

Abstract

A stereoselective synthesis of the C(1)-C(19) fragment of tetrafibricin has been accomplished via a highly diastereoselective double allylboration reaction of 6, 7, and 8, and an iodonium-ion promoted urethane cyclization for the installation of the C(15) alkoxy function in 3.
Tetrafibricin is a polyoxygenated fibrinogen receptor inhibitor, that was isolated in 1993 from the culture broth of Streptomyces neyagawaensis NR0577.1 Fibrinogen binding to the glycoprotein GPIIb/IIIa complex on the platelet surface plays a crucial role in platelet aggregation.2 The ability of tetrafibricin to block fibrinogen from binding to its glycoprotein receptor makes it a viable target for the potential therapeutic intervention of arterial thrombotic diseases such as coronary occlusion.3,4 Kishi and co-workers recently assigned the stereochemistry of tetrafibricin (1) through the use of a NMR databatase method, supplemented by data obtained from NMR measurements in chiral solvents.5 The interesting biological properties and challenging structure, which includes eleven stereocenters of which ten are secondary hydroxyls arrayed as 1,3- and 1,5-diols, renders tetrafibricin an excellent target for synthetic study. Development of an an efficient, convergent synthesis of tetrafibricin (1) will facilitate structure activity relationship studies designed to probe its biological properties. To our knowledge, only Cossy has disclosed efforts towards the total synthesis of tetrafibricin.6 We report herein our initial synthetic studies on 1, culminating in an efficient synthesis of the C(1)-C(19) fragment 3 via application of the double allylboration methodology developed in our laboratory.7
Our retrosynthetic analysis of tetrafibricin is outlined in Figure 1. We envisaged that tetrafibricin can be assembled from a late stage double allylboration sequence involving the coupling of the functionalized aldehydes 2 and 3 with bifunctional allylborane 4.7 It is expected that this reaction will provide the C(19)-C(23) anti-1,5-diol unit of 1 with an embedded trans olefin in a single step. Aldehyde 3 would be assembled in turn from cyclic carbonate 5 through a Horner-Wadsworth-Emmons olefination sequence, followed by oxidation of the C(13) and C(19) alcohols. Further analysis of carbonate 5 suggests it can be accessed via a three component coupling of aldehydes 68 and 79 with bifunctional allylborane 8, followed by installation of C(15)-OH.
Figure 1.

Tetrafibricin Retrosynthetic Analysis.
The synthesis of the C(1)-C(19) fragment 3 commenced with the construction of benzyl carbamate 13 (Scheme 1). Intially, we purused the synthesis of alcohol 12 via a one-pot double allylboration sequence followed by selective protection of the C(17) allylic alcohol.10 However, we were only able to achieve ca. 2 : 1 selectivity in protecting the allylic alcohol of the diol corresponding to 12. Therefore it proved advantageous to synthesize 12 via an interrupted, three-pot double allylboration sequence in order to differentiate the secondary C(17) and C(13) alcohols.11 Thus, treatment of aldehyde 79 with the in situ generated bifunctional (E)-allylborane 87 [derived from the hydroboration of allene 97 with (dIpc)2BH],12 followed by quenching with water, afforded β-hydroxy allylboronate 10 in 82% isolated yield.13 The resultant allylic alcohol was protected by treatment with TBSOTf and 2,6-lutidine to afford allylboronate 11 in 89% yield. Treatment of 11 with 1.1 equiv. of aldehyde 68 at 23 °C for 96 h proceeded in a matched fashion and provided homoallyl alcohol 12 in 73% yield as a 16:1 mixture of diastereomers.14 The homoallyl alcohol was then treated with benzyl isocyanate in toluene at 115 °C to provide key benzyl carbamate 13 in 85% yield.
Scheme 1.
