

Abstract
Neuroactive steroids are among the most efficacious modulators of the mammalian GABA-A receptor. Previous work has proposed that receptor potentiation is mediated by steroid interactions with a site defined by the residues α1N407/Y410 in the M4 transmembrane domain, and residue α1Q241 in the M1 domain. We examined the role of residues in the α1 subunit M1 domain in the modulation of the rat α1β2γ2L GABA-A receptor by neuroactive steroids. The data demonstrate that the region is critical to the actions of potentiating neuroactive steroids. Receptors containing the α1Q241W or α1Q241L mutations were insensitive to (3α,5α)-3-hydroxypregnan-20-one (3α5αP), albeit with different underlying mechanisms. The α1Q241S mutant was potentiated by 3α5αP but the kinetic mode of potentiation was altered by the mutation. Interestingly, the α1Q241L mutation was without effect on channel potentiation by (3α,5α)-3-hydroxymethylpregnan-20-one, but mutation of the neighboring residue, α1S240, prevented channel modulation. A steroid lacking a H-bonding group on C3 (5α-pregnan-20-one) potentiated the wild-type receptor but not the α1Q241L mutant. The findings are consistent with a model where the α1S240 and α1Q241 residues shape the surface to which steroid molecules bind.
INTRODUCTION
Potentiating neurosteroids are among the most efficacious modulators of the mammalian GABA-A receptor having potential applications as anxiolytics, anticonvulsants, sedatives and anesthetics. Recent work has given significant insights into the functional and structural mechanisms of steroid actions. Potentiating steroids, e.g., (3α,5α)-3-hydroxypregnan-20-one (3α5αP) and (3α,5β)-3-hydroxypregnan-20-one (3α5βP) act on the GABA-A receptor by modifying the channel open and closed times, leading to an increase in the open probability of the channel, enhanced macroscopic peak current, and a slower current decay when exposure to agonist is terminated. The putative steroid binding site is located in the membrane-spanning regions of the α subunit of the receptor, extending from the α1Q241 residue in the M1 membrane-spanning region to the residues α1N407 and α1Y410 in the M4 domain (Hosie et al., 2006). Mutations that reduce the H-bonding ability of these residues reduce receptor potentiation by both 5α-reduced and 5β-reduced steroids. It was proposed that a common interaction site mediates the effects of the two classes of steroids, with the α1Q241 residue acting as a H-bond acceptor to the 3α-hydroxyl group of the steroid molecule, and the α1N407/Y410 residues interacting with the ketone group in the side chain on the D ring of steroids (Hosie et al., 2006). Subsequent studies showed that mutations that disrupt channel potentiation by steroids also affect modulation by a tricyclic benz[e]indene neurosteroid analogue (Li et al., 2006), enantiomers of natural steroids (Li et al., 2007a), and the marine cembranoid eupalmerin acetate (Li et al., 2008), suggesting that the site may function as a common interaction site for a number of GABA-A receptor modulators.
In order to fully understand the role of the α1Q241, and the α1N407 and α1Y410 residues in steroid actions, it is essential to establish, first, the role of these residues in normal receptor activity. Many channel modulators act in a state-specific manner. Therefore, a lack of responsiveness to a drug application may result from changes in channel baseline kinetic properties rather than reflect the inability of the drug to interact with the receptor. In addition, previous single-channel work has demonstrated that steroids modify several kinetic parameters, effects that may be mediated by steroid interactions with two or more distinct binding sites (Akk et al., 2004; Li et al., 2007a). Consequently, it is of interest to assess the effect of mutations to the putative steroid site residues on the full spectrum of kinetic variables that are modified in the presence of neurosteroids.
In this study, we have used a combination of whole-cell and single-channel recordings to characterize the effects of mutations to residues in the α1 subunit M1 membrane-spanning domain on channel modulation by neuroactive steroids. A structural model showing the locations of the residues studied is shown in Figure 1. We show that mutations to the α1Q241 site strongly, albeit through different kinetic mechanisms, affect channel modulation by 3α5αP. Channel potentiation by (3α,5α)-3-hydroxymethylpregnan-20-one (3αCH2OH5βP) was disrupted in the α1S240L but not α1Q241L mutant receptor. We also show that the steroid 5α-pregnan-20-one (3deoxy5αP) potentiates the wild-type receptor but not a receptor containing the α1Q241L mutation. The findings are most compatible with a model where the steroids bind to a hydrophobic surface on the receptor α1 subunit with the α1S240 and α1Q241 residues acting to shape the surface to accommodate a variety of structurally distinct steroids.
