

Abstract
Objective
KITL/KIT can elicit diverse sets of signals within lymphoid, myeloid, mast, and erythroid lineages, and exert distinct effects on growth, survival, migration, adhesion, and secretory responses. Presently, we have applied a PY-mutant allele knockin approach to specifically assess possible roles for KIT-PY567 and KIT-PY719 sites, and coupled pathways, during erythropoiesis.
Materials and Methods
Mouse models used to investigate this problem include those harboring knocked-in KITY567F/Y567F, KITY569F/Y569F, KITY719F,Y719F, and KITY567F/Y567F:Y569F/Y569F alleles. The erythron was stressed by myelosuppression using 5-fluorouracil, and by phenylhydrazine- induced hemolysis. In addition, optimized systems for ex vivo analyses of bone marrow and splenic erythropoiesis were employed to more directly analyze possible stage-specific effects on erythroid cell growth, survival, development and KIT signaling events.
Results
In KitY567F/Y567F mice, steady-state erythropoiesis was unperturbed while recovery from anemia due to 5-fluorouracil or phenylhydrazine was markedly impaired. Deficiencies in erythroid progenitor expansion occurred both in the bone marrow and the spleen. Responses to chronic erythropoietin dosing were also compromised. Ex vivo, KitY567F/Y567F (pro)erythroblast development was skewed from a KitposCD71high stage toward a subsequent KitnegCD71high compartment. Proliferation and, to an extent, survival capacities were also compromised. Similar stage-specific defects existed for erythroid progenitors from KitY567F/ Y567F:Y569F/Y569F but not KITY719F/Y719F mice. KitY567F/Y567F erythroblasts were used further to analyze KIT-PY567–dependent signals. MEK-1,2/ERK-1,2 signaling was unaffected while AKT, p70S6K, and especially JNK2/p54 pathways were selectively attenuated.
Conclusions
Nonredundant KIT-PY567–directed erythroblast-intrinsic signals are selectively critical for stress erythropoiesis. Investigations also add to an understanding of how KIT directs distinct outcomes among diverse progenitors and lineages.
Erythropoiesis is subject to regulation via several cytokine receptor systems that affect erythroid progenitor growth and/or development [1,2]. Erythropoietin (EPO) is one essential factor that acts as a colony-forming unit erythroid (CFU-E) survival factor [3–5], and can also modulate proerythroblast proliferation [6]. For earlier erythroid progenitors, expansion can be promoted via interleukins (IL) as IL-3, IL-7, IL-11, IL-4, and IL-12 [7–11]. During stress erythropoiesis, bone morphogenetic protein-4 also can provide priming effects for splenic erythroid progenitors [12], while transforming growth factor–β can promote late-stage erythroblast differentiation [13]. FasL, TRAIL, tumor necrosis factor–α, and interferon–γ, in contrast, can limit erythroid cell production [14–17].
Erythropoiesis is regulated further by several receptor tyrosine kinases. In primary murine and human systems, RON (c-met-related tyrosine kinase) and ligand can enhance burst-forming unit erythroid (BFU-E) formation [18,19]. Insulin- like growth factors-1 and -2C also have been shown to support late-stage erythroblast development [20,21]. For CD34pos progenitors, EPHB4 also promotes erythropoiesis [22,23]. Finally, KIT and its ligand (KITL) also exert clear erythropoietic roles, including effects of KITL on BFU-E growth and/or survival [24–26]. In CFU-E like cells, KITL also has been shown to intensify EPO-dependent growth and survival [26], and cross-talk between the EPO receptor (EPOR) and KIT has been demonstrated [27].
Insight into KIT’s erythropoietic roles originally was provided via the discoveries of murine white spotting (W) and Steel (Sl) loci. Mice carrying either viable W Kit or Sl KITL alleles as partial loss of function mutations exhibit macrocytic anemia, while Kit and KITL null mutations die perinatally due to overall essential roles for KIT in erythropoiesis [28–31]. Kit and KITL loss of function mutations further have revealed roles for KIT/KITL in melanogenesis, gametogenesis and intestinal tract motility [32–35].
Mechanistically, KITL–KIT interactions lead to KIT autophosphorylation at up to 21 cytoplasmic KIT PY docking sites for SH2 or PTB domain proteins [36,37]. In cell line models, studies of KIT PY mutants have defined several PY motif binding specificities. These include p85-PI3K at Y719; phospholipase C-γ at Y728; and Grb2 plus Grb7 adaptor protein binding at Y702 and Y934 [38–40]. Juxtamembrane Y567 and Y569 sites further have been demonstrated to bind to SRC family kinases (SFKs); SHP-1, and/or -2 [41–45] and APS (adapter protein with Pleckstrin homology and Src homology 2 domains) plus Csk-homologous kinase [46,47]. PY567 and PY569 sites functionally have therefore been argued to regulate key survival and/or proliferation pathways [41,45].
The diverse roles exerted by KIT among various cell systems raise questions concerning how KIT mediates distinct cellular responses within different cell types. To address these questions, mice carrying tyrosine-to-phenylalanine substitution mutations as KitY719F/Y719F, KitY567F/Y567F, KitY569F/Y569F, and KitY567F/Y567F:Y569F/Y569F have been developed using knockin strategies to selectively disrupt signaling cascades in vivo [33,48–50]. These mutated alleles interestingly demonstrate that cellular context critically determines the consequences of these KIT mutations in vivo. Whereas KIT-Y719 phosphorylation plus phosphatidylinositol 3 (PI3) kinase signaling are critical for spermatogenesis, but of no consequence to steady-state hematopoiesis and lymphocyte development, KIT Y567 phosphorylation is dispensable for spermatogenesis, but is critical for lymphocyte development [49]. In KitY719F/Y719F, KitY567F/Y567F, and KitY567F/Y567F:Y569F/Y569F mice, mast cell populations are similarly affected, with normal dorsal skin mast cell numbers, but reduced peritoneal mast cell populations [33,49,50]. Erythropoiesis in these mice, however, is unperturbed [33,48–50].
In the present report, we have specifically investigated the role of distinct KIT-activated signaling pathways in stress hematopoiesis and stress erythropoiesis. In 5-fluorouracil (5-FU) and phenylhydrazine (PHZ) anemia models, Kit Y567F/Y567F mice exhibited deficient hematocrits, decreased erythroid progenitor populations, and defective erythrosplenomegaly. Ex vivo bone marrow expansion studies interestingly indicated enhanced formation of KitnegCD71pos Kit Y567F/Y567F erythroblasts, but with marked overall defects in progenitor cell growth and survival. In primary bone marrow–derived KitY567F/Y567F erythroblasts, signal transduction factor pathways also were analyzed. Here, AKTand JNK2/p54 signaling proved to be selectively attenuated. Findings are discussed in the context of an apparently erythroid-selective set of KIT-mediated signals, which are essential for efficient erythropoiesis during anemia.
Materials and methods
Mouse models
KitY567F/Y567F mice were generated as described by Agosti et al. [49]. In brief, codon Y567 was mutated to phenylalanine within a Kit exon 8–13 genomic fragment to generate a targeting construct. A neomycin-resistance gene expression cassette flanked by loxP was inserted within intron 9, and a diphtheria toxin A gene cassette (for negative selection) was sited at the 3′ end. 129/SvJ embryonic stem cells were electroporated with the linearized construct, and neomycin-resistant clones were screened by polymerase chain reaction. Recombined clones were further analyzed by Southern blotting and sequencing. Correctly targeted clones were microinjected into C57BL/6J blastocysts and male mice displaying 85% to 100% chimerism were backcrossed to C57BL/6J females for germline transmission. The floxed neocassette was excised in vivo by mating heterozygous mutant males with EIIa-cre transgenic females. Kit+/Y567F mice were then used to generate KitY567F/Y567F mice. KitY567F/Y567F mice subsequently backcrossed at least six times to C57BL/6J mice were used for experiments. Kit+/+ littermates (or C57BL/J6 mice, Jackson Laboratories Bar Harbor, ME, USA) were used as controls. Lyn−/− and KitF567/F567:F569/F569 mice were as described [50,51]. All procedures described here, and later, were reviewed and approved by Institutional Animal Care and Use Committees.