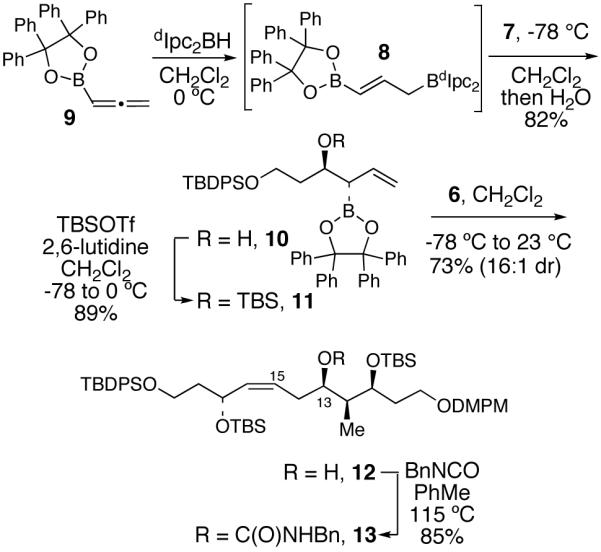
Synthesis of Benzyl Carbamate 13
Attention next focused on the diastereoselective installation of the C(15)-β alkoxy group of fragment 3 via an iodonium-ion promoted urethane cyclization. Initial attempts to affect this cyclization by treatment of 13 with iodine monochloride in CH2Cl2 at -78 °C15 afforded a mixture of the desired iodo carbonate 14 along with an inseparable furan side product 15 (Table 1, entry 1). This side product arises from a 5-exo type cyclization with concomitant deprotection of the TBDPS ether. We anticipated that use of a pyridine-ICl complex would slow the rate of the competing furan formation by moderating the reactivity of the iodonium ion. Accordingly, treatment of carbamate 13 with a preformed pyridine-ICl complex in CH2Cl2 at -78 °C and warming to 23 °C provided a small amount of the targeted iodo carbonate 14 (17%, entry 2); however, more importantly, formation of the furan side product was completely suppresed. This observation prompted an investigation of NIS as the iodine source due to its stability and ease of handling. Gratifyingly, treatment of benzyl carbamate 13 in CHCl3 at 23 °C with NIS (added portionwise) effected a clean cyclization which provided carbonate 14 as the sole product in 70% yield and >20:1 diastereoselectivity as judged by 1H NMR analysis.16
Table 1.
Installation of the C(15) Oxygen by an Iodonium Ion Promoted Urethane Cyclization 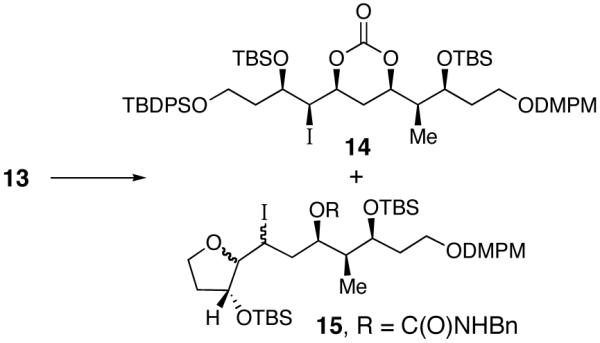
| entry | conditions | 13 : 14 : 15a | yield of 14 (%) |
|---|---|---|---|
| 1 | ICl/CH2Cl2, -78 °C | 0 : 80 : 20 | |
| 2 | ICl-Pyr/CH2Cl2,-78 to 0 °C | 50 : 20 : 0 | 17 |
| 3 | NIS/CHCl3, 0 to 23 °C | 0 : 100 : 0 | 70 |
Ratio determined by 1H NMR analysis
Deiodination of 14 by treatment with Et3B and n-Bu3SnH generated desired intermediate 5 in 88% yield.17 To verify the C(13)-C(15) syn-stereochemistry, intermediate 5 was converted to acetonide analog 17 via a two step-sequence.18 13C NMR analysis of the acetonide according to the Rychnovsky method18 confirmed the presence of a syn-1,3-diol relationship (see Supporting Information for details). Deprotection of the primary DMPM group of 5 with DDQ followed by Dess-Martin oxidation19 yielded sensitive aldehyde 16, the substrate for the subsequent Horner-Wadsworth-Emmons olefination with phosphonate 20.