Figure 1.
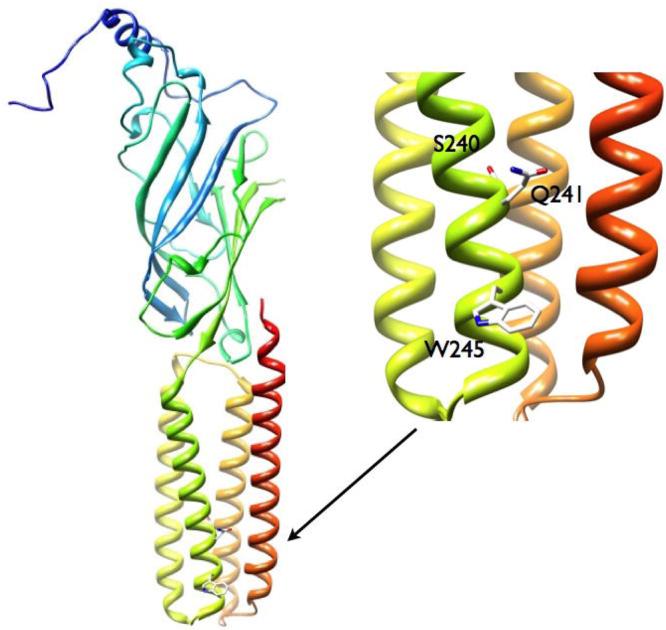
Structural model of the rat α1 subunit, the expanded view shows the locations of the M1 residues studied in this work (M1 is in green and M4 is in red).
MATERIALS AND METHODS
All experiments were carried out on HEK 293 cells expressing rat wild-type and mutant α1β2γ2L GABA-A receptors. The details of receptor expression and electrophysiology have been described in detail previously (Akk et al., 2001; 2004; Li et al., 2006). The mutations were generated using QuikChange (Stratagene, San Diego, CA), and the mutated subunits were fully sequenced to confirm that only the desired mutation had been produced. The α1 subunit is epitope (FLAG) tagged (Ueno et al., 1996) in the aminoterminal end of the subunit. Cells expressing high levels of receptors were determined using a bead-binding technique where the presence of the FLAG peptide was detected with a mouse monoclonal antibody to the FLAG epitope (M2, Sigma-Aldrich, St. Louis, MO), which had been adsorbed to beads with a covalently attached goat anti-mouse IgG antibody (Dynal, Great Neck, NY).
The electrophysiological experiments were carried out using standard single-channel patch clamp and whole-cell voltage clamp methods. The bath solution contained (in mM): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose and 10 HEPES; pH 7.4. In single-channel recordings, the pipet solution contained (in mM): 120 NaCl, 5 KCl, 10 MgCl2, 0.1 CaCl2, 20 tetraethylammonium chloride, 5 4-aminopyridine, 10 glucose, 10 HEPES; pH 7.4. In whole-cell recordings, the pipet solution contained (in mM): 140 CsCl, 4 NaCl, 4 MgCl2, 0.5 CaCl2, 5 EGTA, 10 HEPES, pH 7.4.
The agonist (GABA or piperidine-4-sulfonic acid) and steroid modulators were added to the pipet solution in single-channel recordings, or applied through the bath using an SF-77B fast perfusion stepper system (Warner Instruments, Hamden, CT) in whole-cell experiments. The steroids were initially dissolved in DMSO at 5-10 mM concentration, and diluted immediately before the experiment. The maximal DMSO concentration in diluted steroid solutions was 0.1 %. We have previously found that channel activation by GABA is not affected by the presence of up to 0.3 % DMSO (Li et al., 2007a). All experiments were carried out at room temperature (19-22 °C).