Primary erythroblast preparations
Iscove’s modified Dulbecco’s medium (IMDM; Invitrogen, Carlsbad, CA, USA; #12440-053) plus 2% fetal bovine serum (FBS), was used to flush marrow from long bones. Cells were passed through a 40-µm strainer, washed, resuspended in 1 mL phosphate-buffered saline (PBS) (Invitrogen #14190-144) and exposed for 2 minutes to 9 mL buffered 0.8% ammonium chloride (Stem Cell Technologies, Vancouver, BC, Canada; # 07850). Then 10× PBS (1.1 mL) was added, and cells were collected through 50% FBS in PBS and washed in IMDM. Kitpos erythroblasts were isolated by Linpos cell depletion (Stem Cell Technologies; cat#: 17066) and subsequent positive magnetic-activated cell sorting (MACS) selection using a biotinylated anti-CD117 antibody (Miltenyi Biotech, Auburn, CA, USA; #553353). Cells then were plated at 8 × 105 cells/mL in “SP34-ex medium” as Stem-Pro-34 base medium (GIBCO Carlsbad, CA, USA; #10640) supplemented with 2.5 U/mL EPO, 100 ng/mL mKITL, 1 uM dexamethasone, 1.5 mM l-glutamine, 1 uM β-estradiol, 100 ug/ mL h-transferrin (Sigma, St Louis, MO, USA; #T0665), 0.5% bovine serum albumin (BSA; Stem Cell Technologies; #9300), and 0.1 mM 2-mercaptoethanol. At day 3 of expansion, Kitpos erythroblasts were isolated by Linpos cell depletion (Stem Cell Technologies; #17066) and subsequent positive MACS selection using a biotinylated anti-CD117 antibody (Miltenyi Biotech; #553353). Splenic erythroid progenitors from PHZ-treated mice were similarly prepared, and isolated via Linpos depletion and MACS selection procedures.
Stress hematopoiesis
5-FU was administered intraperitoneally at 150 or 300 mg/kg. Phenylhydrazine was administered subcutaneously at 1 and 24 hours (60 mg/kg). Hematocrits were assayed via microcapillary centrifugation, and reticulocytes by flow cytometry (thiazole orange). Hematological parameters (red blood cell count, hematocrit, red blood cell mean cell volume, platelet count, white blood cell count) were assayed using an Advia 120 multispecies blood analyzer (Bayer, Tarrytown, NY, USA). Splenocytes were isolated by gentle disruption and passage through a 40-µm sieve.
Flow cytometry and cytospins
Washed cells (1 × 106) were incubated at 4°C for 15 minutes in 0.2 mL of PBS, 1% BSA plus 1 µg rat immunoglobulin G, and subsequently with phycoerythrin (PE)-Ter119 (2 µg), fluorescein isothiocyanate–CD71 (1 µg), allophycocyanin–KIT (1 µg) and/or PE-CD41 as indicated (BD Biosciences, San Jose, CA USA). PE/fluorescein isothiocyanate–Annexin-V binding assays (#556 420 and #556 422, respectively; BD Biosciences) were performed in 140 mM NaCl, 2.5 mM CaCl2, 10 mM HEPES
(pH 7.4) (20 minutes at 25°C). Washed cells then were analyzed using a BD FACSCalibur flow cytometer. For staining of incorporated bromodeoxyuridine (BrdU), an allophycocyanin-conjugated antibody was used per established protocols (BD Pharmingen; #51-23619L). In cell cycle analyses, DRAQ5 (5 µM, BOS-889-002; Biostatus, Shepshed, UK) was used together with Modfit (Verity Software, Topsham, ME, USA) as described previously [6]. Cytospin preparations were stained using a standard May-Giemsa protocol.
Signal transduction assays
KitposCD71high erythroblasts were isolated by MACS from expansion cultures. Washed cells were then cultured for 5 hours in IMDM, 0.5% BSA, 50 ug/mL transferrin, 10 ng/mL insulin, and 0.1 mM 2-mercaptoethanol. Erythroblasts then were exposed to KITL at the indicated concentrations and intervals. Cells were washed at 2°C in PBS and lysed in 0.2 mL 1% Igepal, 150 mM NaCl, 50 mM NaF, 2 mM Na2EDTA, 0.1 mM NaVO3, 1 mM dithiothreitol, 10mM sodium pyruvate, 25 mM β-glycerol phosphate, 10% glycerol, 50 mM HEPES (pH 7.5) plus 0.25 mg/mL phenylmethylsulfonylfluoride, 1× protease and phosphatase inhibitor cocktails (Sigma-Aldrich; #P8340, #P5726). As an additional step, 1% Triton-X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 112.5 mM NaCl, 37.5 mM Tris-HCL (pH 7.4) was then added (0.2 mL). Extracts were centrifuged (12,000 rpm), assayed for protein content, and denatured. Proteins were electrophoresed, and transferred to polyvinylidine difluoride membranes. Membranes were blocked with 0.05% Tween-20, 3% fat-free milk, 1% BSA, 0.15M NaCl, 20 mM Tris (pH 7.4), and were incubated with antibodies to AKT (Santa Cruz Biotechnology; #sc-1618, Santa Cruz, CA, USA) and the following antibodies from Cell Signaling (Danvers, MA, USA): PS-AKT (#9271), ERK1,2 (#9102), PY/T ERK1,2 (#4375), SAPK/JNK (#9252), PY/T-SAPK/JNK (#9251), p70S6-kinase (#9202), and PT/S p70S6-kinase (#9204). Horseradish peroxidase–conjugated secondary antibodies were from Jackson Immunoresearch (Westgrove, PA, USA). Enhanced chemiluminescence utilized Dura reagent (#34076; Pierce, Rockford, IL, USA), and Hyperfilm (#RPN2114k; Amersham Biosciences, Piscataway, NJ, USA). Densitometry was via ImageQuant-TL (Amersham Biosciences).
3HdT incorporation assays
In 3HdT-incorporation assays, Kitpos erythroblasts were isolated from day-5 expansion cultures (via lineage depletion and MACS). Following lineage depletion from day-5 expansion cultures, cells were incubated (at 1 × 105 cells/mL) in triplicate in SP34-ex medium with EPO and/or m-KITL as indicated. At 24 hours, 3HdT was added (1 µCi [0.037 MBq]/assay, NET-027A, 2Ci [0.074 MBq]/mol; Perkin Elmer, Boston, MA, USA), and incorporation rates (at 6 hours) were determined.
Cell lines
G1E2 cells are an EPO- and KITL-dependent erythroid line derived from murine ES cells with a disrupted endogenous Gata1 gene, and Gata1/estrogen receptor fusion construct [52]. This line was maintained in IMDM, 15% FBS, 0.1 mM monothioglycerol, 50 ng/mL KITL, 2 U/mL EPO plus penicillin, streptomycin, fungizome. Erythroid myeloid lymphoid cells [53] were maintained in IMDM, 20% horse serum (GIBCO-BRL, Gaithersburg, MD, USA), 2 mM l-glutamine, PSF, and 100 ng/mL recombinant KITL (# 250-03, Peprotech, Rocky Hills, NJ, USA).
Retroviral vectors
To study KIT PY signaling pathways in G1E2 and EML cells, a hMCSF/mKIT chimeric receptor approach was used [54]. Specifically, a chimeric “M/K” construct with phenylalanine mutations at PY 567, 569, 702, 719, 728, and 745 was used to back mutate (i.e., restore) PY567 or PY567 plus PY569 sites (within a MIEG3 vector backbone). From these templates, vesicular stomatitis virus packaged retroviruses were prepared in 293-FT cells. Transduced cells then were isolated by fluorescent activated cell sorting (at matched green fluorescent protein intensities).
Results
Erythropoiesis in KitY567F/Y567F mice is defective during anemia
In KitY567F/Y567F mice, steady-state hematopoiesis is largely unaffected [49]. To assess the ability of the hematopoietic system in these mutant mice to cope with hematopoietic stress, KitY567F/Y567F mice and wild-type littermate controls first were exposed to 5-FU at a dose known to be fully myelosuppressive (300 mg/kg) [51]. In controls, steady-state erythropoiesis was restored between days 12 and18 post 5-FU. In KitY567F/Y567F mice, however, hematocrits fell markedly (to <10%) and lethality frequently was incurred between 6 and 16 days post 5-FU (Fig. 1A). In addition, KitY567F/Y567F mice failed to respond with splenomegaly, and representation of Ter119pos erythroblasts in spleen was markedly decreased (Fig. 1B). Within the bone marrow of KitY567F/Y567F 5-FU–treated mice, Ter119pos erythroblast levels likewise were decreased, and cytospin preparations revealed significant underrepresentation of nucleated erythroid progenitor cells as compared to Kit+/+ wild-type controls (Fig. 1C). By comparison, no such limiting defects were observed following high-dose 5-FU administration to KitY719F/Y719F mice (see Suppl. Fig. S1A).
Figure 1. In KitY567F/Y567F mice, high-dose 5-fluorouracil (5-FU) administration leads to fatal anemia.

(A) Following a single dose of 5-FU at 300 mg/kg, KitY567F/Y567F mice exhibited sustained low-level hematocrits, and compromised survival (mean ± standard error; n = 5). (B) Failed splenic erythropoiesis in KitY567F/Y567F mice following 5-FU dosing—As analyzed at day 15 post 5-FU, splenic hypertrophy faltered in KitY567F/Y567F mice, and Ter119pos erythroblasts were underrepresented. (C) Cytospin analyses of wild-type Kit+/+ (wt) and KitY567F/Y567F bone marrow preparations (day 15 post 5-FU injection, upper panels). As assessed by flow cytometry, Ter119pos cells also were notably underrepresented in bone marrow among KitY567F/Y567F mice. Data shown are representative of two independent analyses.