Horner-Wadsworth-Emmons reagent 20 was synthesized starting with the MnO2 oxidation of the known alcohol 1820 (Scheme 3). Subjection of the derived aldehyde to a Horner-Wadsworth-Emmons21 olefination with triethyl phosphonoacetate afforded intermediate 19 in 70% yield. Deprotection of 19 by treatment with TBAF, followed by conversion of the allylic alcohol to the corresponding allylic bromide by treatment with PBr3 and pyridine, and then treatment of the allylic bromide with P(OEt)3 in refluxing toluene yielded the phosphonate coupling partner 20 in 76% yield over three steps.22 Gratifyingly, treatment of 20 with LHMDS followed by addition of a solution of aldehyde 16 at -78 °C with warming to 0 °C provided intermediate 21 in 77% yield.23
Scheme 3.

Completion of the Synthesis of the C(1)-C(19) Fragment 3 of Tetrafibricin
Tetraenoate 21 contains all the carbon atoms of the C(1)-C(19) fragment 3 of tetrafibricin; all that remained to access keto-aldehyde 3 was to adjust the oxidation state of C(13) and C(19) alcohols. This was achieved by cleavage of the cyclic carbonate in 21, followed by a selective silylation of the less hindered C(15)-OH, which provided 22 with 10:1 regioselectivity. The crucial and selective deprotection of the C(19) TBDPS ether was accomplished in 55% yield by using TAS-F in a DMF-AcOH solvent mixture.24 Finally, oxidation of the C(13)-C(19) diol with the Dess-Martin reagent19 provided the fully elaborated C(1)-C(19) fragment 3 of tetrafibricin.
In summary, we have accomplished an efficient and highly diastereoselective synthesis of 3 corresponding to the C(1)-C(19) fragment of tetrafibricin. This synthesis proceeds in 13 steps from aldehyde 7. The highlights of this synthesis include the highly diastereoselective interrupted double allylboration sequence used to prepare intermediate 12, the diastereoselective iodonium-ion promoted urethane cyclization of 13 for the installation of the syn-C(15) alkoxy group in 14, and a Horner-Wadsworth-Emmons olefination of 16 for introduction of the tetraenoate moeity. Further progress towards the completion of the total synthesis of tetrafibricin will be reported in due course.
Supplementary Material
Scheme 2.
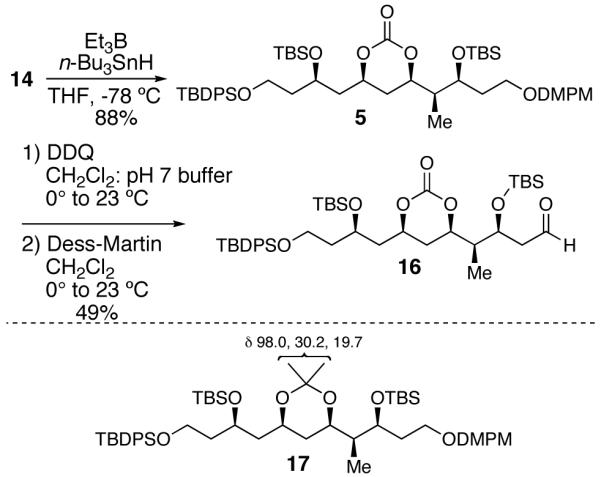
Synthesis of Aldehyde 16
Acknowledgment
This work was supported by the National Institutes of Health (GM 38436).