The recording and analysis of single-channel currents have been described in detail previously (Akk et al., 2001; 2004). The pipet potential was held at +60 to +80 mV, which translates to an approximately −120 to −100 mV potential difference across the patch membrane. The channel activity was recorded using an Axopatch 200B amplifier (Molecular Devices, Union City, CA), low-pass filtered at 10 kHz, and acquired with a Digidata 1320 series interface at 50 kHz using pClamp software (Molecular Devices). The key features of the analysis of single-channel currents are the following. When possible, the analysis was limited to clusters, i.e., episodes of intense activity originating from the activation of a single ion channel, or fragments of clusters containing no overlapping currents. The currents were low-pass filtered at 2-3 kHz, and the data were idealized using the segmented-k-means algorithm (Qin et al., 1996). The open and closed times were estimated from the idealized currents using a maximum likelihood method which incorporates a correction for missed events (QuB Suite; www.qub.buffalo.edu). Under certain conditions (e.g., in the presence of P4S or low concentrations of GABA) no clear-cut clusters were observed. In these cases, episodes of activity containing no overlapping currents were used for analysis, and the analysis was limited to estimation of channel open time durations.
Throughout the manuscript, the open and closed time components as determined from the respective histograms are referrred to as OT1-3 and CT1-3. The numerical designation applies to the lifetime of the component (i.e., OT1 and CT1 are the briefest components in the respective histograms) but does not necessarily connote a specific state in the activation scheme. For example, the CT3 component at one GABA concentration does not have to match up with the CT3 component at another GABA concentration, and the components may involve dwells in different activation states. Thus, no mechanism is implied. In some cases, when we believe that a particular closed time component can be associated with sojourns in a particular activation state, we have used additional nomenclature to illustrate the mechanistic implications. For example, a closed time component whose duration inversely correlates with agonist concentration likely results from sojourns in the un-, mono- and diliganded closed states. We have designated the corresponding closed time component CTβ.
The recording and analysis of whole-cell currents was carried out as described previously (Li et al., 2006). The cells were clamped at −60 mV. The cells were exposed to GABA and steroids for 4 s with 30 s washouts separating successive applications. The current traces were low-pass filtered at 2 kHz and digitized at 10 kHz. The analysis of whole-cell currents was carried out using the pClamp 9.0 software package, and was aimed at determining the peak amplitude. Each cell was, prior to testing the effects of steroids, examined using two GABA concentrations to determine the approximate GABA EC50 for the cell in order to verify the expression of γ subunit in the receptor complexes (Boileau et al., 2003).
Comparative structural models of the wild-type and mutant α1 subunits were developed using the program Modeller (Sali and Blundell, 1993). The alignments used for the models were produced using the program MUSCLE (Edgar, 2004) by aligning the rat α1 sequence with the acetylcholine binding protein of Lymanea stagnalis (PDB code 1I9B) (Brejc et al., 2001) and the nicotinic acetylcholine receptor of Torpedo marmorata (PDB code 2BG9) (Unwin, 2005). In order to improve the quality of the models, the 72 residue cytoplasmic loop connecting the M3 and M4 domains was replaced with seven glycine residues. There are no conserved residues in the cytoplasmic loop and only 30 amino acid residues could be aligned to positions on the templates. By replacing this region with seven glycines, optimal for stabilizing two α helices (Liu et al., 2003), a large region of conformational uncertainty was removed. For each protein, a total of ten models were produced using molecular dynamics based refinement for each model. The model quality was then assessed using the default penalty function, and further scored with “DOPE” (Discrete Optimized Protein Energy), and the best model then used for further analysis (Shen and Sali, 2006). While DOPE was developed from a non-redundant set of 1472 structures from the PDB, perhaps biasing towards soluble proteins rather than membrane bound proteins, the idealized reference state is independent of the composition of the protein and the scoring function represents the current state of the art. The molecular graphics images shown in this work were produced with the UCSF Chimera package (Pettersen et al., 2004) from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
The synthesis of 3αCH2OH5βP will be described in detail in future publications. The compound had spectroscopic properties consistent with the assigned structure. It was chromatographically pure and gave the correct elemental analysis. Steroid 3deoxy5αP was obtained from Steraloids, Inc (Newport, RI). Other chemicals including 3α5αP, 3α5βP, and pregnenolone sulfate were purchased from Sigma-Aldrich (St. Louis, MO).
RESULTS
The effects of the α1Q241W mutation on channel activation by GABA
We first examined the activation of the α1Q241W mutant receptor by GABA. The goal of the experiments was to establish the baseline kinetic properties of the mutant receptor for subsequent studies of steroid modulation.