In order to avoid lethality, and to better understand the hematopoietic defects in KitY567F/Y567F mice, 5-FU next was administered at a standard dose of 150 mg/kg. As compared to Kit+/+ mice, KitY567F/Y567F mice responded with low-level hematocrits and red blood cell counts, and delayed recovery (Fig. 2). All mice survived, however, and restored their erythroid compartment by day 25. During this recovery period (days 18–25), KitY567F/Y567F erythrocytes nonetheless exhibited significantly increased mean cell volumes as compared directly to erythrocytes from 5-FU–treated Kit+/+ mice (Fig. 2, lower left panel). In contrast, platelet and white blood cell production in KitY567F/Y567F mice was not markedly affected (Fig. 2, lower right panel and data not shown). Following the 5-FU–mediated depletion of cycling hematopoietic progenitor cells, KitY567F/Y567F deficiencies, therefore, appear to be predominant during erythroid development (while other lineages are affected to notably lesser extents).
Figure 2. Lower level 5-fluorouracil (5-FU) dosing of KitY567F/Y567F mice leads to sustained macrocytic anemia.
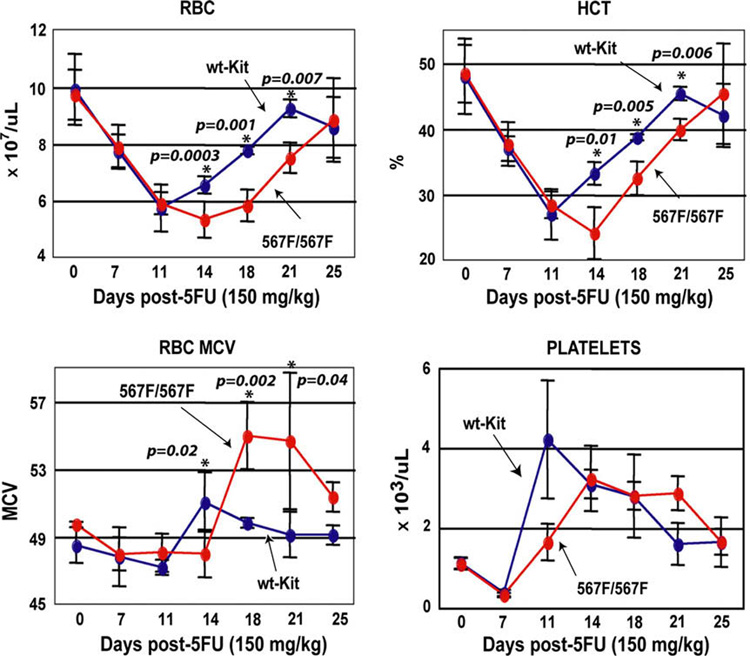
Following a single injection of 5-FU at 150 mg/kg, red blood cell numbers (RBC), hematocrits, mean red cell volumes (MCV) and platelet levels were determined. From day 14 through 21, KitY567F/Y567F mice exhibited decreased RBCs and lowered hematocrits. For KitY567F/Y567F red cells, also note the increase in MCV at day 18. Graphed values are means ± standard error (n = 10 mice per group). The Student’s t-test assuming equal variances between the two samples was applied to determine statistical significance.
To more specifically target the erythroid lineage, PHZ was administered to KitY567F/Y567F mice to induce hemolytic anemia. Specifically, KitY567F/Y567F mice (and Kit+/+ controls) were treated with PHZ at 60 mg/kg, and hematocrits were monitored over a 12-day period. Among KitY567F/Y567F mice, delayed recovery and deficient hematocrits were observed (Fig. 3A). In particular, in both mutant and wild-type Kit+/+ mice, hematocrits initially fell at day 2 to ~25%. In Kit+/+ mice, hematocrits thereafter increased to ~35%, while in KitY567F/Y567F mice, hematocrits remained below 25% until day 5. In addition, KitY567F/Y567F mice did not support efficient erythrosplenomegaly (Fig. 3A), and exhibited decreased splenic cellularity (data not shown). All mice survived, however, and by day 11, hematocrits recovered to baseline levels. Flow cytometric analyses as performed at day 5 also revealed deficient erythroid progenitor pools in KitY567F/Y567F spleens, including a 10-fold decrease in Ter119posCD71pos erythroblasts (Fig. 3B). By comparison, recovery from PHZ-induced anemia among KitY719F/Y719F mice was largely unimpaired (see Suppl. Fig. S1B). As expected cellularity in KitY567F/Y567F bone marrow in this hemolytic model was not substantially altered, but frequencies of bone marrow CD71highTer119pos erythroid cells were impaired, although less dramatically than in spleen (data not shown).
Figure 3. Defective erythropoiesis in KitY567F/Y567F mice during phenylhydrazine-induced anemia.

(A) Wild-type Kit+/+ (wt) and KitY567F/Y567F mice were treated with phenylhydrazine (PHZ) (60 mg/kg at 0 and 24 hours). At the indicated time points, hematocrits (left panel) and spleen size (right panel) were assessed. To determine statistical significance between samples the Student’s t-test assuming equal variances was used. (B) At day 5 post-PHZ treatment, frequencies of CD71posTer119pos erythroblasts in spleen were analyzed (by flow cytometry). For KitY567F/Y567F erythroblasts, note the approximate fivefold underrepresentation. Data are representative of three independent analyses.
KitKitY567F/Y567F erythroid progenitor cells exhibit altered stage-specific development and decreased growth and survival potentials
To study additional specific properties of KitY567F/Y567F (pro)-erythroblasts, a recently optimized ex vivo expansion system was used [55]. Specifically, this involved isolation of Kitpos progenitors from bone marrow, and their expansion in serum-free SP34-ex medium. In these ex vivo analyses, KitY567F/Y567F erythroid progenitor cells unexpectedly were observed to develop more rapidly from a KitposCD71high to a subsequent KitnegCD71high stage. Specifically, flow cytometric analyses at days 3, 4, and 5 interestingly revealed approximate twofold increases in KitnegCD71high erythroblast formation as compared directly to Kit+/+ controls (Fig. 4).
Figure 4. Ex vivo, KitY567F/Y567F erythroid progenitors are underrepresented at a KitposCD71high erythroblast stage, and become overrepresented within a KitnegCD71high compartment.
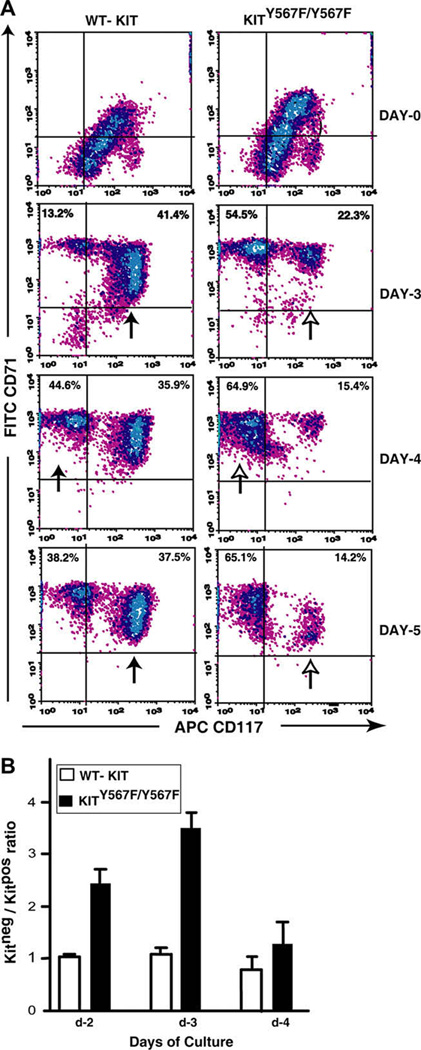
(A) Kitpos progenitor cells were isolated from bone marrow preparations from KitY567F/Y567F mice and wild-type Kit+/+ (wt) littermates, and expanded in SP34-ex medium. Among developing erythroblasts, CD117/KIT and CD71 marker expression was assayed over 6-days of culture. For KitY567F/Y567F cells, note the limited representation of KitposCD71high progenitors, and increased relative frequencies of KitnegCD71high erythroblasts (especially at days 4 and 5). Data shown are representative of three independent experiments. (B) Also graphed (as mean ± standard error, n = 3) are ratios of Kitneg/Kitpos erythroblasts for wild-type and KitY567F/Y567F cells at 48, 72, and 96 hours of culture.