References
- 1.Kamiyama T, Umino T, Fujisaki N, Satoh T, Yamashita Y, Ohshima S, Watanabe J, Yokose K. J. Antibiot. 1993;46:10392. doi: 10.7164/antibiotics.46.1039. [DOI] [PubMed] [Google Scholar]
- 2.Kamiyama T, Itezono Y, Umino T, Satoh T, Nakayama N, Yokose K. J. Antibiot. 1993;46:1047. doi: 10.7164/antibiotics.46.1047. [DOI] [PubMed] [Google Scholar]
- 3.Satoh T, Kouns WC, Yamashita Y, Kamiyama T, Steiner B. Biochem. Biophys. Res. Commun. 1994;204:325. doi: 10.1006/bbrc.1994.2463. [DOI] [PubMed] [Google Scholar]
- 4.Satoh T, Yamashita Y, Kamiyama T, Arisawa M. Thromb. Res. 1993;72:401. doi: 10.1016/0049-3848(93)90240-o. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi Y, Czechtizky W, Kishi Y. Org. Lett. 2003;5:93. doi: 10.1021/ol0272895. [DOI] [PubMed] [Google Scholar]
- 6.BouzBouz S, Cossy J. Org. Lett. 2004;6:3469. doi: 10.1021/ol048862i. [DOI] [PubMed] [Google Scholar]
- 7.Flamme EM, Roush WR. J. Am. Chem. Soc. 2002;124:13644. doi: 10.1021/ja028055j. [DOI] [PubMed] [Google Scholar]
- 8.Prepared as reported for the synthesis of ent-6: Anderson OP, Barrett AGM, Edmunds JM, Hachiya S-I, Hendrix JA, Horita K, Malecha JW, Parkinson CJ, VanSickle A. Can. J. Chem. 2001;79:1562.
- 9.Holmes AB, Hughes AB, Smithe AL. J. Chem. Soc. Perkin Trans. 1. 1993:633. [Google Scholar]
- 10(a).Flamme EM, Roush WR. Beilstein J. Org. Chem. 2005;1:7. doi: 10.1186/1860-5397-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hicks JD, Flamme EM, Roush WR. Org. Lett. 2005;7:5509. doi: 10.1021/ol052322j. [DOI] [PubMed] [Google Scholar]
- 11.Flamme EM, Roush WR. Org. Lett. 2005;7:1411. doi: 10.1021/ol050250q. [DOI] [PubMed] [Google Scholar]
- 12.Brown HC, Singaram B. J. Org. Chem. 1984;49:945. [Google Scholar]
- 13.Absolute stereochemistry of C(17)-OH of 10 was assigned by Mosher ester analysis of an analog of 13; see Supporting Information for details.
- 14.The absolute stereochemistry of C(13)-OH of 12 was assigned by 1H NMR analysis of the corresponding Mosher esters; see Supporting Information for details.
- 15.Duan JJ-W, Smith AB., III J. Org. Chem. 1993;58:3703. [Google Scholar]
- 16.These conditions have previously been employed for the cyclization of allylic trichloroacetimidates: Bongini A, Cardillo G, Orena M, Sandri S, Tomasini C. J. Org. Chem. 1986;51:4905.
- 17.Miura K, Ichinose Y, Nozaki K, Fugami K, Oshima K, Utimoto K. Bull. Chem. Soc. Jpn. 1989;62:143. [Google Scholar]
- 18.Rychnovsky SD, Rogers BN, Richarson TI. Acc. Chem. Res. 1998;31:9. [Google Scholar]
- 19.Dess PB, Martin JC. J. Am. Chem. Soc. 1978;100:300. [Google Scholar]
- 20.Berube G, Deslongchamps P. Can. J. Chem. 1990;68:404. [Google Scholar]
- 21(a).Horner L, Hoffmann H, Wippel HG. Chem. Ber. 1958;91:61. [Google Scholar]; (b) Wadsworth WS, Emmons WD. J. Am. Chem. Soc. 1961;83:1733. [Google Scholar]; (c) Wadsworth WS. Org. Reactions. 1977;25:1. [Google Scholar]
- 22.Mori Y, Asai M, Kawade J-I, Furukawa H. Tetrahedron. 1995;51:5315. [Google Scholar]
- 23.Roush WR. J. Am. Chem. Soc. 1980;102:1390. [Google Scholar]
- 24.Scheidt KA, Chen H, Follows BC, Chemler SR, Coffey DS, Roush WR. J. Org. Chem. 1998;63:6436. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.