In whole-cell recordings, the GABA dose-response relationship of the α1Q241W receptor was shifted to lower agonist concentrations compared to the wild-type receptor. The midpoint of the dose-response curve was at 2.5 ± 0.2 μM and the Hill slope was 1.0 ± 0.1 (Figure 2A).
Figure 2. Activation and modulation of the wild-type and α1Q241W, α1Q241L and α1Q241S mutant receptors.

(A) Wild-type and mutant receptors were activated by GABA, and the mean fractional peak responses (± SEM) from 5-15 cells are plotted as a function of GABA concentration. Curve fitting to the Hill equation yields the following parameters. Wild-type: EC50 = 9.4 ± 1.1 μM, nH = 0.9 ± 0.1; α1Q241W: EC50 = 2.5 ± 0.2 μM, nH = 1.0 ± 0.1; α1Q241L: EC50 = 43.6 ± 1.7 μM, nH = 1.5 ± 0.1; α1Q241S: EC50 = 36.6 ± 1.5 μM, nH = 1.2 ± 0.1. The wild-type receptor data are from Li et al., 2006. (B) Wild-type and mutant receptors were activated by GABA in the presence of 10-1000 nM 3α5αP. The GABA concentration used corresponded to EC20-25 in the macroscopic dose-response curve, and was 5 μM for the wild-type receptor, 1 μM for α1Q241W, 20 μM for α1Q241L and 15 μM for the α1Q241S mutant receptor. The data points show mean ± SEM from 4-6 cells. Fitting to the Hill equation was conducted for data from the wild-type and α1Q241S receptors. The best-fit parameters for the wild-type are: maximal potentiation = 351 ± 4 %, EC50 = 41 ± 2 nM, nH = 1.2 ± 0.1. The best-fit parameters for the α1Q241S mutant receptor are: maximal potentiation = 350 ± 85 %, EC50 = 142 ± 173 nM, nH = 0.7 ± 0.3. Offset was fixed at 100 %. The test applications lasted 4 s and were separated from flanking control (GABA alone) applications by 30 s washouts.
The principal characteristics of single-channel activity were unchanged by the mutation. Similar to what we have observed previously for wild-type receptors (e.g., Steinbach and Akk, 2001; Li et al., 2007b), exposure to high concentrations of GABA led to channel activity in easily identifiable single-channel clusters. Figure 3 shows sample currents from the mutant receptor exposed to 2, 10, or 50 μM GABA. At 2 μM GABA, channel activity was characterized by isolated openings interspersing brief bursts of activity, but no single-channel clusters were evident. When channel activity was elicited by 10 or 50 μM GABA, the openings were condensed into 1-10 s long clusters of activity.
Figure 3. Single-channel activity from the α1Q241W mutant receptor.

Each section shows three consecutive 10 s segments at a specific concentration of GABA. Channel openings are shown as downward deflections. (A) Channel activation by 2 μM GABA resulted in isolated openings and brief (<0.5 s) bursts of openings. No clusters were evident. (B) Channel activation by 10 μM GABA resulted in high open probability clusters of activity (shown with lines underneath the current traces). In addition to the clusters, the record contained brief bursts of activity and isolated openings of unknown origin, which were not included in the kinetic analysis. (C) Channel activation by 50 μM GABA resulted in clusters of activity and isolated openings. The clusters were defined as long-lived (>0.5 s) groups of activity separated from each other by periods of inactivity, ignoring isolated openings, greater than 1 s.
We examined the intracluster open and closed time distributions to gain mechanistic insight into the activation properties of the mutant receptor. The open time histograms were best-fitted to sums of three exponentials. In the presence of 100 μM GABA (a saturating concentration), the mean durations of the open time components (averaged from 4 patches) were 0.36 ± 0.10 ms (33 %), 3.4 ± 1.7 ms (34 %) and 9.0 ± 2.1 ms (32 %). Reduction of the GABA concentration to 10 μM had had little effect on the intracluster open time distributions (see Table 1). However, the relative number of brief openings (OT1) in the record was increased to 48 ± 1 % (n = 2 patches) when 1 μM GABA was used to activate the receptors. This indicates that a portion of the brief openings originates from un- or monoliganded receptors.
Table 1. The summary of the effects of 1 μM 3α5αP on the intracluster open times from the α1β2γ2L wild-type receptor, and α1Q241W, α1Q241L, and α1Q241S mutant receptors.