SRC family kinases are one prime candidate effector of KIT PY567 coupled signals [49,54]. In addition, LYN is a predominant SRC kinase in erythroid cells, and also is activated via both KIT and EPO receptors [56–58]. Therefore, Therefore, ex vivo development of bone marrow erythroblasts from Lyn−/− and KitY567F/Y567F mice was directly compared. In particular, erythroid progenitors from wild-type mice, KitY567F/Y567F mice, and Lyn−/− mice were expanded in SP34-ex medium. At the indicated time points, frequencies of Kitpos- to- KitnegCD71high proerythroblasts were assayed (via flow cytometry). As reported recently [51], Lyn−/− erythroid cells exhibited increased Kitpos/Kitneg ratios (Fig. 5, lower panel). This outcome contrasted with KitY567F/Y567F erythroblasts, for which Kitpos/Kitneg erythroblast ratios were decreased, and for which overall frequencies of Kitneg erythroblasts again were increased (Fig. 5, upper panel). Defective coupling of KitY567F/Y567F to LYN, per se, may only in part explain observed defects exhibited during KitY567F/Y567F erythroblast development.
Figure 5. Unlike KitY567F/Y567F erythroblasts, Lyn−/− erythroid progenitors accumulate ex vivo within a KitposCD71high compartment.

Within SP34-ex cultures of KitY567F/Y567F, Lyn−/−, and wild-type Kit+/+ (wt) erythroid progenitor cells, the development of KitposCD71high and Kit-negCD71high erythroblasts was assayed. For Lyn−/− cells, note the increased frequencies of KitposCD71high erythroblasts (lower subpanel).
For erythroid progenitor cells expanded from Kit+/+ and KitY567F/Y567F bone marrow preparations, despite an observed acceleration of KitY567F/Y567F late erythroid cell formation (upper panels, Fig. 4 and Fig. 5), overall cell counts proved to be decreased two- to fivefold, especially at later time points (Fig. 6A). This result suggested a disadvantage in erythroid progenitor cell growth and/or survival. Possible apoptosis among bone marrow KitY567F/Y567F erythroblasts as they developed ex vivo, therefore, was investigated. Interestingly, flow cytometric analyses of KitnegCD71high erythroblasts revealed decreased survival for KitY567F/Y567F cells, especially within a KitnegCD71high compartment (Fig. 6B, left panel). Annexin-V staining analyses also were performed for splenic erythroid cells from PHZ-treated KitY567F/Y567F and wild-type Kit+/+ mice (at day-5 post PHZ dosing). Significantly higher frequencies of apoptotic erythroblasts were again observed for KitY567F/Y567F splenocytes (Fig. 6B, right panel). To further analyze apparent KitY567F/Y567F cell growth defects, two additional analyses were performed. First, at day 3.5 post-PHZ treatment of KitY567F/Y567F and Kit+/+ mice, BrdU was injected (tail vein). At 1.5 hours, spleens were isolated and frequencies of S-phase CD71high erythroid progenitors were estimated (via flow cytometry) post Ter119pos cell depletion and CD71high cell retrieval (via MACS). As shown in Figure 6C (left panel), frequencies of S-phase (proliferating) KitY567F/Y567F erythroid progenitors were decreased to approximately 35% of Kit+/+ control progenitors. Second, erythroid progenitors from spleen of PHZ-treated mice also were prepared and cultured (at day 3.5 post-PHZ) in the presence of KIT-L at 50 ng/mL (and EPO at 2.5 U/mL). At 16 hours of culture, cells were stained with YoPro1 to determine survival status. KitY567F/Y567F cells proved to be disadvantaged approximately twofold in survival (Fig. 6C, right panel).
Figure 6. KitY567F/Y567F proerythroblasts exhibit decreased proliferation and survival capacities.
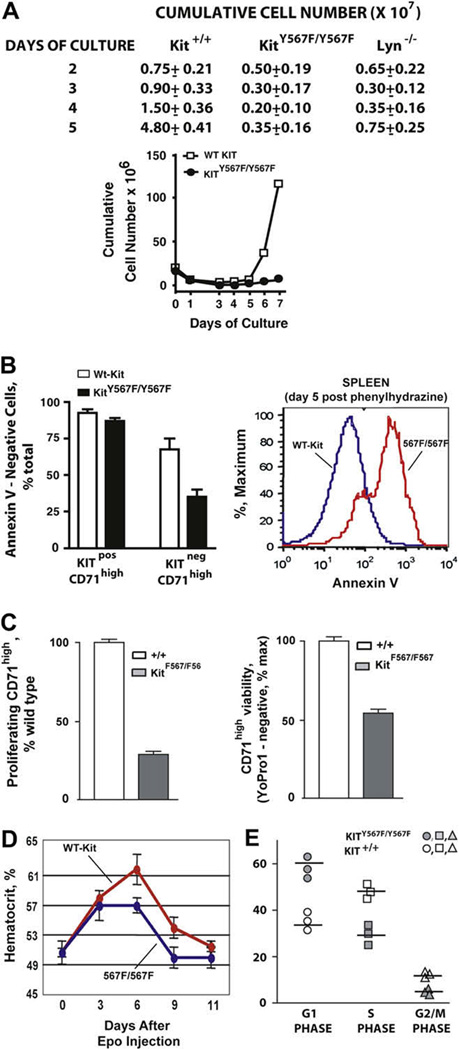
(A) Ex vivo growth profiles are illustrated for bone marrow–derived (pro)erythroblasts from Kit+/+, KitY567F/Y567F, and Lyn−/− mice. Kitpos progenitors (as isolated from bone marrow of KitY567F/Y567F and congenic control mice) were cultured in SP34-ex media in the presence of stem cell factor (SCF; 100 ng/mL) and erythropoietin (Epo; 2.5 U/mL). Viable cells counts were performed daily. Note the marked defect in expansion potential of KitY567F/Y567F erythroblasts. (B) Left panel: bone marrow–derived progenitor cells were prepared from wild-type Kit+/+ (wt-KIT) and KitY567F/Y567F mice, and expanded in SP34-ex medium. At day 5, frequencies of Annexin-V positive vs negative cells among KitposCD71high and KitnegCD71high erythroblasts were analyzed. (B) Right panel: For splenic erythroblasts produced in vivo in response to phenylhydrazine-induced anemia, substantial increases in frequencies of Annexin-V–positive apoptotic erythroblasts also were observed. (C) Left panel: At day 3.5 post-phenylhydrazine treatment of KitY567F/Y567F and Kit+/+ mice, BrdU was injected (tail vein). At 1.5 hours spleens were isolated. Post Ter119pos cell depletion, CD71high erythroid progenitors were retrieved via magnetic-activated cell sorting, and frequencies of S-phase (proliferating) CD71high erythroid progenitors were estimated via flow cytometry. (C) Right panel: At day 3.5 post-phenylhydrazine treatment of KitY567F/Y567F and Kit+/+ mice, erythroid progenitors from spleen were prepared and cultured in the presence of KIT-L at 50 ng/mL (and Epo at 2.5 U/mL). At 16 hours of culture, cells were stained with YoPro1 to determine cell survival. (D) To test whether defects in KitY567F/Y567F erythroblast expansion are exhibited during Epo-induced erythropoiesis, mice were injected with Epo (300 U/kg for 5 days) and increases in hematocrits were assayed over an 11-day period. Mean ± standard error are graphed (n = 4). Note the deficit response among KitY567F/Y567F mice. (E) KitY567F/Y567F and Kit+/+ mice were dosed with both Epo (1800 U/kg) and stem cell factor (SCF) (100 µg/kg). At day 3 post dosing, bone marrow KitposCD71high erythroid progenitors were isolated and cultured in SP34-ex media. Hematopoietic cytokines were withdrawn for 4 hours, and cells were then exposed to KIT-L at 50 ng/mL. At 8 hours, cell cycle phase distributions were determined via staining with DRAQ5.
Because KITand the EPO receptor have been indicated to act in cooperative ways [2,59,60], EPO’s capacity to stimulate erythropoiesis in KitY567F/Y567F and Kit+/+ mice also was tested. Specifically, mice were injected with EPO (300 U/kg) at 24-hour intervals for 5 days, and hematocrits were monitored over an 11-day period (Fig. 6D). Overall, time courses for EPO-dependent increases in hematocrits were similar, but in KitY567F/Y567F mice, red cell levels generated at days 6 to 9 were deficient. Finally, for mice dosed with both EPO and stem cell factor, bone marrow erythroid progenitors were isolated (at day 3 postdosing) and were cultured in SP34-ex media containing KIT-L at 50 ng/mL. For KitposCD71high cells, cell cycle phase distributions then were analyzed via staining with DRAQ5. As shown in Figure 6E, KitY567F/Y567F progenitors were significantly underrepresented in S-phase, and overrepresented in G1.
