Core Complex and the Role of the Immunophilin-Related Protein XAP2 in AhR Stabilization. Biochemistry. 1999;38:8907–8917. doi: 10.1021/bi982223w. [DOI] [PubMed] [Google Scholar]
- 10.Bell DR, Poland A. Binding of Aryl Hydrocarbon Receptor (AhR) to AhR-Interacting Protein. the Role of hsp90. J. Biol. Chem. 2000;275:36407–36414. doi: 10.1074/jbc.M004236200. [DOI] [PubMed] [Google Scholar]
- 11.Chen HS, Perdew GH. Subunit Composition of the Heteromeric Cytosolic Aryl Hydrocarbon Receptor Complex. J. Biol. Chem. 1994;269:27554–27558. [PubMed] [Google Scholar]
- 12.Rowlands JC, Gustafsson JA. Aryl Hydrocarbon Receptor-Mediated Signal Transduction. Crit. Rev. Toxicol. 1997;27:109–134. doi: 10.3109/10408449709021615. [DOI] [PubMed] [Google Scholar]
- 13.Bagatell R, Whitesell L. Altered Hsp90 Function in Cancer: A Unique Therapeutic Opportunity. Mol. Cancer. Ther. 2004;3:1021–1030. [PubMed] [Google Scholar]
- 14.Terasawa K, Minami M, Minami Y. Constantly Updated Knowledge of Hsp90. J. Biochem. (Tokyo) 2005;137:443–447. doi: 10.1093/jb/mvi056. [DOI] [PubMed] [Google Scholar]
- 15.Chiosis G, Huezo H, Rosen N, Mimnaugh E, Whitesell L, Neckers L. 17AAG: Low Target Binding Affinity and Potent Cell Activity--Finding an Explanation. Mol. Cancer. Ther. 2003;2:123–129. [PubMed] [Google Scholar]
- 16.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A High-Affinity Conformation of Hsp90 Confers Tumour Selectivity on Hsp90 Inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 17.Ahmad N, Feyes DK, Nieminen AL, Agarwal R, Mukhtar H. Green Tea Constituent Epigallocatechin-3-Gallate and Induction of Apoptosis and Cell Cycle Arrest in Human Carcinoma Cells. J. Natl. Cancer Inst. 1997;89:1881–1886. doi: 10.1093/jnci/89.24.1881. [DOI] [PubMed] [Google Scholar]
- 18.Neckers L, Ivy SP. Heat Shock Protein 90. Curr. Opin. Oncol. 2003;15:419–424. doi: 10.1097/00001622-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Vanden Berghe T, Kalai M, van Loo G, Declercq W, Vandenabeele P. Disruption of HSP90 Function Reverts Tumor Necrosis Factor-Induced Necrosis to Apoptosis. J. Biol. Chem. 2003;278:5622–5629. doi: 10.1074/jbc.M208925200. [DOI] [PubMed] [Google Scholar]
- 20.Ramanathan RK, Egorin MJ, Eiseman JL, Ramalingam S, Friedland D, Agarwala SS, Ivy SP, Potter DM, Chatta G, Zuhowski EG, Stoller RG, Naret C, Guo J, Belani CP. Phase I and Pharmacodynamic Study of 17-(Allylamino)-17-Demethoxygeldanamycin in Adult Patients with Refractory Advanced Cancers. Clin. Cancer Res. 2007;13:1769–1774. doi: 10.1158/1078-0432.CCR-06-2233. [DOI] [PubMed] [Google Scholar]
- 21.Henry EC, Bemis JC, Henry O, Kende AS, Gasiewicz TA. A Potential Endogenous Ligand for the Aryl Hydrocarbon Receptor has Potent Agonist Activity in Vitro and in Vivo. Arch. Biochem. Biophys. 2006;450:67–77. doi: 10.1016/j.abb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 22.Yun BG, Huang W, Leach N, Hartson SD, Matts RL. Novobiocin Induces a Distinct Conformation of Hsp90 and Alters Hsp90-Cochaperone-Client Interactions. Biochemistry. 2004;43:8217–8229. doi: 10.1021/bi0497998. [DOI] [PubMed] [Google Scholar]
- 23.Turbyville TJ, Wijeratne EM, Liu MX, Burns AM, Seliga CJ, Luevano LA, David CL, Faeth SH, Whitesell L, Gunatilaka AA. Search for Hsp90 Inhibitors with Potential Anticancer Activity: Isolation and SAR Studies of Radicicol and Monocillin I from Two Plant-Associated Fungi of the Sonoran Desert. J. Nat. Prod. 2006;69:178–184. doi: 10.1021/np058095b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartson SD, Thulasiraman V, Huang W, Whitesell L, Matts RL. Molybdate Inhibits hsp90, Induces Structural Changes in its C-Terminal Domain, and Alters its Interactions with Substrates. Biochemistry. 1999;38:3837–3849. doi: 10.1021/bi983027s. [DOI] [PubMed] [Google Scholar]