The intracluster open time histograms were fitted to sums of three exponentials (wild-type and α1Q241W) or two exponentials (α1Q241L and α1Q241S). In the wild-type receptor, 3α5αP potentiates the currents by enhancing the mean duration and prevalence of OT3. Additionally, the presence of steroid influences the mean duration and prevalence of OT1, and the prevalence of OT2. The presence of steroid was without effect on the properties (mean durations and fractions) of channel openings from the α1Q241W and α1Q241L receptors, but prolonged the mean duration of OT2 in the α1Q241S receptor. Wild-type data are from Li et al. (2007b). The statistical analysis applies to the effect of the steroid for a given receptor (wild-type or mutant).
| Mutation | GABA (μM) |
3α5αP (μM) |
OT1 (ms) | Fraction OT1 |
OT2 (ms) | Fraction OT2 |
OT3 (ms) | Fraction OT3 |
n |
|---|---|---|---|---|---|---|---|---|---|
| None | 50 | - | 0.28±0.05 | 0.22±0.02 | 3.0±0.7 | 0.65±0.06 | 7.3±3.2 | 0.13±0.07 | 4 |
| None | 50 | 1 | 0.41±0.04* | 0.39±0.07** | 2.4±0.9 | 0.23±0.03*** | 14.1±2.1* | 0.38±0.04* | 3 |
| α1Q241W | 10 | - | 0.33±0.08 | 0.26±0.09 | 2.2±1.5 | 0.22±0.07 | 9.6±4.1 | 0.52±0.12 | 4 |
| α1Q241W | 10 | 1 | 0.30±0.04 | 0.33±0.06 | 3.1±2.3 | 0.27±0.19 | 8.5±3.2 | 0.40±0.17 | 3 |
| α1Q241L | 50 | - | 0.60±0.25 | 0.47±0.32 | 1.9±0.7 | 0.53±0.32 | - | - | 4 |
| α1Q241L | 50 | 1 | 0.54±0.14 | 0.23±0.03 | 2.2±0.1 | 0.77±0.03 | - | - | 3 |
| α1Q241S | 50 | - | 0.69±0.21 | 0.60±0.21 | 1.8±0.6 | 0.40±0.21 | - | - | 7 |
| α1Q241S | 50 | 1 | 0.66±0.16 | 0.40±0.13 | 5.7±1.5*** | 0.60±0.13 | - | - | 6 |
P < 0.05
P < 0.01
P < 0.001.
The α1Q241W mutation resulted in an increase in the relative frequency of the longest-lived open time component, OT3. At GABA concentrations evoking equivalent responses, the prevalence of OT3 is 13 ± 4 % in the wild-type receptor (Li et al., 2007b), and 52 ± 12 % in the α1Q241W mutant receptor (Table 1). In addition, there was a trend towards an increase in the duration of OT3 in the mutant receptor. As a result, the mean open duration was enhanced from 2.1 ± 0.3 to 5.6 ± 0.6 ms in the presence of the α1Q241W mutation.
In the presence of 10-100 μM GABA, the intracluster closed time distributions contained three components. At 10 μM GABA, the mean durations of the closed time components (see also Table 2) were 0.13 ± 0.01 ms (58 %; CT1), 1.9 ± 0.4 ms (27 %; CT2) and 20.3 ± 2.0 ms (15 %; CT3). In the presence of 100 μM GABA, the mean durations and fractions of the closed times (n = 4 patches) were 0.18 ± 0.04 ms (63 %; CT1), 1.7 ± 0.2 ms (29 %; CT2) and 8.8 ± 2.7 ms (8 %; CT3).
Table 2. The summary of the effects of 1 μM 3α5αP on intracluster closed times from the α1β2γ2L wild-type receptor, and α1Q241W, α1Q241L, and α1Q241S mutant receptors.