Regulation of AKT and JNK2/p54 signaling is selectively attenuated in primary KitY567F/Y567F erythroid progenitor cells
Signals relayed via KIT Y567 previously have been associated, in part, with mitogen-activated protein kinase and/or SRC kinase pathways [50,54]. In primary KitY567F/Y567F and Kit+/+ erythroid cells, select signal transduction events, therefore, were analyzed. Here, bone marrow progenitor cells were expanded in SP34-ex medium. KitposCD71high erythroblasts then were isolated (via lineage depletion and MACS selection) and were cultured without KITL (or EPO) for 5.5 hours. Cells were then exposed to KITL (150 ng/mL) for the indicated intervals, and levels of phospho (and total) AKT, p70S6K, JNK, and ERK proteins were assayed via Western blotting. In these analyses, levels of phospho-AKT and its downstream target phospho-p70S6K were diminished in KitY567F/Y567F erythroblasts (Fig. 7A). Quantitative densitometric analyses confirmed observed outcomes with phospho-AKT levels deficient at 5 minutes of KITL stimulation, and phospho-p70S6K levels diminished overall (Fig. 7A). In KitY567F/Y567F erythroblasts, activation of JNK2/p54 (but not JNK1/p46), interestingly, was also selectively attenuated (Fig. 7B). ERKs, in contrast, were activated at essentially equivalent levels (and time courses) in Kit+/+ and KitY567F/Y567F erythroblasts (Fig. 7B). Therefore, these analyses point overall (and specifically) to AKT and JNK2/p54 as prime KIT PY567-regulated responses in primary bone marrow–derived erythroblasts.
Figure 7. KIT activation of AKT, p70S6K, and JNK2/p54 is attenuated in KitY567F/Y567F erythroblasts.

(A) Attenuated AKT and p70S6K activation in KitY567F/Y567F erythroblasts—Bone marrow–derived progenitor cells from wild-type Kit+/+ (wt-Kit) and KitY567F/Y567F mice were expanded in SP34-ex medium. KitposCD71high erythroblasts were then isolated (by magnetic-activated cell sorting) and incubated for 5.5 hours in the absence of hematopoietic cytokines. Following exposure to KITL (150 ng/mL) for the indicated intervals, levels of phospho- (and total) AKT (A1) and p70-S6K (A2) were assayed by Western blotting. For KitY567F/Y567F erythroblasts, note the attenuated phosphorylation/activation of AKT (especially 5 minutes). Phospho-p70S6K levels also were diminished in KitY567F/ Y567F erythroblasts (especially at 15 and 30 minutes). Signal quantitation was by densitometry (A3, A4). (B) Attenuated JNK2/p54 activation in KitY567F/Y567F erythroblasts—Primary erythroblasts were expanded and prepared as above (A). KITL-induced levels of phospho- JNK2/p54 and JNK1/p46 were assayed by immunoblotting (B1). In KitY567F/Y567F erythroblasts, note the selectively attenuated activation of JNK2/p54. In parallel, levels of phospho-ERK1,2 were determined (B2), including signal quantitation (B3, B4).
Discussion
KIT is an established regulator of hematopoietic stem and progenitor cell growth and survival [2,61–63]. In certain committed progenitors, KIT expression persists and can regulate lineage-selective growth, survival, and/or differentiation [49,64–67]. In early T-cell development for example, KIT is required for progression from TN1 to TN2 stages [49], and in early B-cell development supports pro-B to pre-B formation [67]. KIT also is expressed on mature mast cells and can modulate their growth, survival, differentiation, adhesion, secretory response, and migration [64]. As illustrated by macrocytic anemia in KIT loss-of-function mice, KIT also significantly affects erythropoiesis.
As an in vivo approach to investigate the significance of select KIT- mediated events, mice with mutations in KIT PY motifs with critical roles in the activation of PI3-kinase, and/or SFK signaling have been developed [33,49,50]. Mice carrying a KitY719F/Y719F mutation, which abolishes p85/PI3K docking, fail to support spermatogenesis [33]. In contrast, mice carrying a KitY567F mutation support normal gametogenesis, but are impaired in lymphopoiesis at pro-T and pro-B cell stages [49]. In each line, mast cell formation is compromised [49,65]. Finally, mutation of both this predominant Y567 SFK binding site plus a Y569 SHP1 tyrosine phosphatase binding site, leads to defects in B-cell development and megakaryopoiesis [50]. Despite the well-known critical role for KIT in erythropoiesis, erythropoiesis in these KIT mutant models remained unperturbed [33,49,50]. This provoked the concept that disruption of signaling in such PY site KIT mutations might become selectively important during stress erythropoiesis. Evidence presented in this report supports this hypothesis in several ways.
First, ablation of cycling hematopoietic progenitor cells by high-dose 5-FU resulted in lethality in KitY567F/Y567F mice in contrast to normal controls. This was associated with deficient erythrosplenomegaly, and markedly decreased Ter119pos erythroblast representation in bone marrow and spleen. By direct comparison, this defect was not exhibited by KitY719F/Y719F mice, for which 5-FU recovery parameters were comparable to wild-type controls (see Suppl. Fig. S1). To better understand the dynamics of anemia and recovery, KitY567F/Y567F mice were dosed with 5-FU at 150 mg/kg. Here, lower hematocrits, delayed recovery, and macrocytosis were observed. Macrocytic anemia is associated with mutations at Kit and KITL loci [68] (and KitW/W mice have been considered as a Diamond Blackfan anemia model) [69]. One factor linked to macrocytic anemia is perturbed DNA synthesis (i.e., due to folic acid deficiency) [70]. In erythroid progenitor cells, both KITL and EPO promote cell cycle progression [6,71]. Among several compromised EPO receptor mutants studied in vivo, however, none have associated macrocytic anemia [4,72,73]. This observation distinguishes KIT (and KITY567F/Y567F) pathways from EPOR actions, which may relate to discrete signaling pathways, or possible stage-specific developmental effects. In a related context, crosstalk between KIT and the EPOR has been reported, including KIT-mediated EPOR phosphorylation [27,74]. Using SP34-ex- expanded proerythroblasts, we therefore sought possible skewing effects of KitY567F/Y567F on EPO-induced STAT5, ERK1,2, and/or AKT phosphorylation. Overall, however, no such effects were obvious (with the exception of an apparent modest increase in EPO-induced levels of phospho-STAT5) (negative data, not shown).
In KitY567F/Y567F mice, induction of hemolytic anemia by PHZ led to decreased red cell production and delayed recovery, again in contrast to KitY719F/Y719F mice. This was accompanied by defective erythrosplenomegaly, decreased cellularity, and underrepresentation of Ter119pos erythroblasts. In mice, the spleen is a stress erythropoietic tissue and KIT has been shown to play a key role in this process [75]. Impaired erythropoiesis during PHZ-induced anemia might therefore reflect involvement of KIT PY567-directed signals in splenic erythroid progenitor cells. By speculation, this could involve erythroid progenitor cell-intrinsic effects, or possibly proposed migration events for marrow progenitors to spleen [76]. In addition, possible effects of differential mouse strain sensitivity to anemia should be considered.
To better study possible alterations in bone marrow erythropoiesis in KitY567F/Y567F mice, an ex vivo erythroid expansion system was used [4]. Studies interestingly indicated that at a KitnegCD71high stage KitY567F/Y567F proerythroblasts developed at increased relative frequencies. This, however, was paralleled by a several-fold decrease in erythroid progenitor cell proliferative capacity. Observed increases in the formation of KitnegCD71high KitY567F/Y567F erythroblasts might therefore relate to an attenuation of the inhibitory effects of KITL on erythroid differentiation. In particular, KITL can inhibit CFU-E differentiation [26] and sustain an undifferentiated state via cyclin/CDK effects [77]. In KitY567F/Y567F erythroblasts, such pathways may be weakened (or lost). Interestingly, KitY567F/Y567F erythroblasts at a KitnegCD71high stage also were more prone to apoptosis. Here, death may occur subsequent to failed survival signals at a preceding KitposCD71high stage.
In several tissues and contexts, KIT PY567 has been demonstrated to couple to SRC family kinases [41,42]. In erythroblasts, LYN is a predominant SRC kinase [51]. It, therefore, was of interest to compare the ex vivo development of KitY567F/Y567F and Lyn−/− proerythroblasts. Unlike KitY567F/Y567F erythroblasts, which were overrepresented at a KitnegCD71high stage, Lyn−/− erythroblasts were found to accumulate at the KitposCD71high stage. Uncoupling from LYN, therefore, appears to limit erythroid development at an earlier stage. In KitY567F/Y567F cells, LYN also might be activated by other potentially compensatory signaling pathways. We also obtained KitY567F/Y567F:Y569F/ Y569F mice, and analyzed their ex vivo developmental profiles for bone marrow–derived erythroblasts. As shown in Supplemental Figure S2, KitY567F/Y567F:Y569F/Y569F erythroblasts proved to exhibit properties similar to KitY567F/Y567F progenitors (and growth capacities of Kit Y567F/Y567F:Y569F/Y569F erythroid progenitors also were attenuated, data not shown). These latter findings suggest that KIT-PY567 coupled events may critically mediate stress erythropoiesis, and that KIT-PY569 may not contribute to this in a major capacity.