- 25.Chen HS, Singh SS, Perdew GH. The Ah Receptor is a Sensitive Target of Geldanamycin-Induced Protein Turnover. Arch. Biochem. Biophys. 1997;348:190–198. doi: 10.1006/abbi.1997.0398. [DOI] [PubMed] [Google Scholar]
- 26.Pratt WB, Toft DO. Steroid Receptor Interactions with Heat Shock Protein and Immunophilin Chaperones. Endocr. Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 27.Garnier C, Lafitte D, Tsvetkov PO, Barbier P, Leclerc-Devin J, Millot JM, Briand C, Makarov AA, Catelli MG, Peyrot V. Binding of ATP to Heat Shock Protein 90: Evidence for an ATP-Binding Site in the C-Terminal Domain. J. Biol. Chem. 2002;277:12208–12214. doi: 10.1074/jbc.M111874200. [DOI] [PubMed] [Google Scholar]
- 28.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The Heat Shock Protein 90 Antagonist Novobiocin Interacts with a Previously Unrecognized ATP-Binding Domain in the Carboxyl Terminus of the Chaperone. J. Biol. Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 29.Pearl LH, Prodromou C. Structure and Mechanism of the Hsp90 Molecular Chaperone Machinery. Annu. Rev. Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 30.Nemoto T, Sato N. Oligomeric Forms of the 90-kDa Heat Shock Protein. Biochem. J. 1998;330(Pt 2):989–995. doi: 10.1042/bj3300989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcu MG, Neckers LM. The C-Terminal Half of Heat Shock Protein 90 Represents a Second Site for Pharmacologic Intervention in Chaperone Function. Curr. Cancer. Drug Targets. 2003;3:343–347. doi: 10.2174/1568009033481804. [DOI] [PubMed] [Google Scholar]
- 32.Schumacher RJ, Hurst R, Sullivan WP, McMahon NJ, Toft DO, Matts RL. ATP-Dependent Chaperoning Activity of Reticulocyte Lysate. J. Biol. Chem. 1994;269:9493–9499. [PubMed] [Google Scholar]
- 33.Thulasiraman V, Matts RL. Effect of Geldanamycin on the Kinetics of Chaperone-Mediated Renaturation of Firefly Luciferase in Rabbit Reticulocyte Lysate. Biochemistry. 1996;35:13443–13450. doi: 10.1021/bi9615396. [DOI] [PubMed] [Google Scholar]
- 34.Carlson DB, Perdew GH. A Dynamic Role for the Ah Receptor in Cell Signaling? Insights from a Diverse Group of Ah Receptor Interacting Proteins. J. Biochem. Mol. Toxicol. 2002;16:317–325. doi: 10.1002/jbt.10051. [DOI] [PubMed] [Google Scholar]
- 35.Henry EC, Kende AS, Rucci G, Totleben MJ, Willey JJ, Dertinger SD, Pollenz RS, Jones JP, Gasiewicz TA. Flavone Antagonists Bind Competitively with 2,3,7, 8-Tetrachlorodibenzo-p-Dioxin (TCDD) to the Aryl Hydrocarbon Receptor but Inhibit Nuclear Uptake and Transformation. Mol. Pharmacol. 1999;55:716–725. [PubMed] [Google Scholar]
- 36.Riggs DL, Cox MB, Cheung-Flynn J, Prapapanich V, Carrigan PE, Smith DF. Functional Specificity of Co-Chaperone Interactions with Hsp90 Client Proteins. Crit. Rev. Biochem. Mol. Biol. 2004;39:279–295. doi: 10.1080/10409230490892513. [DOI] [PubMed] [Google Scholar]
- 37.An WG, Schulte TW, Neckers LM. The Heat Shock Protein 90 Antagonist Geldanamycin Alters Chaperone Association with p210bcr-Abl and v-Src Proteins before their Degradation by the Proteasome. Cell Growth Differ. 2000;11:355–360. [PubMed] [Google Scholar]