The intracluster closed time histograms were fitted to sums of three exponentials. In the wild-type receptor, 3α5αP potentiates the currents by decreasing the prevalence of the longest-lived closed time component, CT3. Additionally, the presence of steroid influences the mean duration of CT1, and the prevalence of CT2. The presence of steroid was without effect on the properties (mean durations and fractions) of intracluster closed times from the α1Q241W receptor. In receptors containing the α1Q241L mutation, 3α5αP affected the fraction of CT1. In receptors containing the α1Q241S mutation, 3α5αP affected the mean duration and fraction of CT1, and the fraction of CT2. Wild-type data are from Li et al. (2007b). The statistical analysis applies to the effect of the steroid for a given receptor (wild-type or mutant).
| Mutation | GABA (μM) |
3α5αP (μM) |
CT1 (ms) | Fraction CT1 |
CT2 (ms) |
Fraction CT2 |
CT3 (ms) | Fraction CT3 |
n |
|---|---|---|---|---|---|---|---|---|---|
| None | 50 | - | 0.15±0.01 | 0.60±0.10 | 1.5±0.2 | 0.13±0.05 | 14.4±4.2 | 0.27±0.06 | 4 |
| None | 50 | 1 | 0.22±0.04* | 0.64±0.12 | 1.4±0.2 | 0.30±0.10* | 14.3±1.2 | 0.05±0.01*** | 4 |
| α1Q241W | 10 | - | 0.13±0.01 | 0.58±0.07 | 1.9±0.4 | 0.27±0.04 | 20.3±2.0 | 0.15±0.04 | 4 |
| α1Q241W | 10 | 1 | 0.16±0.03 | 0.55±0.15 | 1.8±0.3 | 0.24±0.04 | 19.7±9.6 | 0.21±0.11 | 3 |
| α1Q241L | 50 | - | 0.22±0.05 | 0.54±0.02 | 2.0±1.1 | 0.17±0.05 | 11.3±2.7 | 0.29±0.05 | 4 |
| α1Q241L | 50 | 1 | 0.17±0.03 | 0.61±0.05* | 1.2±0.3 | 0.13±0.03 | 13.2±5.6 | 0.25±0.04 | 3 |
| α1Q241S | 50 | - | 0.21±0.03 | 0.70±0.07 | 1.5±0.5 | 0.10±0.02 | 14.4±2.7 | 0.20±0.05 | 7 |
| α1Q241S | 50 | 1 | 0.14±0.03** | 0.63±0.04* | 1.4±0.3 | 0.18±0.05** | 14.6±6.9 | 0.19±0.03 | 6 |
P < 0.05
P < 0.01
P < 0.001.
We have previously postulated that the intracluster closed time histograms from the wild-type receptor contain a component whose duration (but not prevalence) depends on agonist concentration, as well as several components whose durations are not modulated by changes in agonist concentration (Steinbach and Akk, 2001). The agonist concentration-dependent component (CTβ) results from dwells in unliganded, monoliganded and diliganded closed states. The lifetime of the CTβ component inversely correlates with agonist concentration because the movement from states with lower ligation status to the diliganded open state involves the binding of agonist. The agonist concentration-independent closed states likely result from dwells in various blocked or short-lived desensitized states. The relative frequencies of the closed states are not affected by GABA concentration (Steinbach and Akk, 2001).
It is typically not feasible to resolve all states at a given GABA concentration, i.e., the CTβ state may overlap in duration with dwells in the blocked or short-lived desensitized states, but, by altering the agonist concentration, it is possible to manipulate the duration of the CTβ state and separate it from other closed time components. For example, we previously showed that the single-channel clusters from the wild-type receptor contain a long-lived (10-20 ms), but infrequent (2 % of all intracluster closed events) closed time component (Steinbach and Akk, 2001). We associated this closed event with a short-lived desensitized state (CTSD) as it appeared to be related to the long-lived closed times observed among channel openings following a brief pulse of agonist (Jones and Westbrook, 1995). In the presence of 50 μM GABA, where the activation-related closed time component is prolonged, the CTβ and CTSD states have indistinguishable lifetimes and both states contribute to the longest-lived intracluster closed time component (CT3). In contrast, at saturating GABA concentrations, where the activation-related closed times are brief, the infrequent CTSD state is the sole contributor to the long-lived CT3 closed time component.