These findings raise questions concerning the specific nature of KIT-PY567–coupled signaling events in developing erythroblasts, especially during stress erythropoiesis. Previously, this question has been studied in cell line models. One reductionist approach involved the mutation of six key KIT PY sites (Y567F, Y569F, Y702F, Y719F, Y728F, Y745F, and Y934F), followed by the subsequent restoration of select single PY sites. Recovered activities were then assessed using hMCSFR/mKIT chimeric receptor constructs in myeloid 32D cells [78]. Notably, the restoration of either KIT Y567 or Y569 (or both) rescued 32D cell proliferation, and survival. Biochemically, this correlated with an apparent restoration of chimeric receptor- mediated ERK, PI3K-AKT, and JNK kinase activation. To follow-up on this approach, we performed similar analyses, but in erythroid G1E2 and EML cell lines (supplemental studies; see Suppl. Fig. S3). In both cell models, growth and survival activities for the above hMCSFR/mKIT chimeras were largely recovered upon restoration of Y567 (and Y567 plus Y569). These cell line results, therefore, emphasize functional capacities provided by KIT PY567 in erythroid progenitor cells.
In primary cell systems, signaling capacities of KIT-PY–mutated alleles have been studied to date only in bone marrow–derived mast cells [33,41,44]. Presently, opportunities were provided to examine KIT and KIT-PY567 signaling pathways in primary KitposCD71high bone marrow erythroblasts. In contrast to cell line studies [50,78], outcomes in KITY567F/Y567F erythroblasts first indicated apparently normal signaling to ERKs. However, AKT, p70S6K, and especially JNK2 activation, was attenuated in a KIT-PY567–dependent fashion (see Fig. 7). One common pathway mediated by KIT PY567 in bone marrow –derived mast cells [33,41,44] and bone marrow erythroblasts, therefore, appears to be a route to JNK activation, with preferential activation of JNK2 [37]. Previously, differential roles for JNK2 and JNK1 have been proposed based on apparent inhibitory effects of JNK2 in gene disruption experiments (although both can act as positive cell growth regulators) [79,80]. In primary erythroblasts, apparent utilization of JNK2 via KIT-PY567 at least suggests a selective role for JNK2 during KITL-dependent proliferation. Interestingly, JNK inhibitors inhibit BFU-E, but not CFU-E formation [81].
As also observed in KITY567F/Y567F erythroblasts, attenuated AKT and p70S6K activation might relate to uncoupling from SFKs (which can bind p85-α via an SH3 subdomain) [82]. Interestingly, in KITY567F/ Y567F bone marrow–derived mast cells, cell proliferation, and survival responses are increased [49], AKT activation is increased, and activation of SHIP-1 is abolished (Ehlers and Besmer, unpublished observation). These considerations suggest that the tissue and lineage- selection actions of KIT may be differentially affected by negative regulators. In ongoing erythroid-profiling studies, we have observed significant stage-specific modulation of both SHIPs and SHPTPs. As shown in Supplemental Figure S4, early stage Kitpos erythroblasts express SHPTP1 together with SHIP1 and Inpp5b, while SHPTP2 and Inpp5a are induced at later stages. Specific roles for these negative regulators during KIT-dependent stress erythropoiesis, and their targets in an anemia context, remain to be determined. Future investigations of such regulatory networks ultimately may lead to identification of rational new pathways and targets for antianemia agents.
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.exphem.2008.10.009.
Acknowledgments
This work was supported by National Institutes of Health grants R01-HL-44491 (D.M.W.), R01-HL-55748 (P.B.), and MMCRI Core Facilities as sponsored via P20-RR018789 and P20-RR015555. The authors thank Drs. Y. Kimura and A. Bernstein (Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto) for the generous provision of KitY567/Y567F:Y569F/Y569F mice. We also thank Dr. Jan Hendrix for help with FACS analysis; the Tri-Institutional Genetically Engineered Mouse (GEM) Phenotyping core facility for hematological determinations; Yasemine Yozgat for mouse colony maintenance and genotyping; Dr. M.A.S. Moore for discussions; Dr. R. Kapur (Indiana University School of Medicine) for generously providing a chimeric MCSF-R/KIT retroviral construct “F6”; and Dr, M. Weiss (The Children’s Hospital of Philadelphia) for the provision of G1E2 cells. Authorship: V. Agosti and V. Karur performed essentially all of the in vivo and ex vivo investigations described. Dr. P Sathyanarayana assisted with additional experiments, figures and manuscript preparation. D. M. Wojchowski and P. Besmer provided lead efforts for experimental designs. All authors participated in major ways in the interpretation of data, and manuscript construction.
Footnotes
The authors declare no competing financial interests or conflicts of interest.
References
- 1.Richmond TD, Chohan M, Barber DL. Turning cells red: signal transduction mediated by erythropoietin. Trends Cell Biol. 2005;15:146–155. doi: 10.1016/j.tcb.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Munugalavadla V, Kapur R. Role of c-Kit and erythropoietin receptor in erythropoiesis. Crit Rev Oncol Hematol. 2005;54:63–75. doi: 10.1016/j.critrevonc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83:59–67. doi: 10.1016/0092-8674(95)90234-1. [DOI] [PubMed] [Google Scholar]
- 4.Menon MP, Karur V, Bogacheva O, Bogachev O, Cuetara B, Wojchowski DM. Signals for stress erythropoiesis are integrated via an erythropoietin receptor-phosphotyrosine-343-Stat5 axis. J Clin Invest. 2006;116:683–694. doi: 10.1172/JCI25227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sathyanarayana P, Dev A, Fang J, et al. EPO receptor circuits for primary erythroblast survival. Blood. 2008;111:5390–5399. doi: 10.1182/blood-2007-10-119743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang J, Menon M, Kapelle W, et al. EPO modulation of cell-cycle regulatory genes, and cell division, in primary bone marrow erythroblasts. Blood. 2007;110:2361–2370. doi: 10.1182/blood-2006-12-063503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quesniaux VF, Clark SC, Turner K, Fagg B. Interleukin-11 stimulates multiple phases of erythropoiesis in vitro. Blood. 1992;80:1218–1223. [PubMed] [Google Scholar]
- 8.Aiello FB, Keller JR, Klarmann KD, Dranoff G, Mazzucchelli R, Durum SK. IL-7 induces myelopoiesis and erythropoiesis. J Immunol. 2007;178:1553–1563. doi: 10.4049/jimmunol.178.3.1553. [DOI] [PubMed] [Google Scholar]
- 9.Bohmer RM. Erythropoiesis from adult but not fetal blood-derived CD133+ stem cells depends strongly on interleukin-3. Growth Factors. 2004;22:45–50. doi: 10.1080/08977190310001649304. [DOI] [PubMed] [Google Scholar]
- 10.Dybedal I, Larsen S, Jacobsen SE. IL-12 directly enhances in vitro murine erythropoiesis in combination with IL-4 and stem cell factor. J Immunol. 1995;154:4950–4955. [PubMed] [Google Scholar]
- 11.Papayannopoulou T, Brice M, Blau CA. Kit ligand in synergy with interleukin- 3 amplifies the erythropoietin-independent, globin-synthesizing progeny of normal human burst-forming units-erythroid in suspension cultures: physiologic implications. Blood. 1993;81:299–310. [PubMed] [Google Scholar]
- 12.Perry JM, Harandi OF, Paulson RF. BMP4, SCF, and hypoxia cooperatively regulate the expansion of murine stress erythroid progenitors. Blood. 2007;109:4494–4502. doi: 10.1182/blood-2006-04-016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zermati Y, Fichelson S, Valensi F, et al. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp Hematol. 2000;28:885–894. doi: 10.1016/s0301-472x(00)00488-4. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Pop R, Sadegh C, Brugnara C, Haase VH, Socolovsky M. Suppression of Fas-FasL coexpression by erythropoietin mediates erythroblast expansion during the erythropoietic stress response in vivo. Blood. 2006;108:123–133. doi: 10.1182/blood-2005-11-4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silvestris F, Cafforio P, Tucci M, Dammacco F. Negative regulation of erythroblast maturation by Fas-L(+)/TRAIL(+) highly malignant plasma cells: a major pathogenetic mechanism of anemia in multiple myeloma. Blood. 2002;99:1305–1313. doi: 10.1182/blood.v99.4.1305. [DOI] [PubMed] [Google Scholar]