- 38.Zsebik B, Citri A, Isola J, Yarden Y, Szollosi J, Vereb G. Hsp90 Inhibitor 17-AAG Reduces ErbB2 Levels and Inhibits Proliferation of the Trastuzumab Resistant Breast Tumor Cell Line JIMT-1. Immunol. Lett. 2006;104:146–155. doi: 10.1016/j.imlet.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 39.Kweon MH, Adhami VM, Lee JS, Mukhtar H. Constitutive Overexpression of Nrf2-Dependent Heme Oxygenase-1 in A549 Cells Contributes to Resistance to Apoptosis Induced by Epigallocatechin 3-Gallate. J. Biol. Chem. 2006;281:33761–33772. doi: 10.1074/jbc.M604748200. [DOI] [PubMed] [Google Scholar]
- 40.Wu AH, Yu MC, Tseng CC, Hankin J, Pike MC. Green Tea and Risk of Breast Cancer in Asian Americans. Int. J. Cancer. 2003;106:574–579. doi: 10.1002/ijc.11259. [DOI] [PubMed] [Google Scholar]
- 41.Bettuzzi S, Brausi M, Rizzi F, Castagnetti G, Peracchia G, Corti A. Chemoprevention of Human Prostate Cancer by Oral Administration of Green Tea Catechins in Volunteers with High-Grade Prostate Intraepithelial Neoplasia: A Preliminary Report from a One-Year Proof-of-Principle Study. Cancer Res. 2006;66:1234–1240. doi: 10.1158/0008-5472.CAN-05-1145. [DOI] [PubMed] [Google Scholar]
- 42.Henning SM, Niu Y, Lee NH, Thames GD, Minutti RR, Wang H, Go VL, Heber D. Bioavailability and Antioxidant Activity of Tea Flavanols After Consumption of Green Tea, Black Tea, Or a Green Tea Extract Supplement. Am. J. Clin. Nutr. 2004;80:1558–1564. doi: 10.1093/ajcn/80.6.1558. [DOI] [PubMed] [Google Scholar]
- 43.Soti C, Racz A, Csermely P. A Nucleotide-Dependent Molecular Switch Controls ATP Binding at the C-Terminal Domain of Hsp90. N-Terminal Nucleotide Binding Unmasks a C-Terminal Binding Pocket. J. Biol. Chem. 2002;277:7066–7075. doi: 10.1074/jbc.M105568200. [DOI] [PubMed] [Google Scholar]
- 44.Jin JY, Park SH, Bae JH, Cho HC, Lim JG, Park WS, Han J, Lee JH, Song DK. Uncoupling by (-)-Epigallocatechin-3-Gallate of ATP-Sensitive Potassium Channels from Phosphatidylinositol Polyphosphates and ATP. Pharmacol. Res. 2007;56:237–247. doi: 10.1016/j.phrs.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Heid SE, Pollenz RS, Swanson HI. Role of Heat Shock Protein 90 Dissociation in Mediating Agonist-Induced Activation of the Aryl Hydrocarbon Receptor. Mol. Pharmacol. 2000;57:82–92. [PubMed] [Google Scholar]
- 46.Na HK, Kim EH, Jung JH, Lee HH, Hyun JW, Surh YJ. (-)-Epigallocatechin Gallate Induces Nrf2-Mediated Antioxidant Enzyme Expression Via Activation of PI3K and ERK in Human Mammary Epithelial Cells. Arch. Biochem. Biophys. 2008 doi: 10.1016/j.abb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Allan RK, Mok D, Ward BK, Ratajczak T. Modulation of Chaperone Function and Cochaperone Interaction by Novobiocin in the C-Terminal Domain of Hsp90: Evidence that Coumarin Antibiotics Disrupt Hsp90 Dimerization. J. Biol. Chem. 2006;281:7161–7171. doi: 10.1074/jbc.M512406200. [DOI] [PubMed] [Google Scholar]
- 48.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt Forms an Intracellular Complex with Heat Shock Protein 90 (Hsp90) and Cdc37 and is Destabilized by Inhibitors of Hsp90 Function. J. Biol. Chem. 2002;277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 49.Tachibana H, Koga K, Fujimura Y, Yamada K. A Receptor for Green Tea Polyphenol EGCG. Nat. Struct. Mol. Biol. 2004;11:380–381. doi: 10.1038/nsmb743. [DOI] [PubMed] [Google Scholar]
- 50.Zhang MH, Lee JS, Kim HJ, Jin DI, Kim JI, Lee KJ, Seo JS. HSP90 Protects Apoptotic Cleavage of Vimentin in Geldanamycin-Induced Apoptosis. Mol. Cell. Biochem. 2006;281:111–121. doi: 10.1007/s11010-006-0638-x. [DOI] [PubMed] [Google Scholar]