Single-channel currents from the α1Q241W mutant receptor indicated the presence of a similar, long-lived closed state. At 100 μM GABA (a saturating concentration), the longest-lived closed component (CT3) had a relative frequency of 8 %, but when the receptors were activated by 10 μM GABA, the prevalence of CT3 was 15 %. We suggest that the CT3 component at saturating GABA concentrations consists of dwells in the short-lived desensitized state (CTSD), while at 10 μM GABA, the CT3 component contains dwells in the CTSD state as well as sojourns in the mono- and unliganded closed states, i.e., CTβ. We can therefore estimate the prevalence of CTβ by subtracting the relative frequency of CTSD, determined at 100 μM GABA (8 ± 3 %), from the relative frequency of CT3 measured at 10 μM GABA (15 ± 4 %). Assuming that the relative frequencies of the two states are unaffected by changes in the GABA concentration, we get a prevalence of about 7 % for the activation-related closed time component. This value is lower than that in the wild-type receptor (wild-type: 27 ± 6 %; Li et al., 2007b).
In sum, kinetic analysis of the single-channel data from the α1Q241W mutant receptor shows both similarities to and differences from the wild-type data. In the presence of elevated GABA concentrations, the mutant receptor single-channel activity consists of high open probability clusters that contain three open and three closed time components. However, the properties of the open and closed states are dissimilar to those in the wild-type receptor. The prevalence and duration of the longest-lived open time component (OT3) are increased, and the prevalence of the activation-related closed time component (CTβ) is decreased compared to the wild-type receptor. These are the same kinetic parameters that are affected when the wild-type receptor is activated by GABA in the presence of neurosteroid 3α5αP (Akk et al., 2005).
The α1Q241W mutant receptor is not potentiated by 3α5αP
Previous work (Hosie et al., 2006), confirmed by us (Figure 2B), showed that 3α5αP is ineffective at potentiating macroscopic currents from the α1Q241W mutant receptor. To determine the effect of the steroid on single-channel currents, we compared single-channel currents elicited by 10 μM GABA in the absence and presence of 1 μM 3α5αP. This steroid concentration elicits maximal potentiation in the wild-type receptor. As predicted by macroscopic recordings, the presence of steroid did not affect the kinetic properties of single-channel activity. Sample currents are shown in Figures 4A-B, and the data are summarized in Tables 1-2.
Figure 4. The single-channel currents from the α1Q241W mutant receptor are not modulated by 3α5αP.

(A) Sample single-channel currents elicited by 10 μM GABA, and the open and closed time histograms. The lower trace gives a portion of the current trace (shown with a thick line underneath the data trace) at higher resolution. The histograms were fitted to sums of three exponentials. The open times were 0.42 ms (19 %), 3.9 ms (19 %) and 9.3 ms (45 %). The closed times were 0.14 ms (56 %), 1.6 ms (29 %) and 21.7 ms (15 %). (B) Sample single-channel currents elicited by 10 μM GABA + 1 μM 3α5αP, and the open and closed time histograms. The lower trace gives a portion of the current trace at higher resolution. The histograms were fitted to sums of three exponentials. The open times were 0.29 ms (28 %), 5.7 ms (49 %) and 11.9 ms (23 %). The closed times were 0.13 ms (70 %), 2.2 ms (19 %) and 27.0 ms (11 %). The open and closed time parameters apply to the specific patch. Averaged values from multiple patches are given in the text.
In the wild-type α1β2γ2L GABA-A receptor, 3α5αP potentiates the currents by increasing the duration and prevalence of OT3, and by decreasing the prevalence of CT3 (Akk et al., 2005). As a result, the mean open time is enhanced and the mean intracluster closed time is reduced in the presence of steroid. Our findings indicate that none of these changes occur when the glutamine residue in the α241 position is replaced with tryptophan. But the baseline values for the duration and prevalence of OT3, and the prevalence of CT3 in the α1Q241W receptor are similar to those observed with the wild-type receptor in the presence of steroid, suggesting that the amino acid substitution mimics the presence of steroid.
The α1Q241W mutation modifies channel activation by piperidine-4-sulfonic acid
Piperidine-4-sulfonic acid (P4S) is a high-affinity, low-efficacy agonist of the GABA-A receptor. In macroscopic recordings, the concentration producing half-maximal response from wild-type α1β2γ2 receptors is 16.5 ± 0.3 μM, i.e. similar to that for GABA, but the peak current elicited by saturating concentrations of P4S is only 63 ± 13 % of that for GABA (data not shown). Unlike GABA, P4S, even at saturating concentrations, elicits single-channel currents best-characterized as monotonous, low open probability episodes of activity (Figure 5A; also Steinbach and Akk, 2001). The channel open time histograms contain two components, with mean durations resembling those of OT1 and OT2 for receptors activated by GABA. The studies of closed times are typically inconclusive because the number of active receptors in the patch producing the single-channel activity is unknown.