- 16.Dufour C, Corcione A, Svahn J, et al. TNF-alpha and IFN-gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood. 2003;102:2053–2059. doi: 10.1182/blood-2003-01-0114. [DOI] [PubMed] [Google Scholar]
- 17.Thawani N, Tam M, Chang KH, Stevenson MM. Interferon-gamma mediates suppression of erythropoiesis but not reduced red cell survival following CpG-ODN administration in vivo. Exp Hematol. 2006;34:1451–1461. doi: 10.1016/j.exphem.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 18.van den Akker E, van Dijk T, Parren-van Amelsvoort M, et al. Tyrosine kinase receptor RON functions downstream of the erythropoietin receptor to induce expansion of erythroid progenitors. Blood. 2004;103:4457–4465. doi: 10.1182/blood-2003-08-2713. [DOI] [PubMed] [Google Scholar]
- 19.Teal HE, Craici A, Paulson RF, Correll PH. Macrophage-stimulating protein cooperates with erythropoietin to induce colony formation and MAP kinase activation in primary erythroid progenitor cells. J Hematother Stem Cell Res. 2003;12:165–177. doi: 10.1089/152581603321628313. [DOI] [PubMed] [Google Scholar]
- 20.Miyagawa S, Kobayashi M, Konishi N, Sato T, Ueda K. Insulin and insulin-like growth factor I support the proliferation of erythroid progenitor cells in bone marrow through the sharing of receptors. Br J Haematol. 2000;109:555–562. doi: 10.1046/j.1365-2141.2000.02047.x. [DOI] [PubMed] [Google Scholar]
- 21.Miharada K, Hiroyama T, Sudo K, Nagasawa T, Nakamura Y. Refinement of cytokine use in the in vitro expansion of erythroid cells. Hum Cell. 2006;19:30–37. doi: 10.1111/j.1749-0774.2005.00005.x. [DOI] [PubMed] [Google Scholar]
- 22.Suenobu S, Takakura N, Inada T, et al. A role of EphB4 receptor and its ligand, ephrin-B2, in erythropoiesis. Biochem Biophys Res Commun. 2002;293:1124–1131. doi: 10.1016/S0006-291X(02)00330-3. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Miura N, Bonelli A, et al. Receptor tyrosine kinase, EphB4 (HTK), accelerates differentiation of select human hematopoietic cells. Blood. 2002;99:2740–2747. doi: 10.1182/blood.v99.8.2740. [DOI] [PubMed] [Google Scholar]
- 24.Nocka K, Buck J, Levi E, Besmer P. Candidate ligand for the c-kit trans-membrane kinase receptor: KL, a fibroblast derived growth factor stimulates mast cells and erythroid progenitors. EMBO J. 1990;9:3287–3294. doi: 10.1002/j.1460-2075.1990.tb07528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zsebo KM, Williams DA, Geissler EN, et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63:213–224. doi: 10.1016/0092-8674(90)90302-u. [DOI] [PubMed] [Google Scholar]
- 26.Muta K, Krantz SB, Bondurant MC, Dai CH. Stem cell factor retards differentiation of normal human erythroid progenitor cells while stimulating proliferation. Blood. 1995;86:572–580. [PubMed] [Google Scholar]
- 27.Wu H, Klingmuller U, Besmer P, Lodish HF. Interaction of the erythropoietin and stem-cell-factor receptors. Nature. 1995;377:242–246. doi: 10.1038/377242a0. [DOI] [PubMed] [Google Scholar]
- 28.Nocka K, Majumder S, Chabot B, et al. Expression of c-kit gene products in known cellular targets of W mutations in normal and W mutant mice–evidence for an impaired c-kit kinase in mutant mice. Genes Dev. 1989;3:816–826. doi: 10.1101/gad.3.6.816. [DOI] [PubMed] [Google Scholar]
- 29.Besmer P. The kit ligand encoded at the murine Steel locus: a pleiotropic growth and differentiation factor. Curr Opin Cell Biol. 1991;3:939–946. doi: 10.1016/0955-0674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 30.Waskow C, Terszowski G, Costa C, Gassmann M, Rodewald HR. Rescue of lethal c-KitW/W mice by erythropoietin. Blood. 2004;104:1688–1695. doi: 10.1182/blood-2004-04-1247. [DOI] [PubMed] [Google Scholar]
- 31.Russell ES. Hereditary anemias of the mouse: a review for geneticists. Adv Genet. 1979;20:357–459. [PubMed] [Google Scholar]
- 32.Besmer P, Manova K, Duttlinger R, et al. The kit-ligand (steel factor) and its receptor c-kit/W: pleiotropic roles in gametogenesis and melanogenesis. Dev Suppl. 1993:125–137. [PubMed] [Google Scholar]
- 33.Kissel H, Timokhina I, Hardy MP, et al. Point mutation in kit receptor tyrosine kinase reveals essential roles for kit signaling in spermatogenesis and oogenesis without affecting other kit responses. EMBO J. 2000;19:1312–1326. doi: 10.1093/emboj/19.6.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature. 1995;373:347–349. doi: 10.1038/373347a0. [DOI] [PubMed] [Google Scholar]
- 35.Duttlinger R, Manova K, Berrozpe G, et al. The Wsh and Ph mutations affect the c-kit expression profile: c-kit misexpression in embryogenesis impairs melanogenesis in Wsh and Ph mutant mice. Proc Natl Acad Sci U S A. 1995;92:3754–3758. doi: 10.1073/pnas.92.9.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitamura Y, Hirotab S. Kit as a human oncogenic tyrosine kinase. Cell Mol Life Sci. 2004;61:2924–2931. doi: 10.1007/s00018-004-4273-y. [DOI] [PubMed] [Google Scholar]
- 37.Linnekin D. Early signaling pathways activated by c-Kit in hematopoietic cells. Int J Biochem Cell Biol. 1999;31:1053–1074. doi: 10.1016/s1357-2725(99)00078-3. [DOI] [PubMed] [Google Scholar]
- 38.Serve H, Yee NS, Stella G, Sepp-Lorenzino L, Tan JC, Besmer P. Differential roles of PI3-kinase and Kit tyrosine 821 in Kit receptor-mediated proliferation, survival and cell adhesion in mast cells. EMBO J. 1995;14:473–483. doi: 10.1002/j.1460-2075.1995.tb07023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gommerman JL, Sittaro D, Klebasz NZ, Williams DA, Berger SA. Differential stimulation of c-Kit mutants by membrane-bound and soluble Steel Factor correlates with leukemic potential. Blood. 2000;96:3734–3742. [PubMed] [Google Scholar]
- 40.Thommes K, Lennartsson J, Carlberg M, Ronnstrand L. Identification of Tyr-703 and Tyr-936 as the primary association sites forGrb2 andGrb7 in the c-Kit/stem cell factor receptor. Biochem J. 1999;341(Pt 1):211–216. [PMC free article] [PubMed] [Google Scholar]
- 41.Timokhina I, Kissel H, Stella G, Besmer P. Kit signaling through PI 3-kinase and Src kinase pathways: an essential role for Rac1 and JNK activation in mast cell proliferation. EMBO J. 1998;17:6250–6262. doi: 10.1093/emboj/17.21.6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ueda S, Mizuki M, Ikeda H, et al. Critical roles of c-Kit tyrosine residues 567 and 719 in stem cell factor-induced chemotaxis: contribution of src family kinase and PI3-kinase on calcium mobilization and cell migration. Blood. 2002;99:3342–3349. doi: 10.1182/blood.v99.9.3342. [DOI] [PubMed] [Google Scholar]
- 43.Kozlowski M, Larose L, Lee F, Le DM, Rottapel R, Siminovitch KA. SHP-1 binds and negatively modulates the c-Kit receptor by interaction with tyrosine 569 in the c-Kit juxtamembrane domain. Mol Cell Biol. 1998;18:2089–2099. doi: 10.1128/mcb.18.4.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu M, Luo J, Yang W, et al. The scaffolding adapter Gab2, via Shp-2, regulates kit-evoked mast cell proliferation by activating the Rac/JNK pathway. J Biol Chem. 2006;281:28615–28626. doi: 10.1074/jbc.M603742200. [DOI] [PubMed] [Google Scholar]
- 45.Lennartsson J, Blume-Jensen P, Hermanson M, Ponten E, Carlberg M, Ronnstrand L. Phosphorylation of Shc by Src family kinases is necessary for stem cell factor receptor/c-kit mediated activation of the Ras/MAP kinase pathway and c-fos induction. Oncogene. 1999;18:5546–5553. doi: 10.1038/sj.onc.1202929. [DOI] [PubMed] [Google Scholar]
- 46.Price DJ, Rivnay B, Fu Y, Jiang S, Avraham S, Avraham H. Direct association of Csk homologous kinase (CHK) with the diphosphorylated site Tyr568/570 of the activated c-KIT in megakaryocytes. J Biol Chem. 1997;272:5915–5920. doi: 10.1074/jbc.272.9.5915. [DOI] [PubMed] [Google Scholar]
- 47.Wollberg P, Lennartsson J, Gottfridsson E, Yoshimura A, Ronnstrand L. The adapter protein APS associates with the multifunctional docking sites Tyr-568 and Tyr-936 in c-Kit. Biochem J. 2003;370:1033–1038. doi: 10.1042/BJ20020716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blume-Jensen P, Jiang G, Hyman R, Lee KF, O’Gorman S, Hunter T. Kit/stem cell factor receptor-induced activation of phosphatidylinositol 3’-kinase is essential for male fertility. Nat Genet. 2000;24:157–162. doi: 10.1038/72814. [DOI] [PubMed] [Google Scholar]
- 49.Agosti V, Corbacioglu S, Ehlers I, et al. Critical role for Kit-mediated Src kinase but not PI 3-kinase signaling in pro T and pro B cell development. J Exp Med. 2004;199:867–878. doi: 10.1084/jem.20031983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimura Y, Jones N, Kluppel M, et al. Targeted mutations of the juxtamembrane tyrosines in the Kit receptor tyrosine kinase selectively affect multiple cell lineages. Proc Natl Acad Sci U S A. 2004;101:6015–6020. doi: 10.1073/pnas.0305363101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karur VG, Lowell CA, Besmer P, Agosti V, Wojchowski DM. Lyn kinase promotes erythroblast expansion and late-stage development. Blood. 2006;108:1524–1532. doi: 10.1182/blood-2005-09-008243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weiss MJ, Yu C, Orkin SH. Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a genetargeted cell line. Mol Cell Biol. 1997;17:1642–1651. doi: 10.1128/mcb.17.3.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weiler SR, Gooya JM, Ortiz M, Tsai S, Collins SJ, Keller JR. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 1999;93:527–536. [PubMed] [Google Scholar]
- 54.Tan BL, Hong L, Munugalavadla V, Kapur R. Functional and biochemical consequences of abrogating the activation of multiple diverse early signaling pathways in Kit. Role for Src kinase pathway in Kit-induced cooperation with erythropoietin receptor. J Biol Chem. 2003;278:11686–11695. doi: 10.1074/jbc.M207068200. [DOI] [PubMed] [Google Scholar]
- 55.Sathyanarayana P, Menon MP, Bogacheva O, et al. Erythropoietin modulation of podocalyxin, and a proposed erythroblast niche. Blood. 2007;110:509–518. doi: 10.1182/blood-2006-11-056465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linnekin D, DeBerry CS, Mou S. Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J Biol Chem. 1997;272:27450–27455. doi: 10.1074/jbc.272.43.27450. [DOI] [PubMed] [Google Scholar]
- 57.Chin H, Arai A, Wakao H, Kamiyama R, Miyasaka N, Miura O. Lyn physically associates with the erythropoietin receptor and may play a role in activation of the Stat5 pathway. Blood. 1998;91:3734–3745. [PubMed] [Google Scholar]
- 58.Ingley E, Sarna MK, Beaumont JG, et al. HS1 interacts with Lyn and is critical for erythropoietin-induced differentiation of erythroid cells. J Biol Chem. 2000;275:7887–7893. doi: 10.1074/jbc.275.11.7887. [DOI] [PubMed] [Google Scholar]
- 59.Kapur R, Zhang L. A novel mechanism of cooperation between c-Kit and erythropoietin receptor. Stem cell factor induces the expression of Stat5 and erythropoietin receptor, resulting in efficient proliferation and survival by erythropoietin. J Biol Chem. 2001;276:1099–1106. doi: 10.1074/jbc.M007442200. [DOI] [PubMed] [Google Scholar]
- 60.Pircher TJ, Geiger JN, Zhang D, Miller CP, Gaines P, Wojchowski DM. Integrative signaling by minimal erythropoietin receptor forms and c-Kit. J Biol Chem. 2001;276:8995–9002. doi: 10.1074/jbc.M007473200. [DOI] [PubMed] [Google Scholar]
- 61.Andrews RG, Briddell RA, Appelbaum FR, McNiece IK. Stimulation of hematopoiesis in vivo by stem cell factor. Curr Opin Hematol. 1994;1:187–196. [PubMed] [Google Scholar]
- 62.Hassan HT, Zander A. Stem cell factor as a survival and growth factor in human normal and malignant hematopoiesis. Acta Haematol. 1996;95:257–262. doi: 10.1159/000203893. [DOI] [PubMed] [Google Scholar]
- 63.McNiece IK, Briddell RA. Stem cell factor. J Leukoc Biol. 1995;58:14–22. doi: 10.1002/jlb.58.1.14. [DOI] [PubMed] [Google Scholar]
- 64.Okayama Y, Kawakami T. Development, migration, and survival of mast cells. Immunol Res. 2006;34:97–115. doi: 10.1385/IR:34:2:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McNiece IK, Langley KE, Zsebo KM. The role of recombinant stem cell factor in early B cell development. Synergistic interaction with IL-7. J Immunol. 1991;146:3785–3790. [PubMed] [Google Scholar]
- 66.McNiece IK, Langley KE, Zsebo KM. Recombinant human stem cell factor synergises with GM-CSF, G-CSF, IL-3 and epo to stimulate human progenitor cells of the myeloid and erythroid lineages. Exp Hematol. 1991;19:226–231. [PubMed] [Google Scholar]
- 67.Waskow C, Paul S, Haller C, Gassmann M, Rodewald HR. Viable c-Kit(W/W) mutants reveal pivotal role for c-kit in the maintenance of lymphopoiesis. Immunity. 2002;17:277–288. doi: 10.1016/s1074-7613(02)00386-2. [DOI] [PubMed] [Google Scholar]
- 68.Bernstein A, Chabot B, Dubreuil P, et al. The mouse W/c-kit locus. Ciba Found Symp. 1990;148:158–166. [PubMed] [Google Scholar]
- 69.Drachtman RA, Geissler EN, Alter BP. The SCF and c-kit genes in Diamond-Blackfan anemia. Blood. 1992;79:2177–2178. [PubMed] [Google Scholar]
- 70.Streiff RR. Folic acid deficiency anemia. Semin Hematol. 1970;7:23–39. [PubMed] [Google Scholar]
- 71.Kapur R, Chandra S, Cooper R, McCarthy J, Williams DA. Role of p38 and ERK MAP kinase in proliferation of erythroid progenitors in response to stimulation by soluble and membrane isoforms of stem cell factor. Blood. 2002;100:1287–1293. [PubMed] [Google Scholar]
- 72.Zang H, Sato K, Nakajima H, McKay C, Ney PA, Ihle JN. The distal region and receptor tyrosines of the Epo receptor are non-essential for in vivo erythropoiesis. EMBO J. 2001;20:3156–3166. doi: 10.1093/emboj/20.12.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miller CP, Heilman DW, Wojchowski DM. Erythropoietin receptor-dependent erythroid colony-forming unit development: capacities of Y343 and phosphotyrosine-null receptor forms. Blood. 2002;99:898–904. doi: 10.1182/blood.v99.3.898. [DOI] [PubMed] [Google Scholar]
- 74.Wu H, Klingmuller U, Acurio A, Hsiao JG, Lodish HF. Functional interaction of erythropoietin and stem cell factor receptors is essential for erythroid colony formation. Proc Natl Acad Sci U S A. 1997;94:1806–1810. doi: 10.1073/pnas.94.5.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Broudy VC, Lin NL, Priestley GV, Nocka K, Wolf NS. Interaction of stem cell factor and its receptor c-kit mediates lodgment and acute expansion of hematopoietic cells in the murine spleen. Blood. 1996;88:75–81. [PubMed] [Google Scholar]
- 76.Porayette P, Paulson RF. BMP4/Smad5 dependent stress erythropoiesis is required for the expansion of erythroid progenitors during fetal development. Dev Biol. 2008;317:24–35. doi: 10.1016/j.ydbio.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tamir A, Petrocelli T, Stetler K, et al. Stem cell factor inhibits erythroid differentiation by modulating the activity of G1-cyclin-dependent kinase complexes: a role for p27 in erythroid differentiation coupled G1 arrest. Cell Growth Differ. 2000;11:269–277. [PubMed] [Google Scholar]
- 78.Hong L, Munugalavadla V, Kapur R. c-Kit-mediated overlapping and unique functional and biochemical outcomes via diverse signaling pathways. Mol Cell Biol. 2004;24:1401–1410. doi: 10.1128/MCB.24.3.1401-1410.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jaeschke A, Karasarides M, Ventura JJ, et al. JNK2 is a positive regulator of the cJun transcription factor. Mol Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 80.Blonska M, Pappu BP, Matsumoto R, Li H, Su B, Wang D, Lin X. The CARMA1-Bcl10 signaling complex selectively regulates JNK2 kinase in the T cell receptor-signaling pathway. Immunity. 2007;26:55–66. doi: 10.1016/j.immuni.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jacobs-Helber SM, Sawyer ST. Jun N-terminal kinase promotes proliferation of immature erythroid cells and erythropoietin-dependent cell lines. Blood. 2004;104:696–703. doi: 10.1182/blood-2003-05-1754. [DOI] [PubMed] [Google Scholar]
- 82.Pleiman CM, Hertz WM, Cambier JC. Activation of phosphatidylinositol-3’ kinase by Src-family kinase SH3 binding to the p85 subunit. Science. 1994;263:1609–1612. doi: 10.1126/science.8128248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.exphem.2008.10.009.