Figure 5. The α1Q241W mutation affects channel activation by a low-efficacy agonist piperidine-4-sulfonic acid.

(A) Sample single-channel activity from the wild-type receptor elicited by 1 mM P4S. No single-channel clusters were evident. Occasional overlaps seen in the data segment indicate that two or more channels contributed to the single-channel activity shown. The open times, measured from portions of the record without overlaps, were 0.18 ms (32 %) and 1.6 ms (68 %). No long duration openings (OT3) were apparent in the record. The closed times were 0.27 ms (24 %), 8.8 ms (30 %) and 30.3 ms (46 %). (B) Exposure of wild-type receptors to 1 mM P4S + 1 μM 3α5αP results in increased open time durations and the appearance of grouped openings, i.e., single-channel clusters. The intracluster open times were 0.15 ms (22 %), 1.3 ms (40 %) and 13.3 ms (38 %). The intracluster closed times were 0.22 ms (47 %), 1.8 ms (33 %) and 34.5 ms (20 %). (C) Exposure of the α1Q241W mutant receptor to 1 mM P4S elicits currents qualitatively similar to those from the wild-type receptor activated by P4S + 3α5αP. The intracluster open times were 0.55 ms (30 %), 4.3 ms (62 %) and 12.7 ms (8 %). The intracluster closed times were 0.17 ms (53 %), 2.0 ms (21 %) and 13.7 ms (26 %). The open and closed time parameters apply to the specific patch. Averaged values from multiple patches are given in the text.
Coapplication of 3α5αP with P4S cardinally changes the mode of activity. Instead of isolated openings, channel activity in the presence of the steroid takes place in easily identifiable clusters. Sample recordings from the wild-type receptor activated by 1 mM P4S + 1 μM 3α5αP are shown in Figure 5B. The analysis of intracluster open time histograms revealed the emergence of the third, long-lived open state. Averaged from 5 patches, the mean open times were 0.21 ± 0.04 ms (33 %; OT1), 1.4 ± 0.5 ms (28 %; OT2) and 8.4 ± 3.1 ms (40 %; OT3).
The data described above suggested that in the presence of GABA, the α1Q241W mutation acts by mimicking the effects of steroid. We were interested in testing whether channel activation by P4S is similarly modified by the mutation. Accordingly, we next examined the activation of the α1Q241W mutant receptor by P4S. The major finding was that mutant receptors activated by P4S exhibited clear-cut clusters that contained three classes of open events. Sample single-channel activity is shown in Figure 5C. At 1 mM P4S, the intracluster open and closed time histograms contained three components. The mean open times (averaged from 4 patches) were 0.41 ± 0.21 ms (23 %; OT1), 3.1 ± 1.0 ms (66 %; OT2) and 8.3 ± 3.5 ms (11 %; OT3).
The presence of the long-lived (∼8 ms) OT3 component in the α1Q241W receptor activated by P4S is qualitatively similar to the emergence of OT3 when the wild-type receptor is activated by P4S + 3α5αP. We conclude that the α1Q241W mutation and the presence of steroid 3α5αP have qualitatively similar effects on GABA-A receptor activation by P4S.
The α1Q241W mutation does not affect GABA-A receptor inhibition by pregnenolone sulfate
Pregnenolone sulfate is an endogenous neurosteroid which inhibits GABA-A receptor activation (Majewska et al., 1988). In macroscopic recordings, the effect manifests as an increase in the apparent rate of desensitization (Shen et al., 2000). Previous studies have suggested that distinct, non-overlapping sites mediate the inhibitory effect of pregnenolone sulfate and the effects of potentiating neuroactive steroids (Park-Chung et al., 1999; Akk et al., 2001). Here, we sought to confirm this by probing the effect of the α1Q241W mutation on channel modulation by pregnenolone sulfate. We hypothesized that if the site mediating the effect of pregnenolone sulfate is distinct from the 3α5αP binding site, then the α1Q241W mutation is likely to be without effect on channel modulation by the inhibitory steroid.
Whole-cell recordings were conducted on cells expressing wild-type or α1Q241W mutant receptors. The receptors were activated by a saturating concentration of GABA (1 mM for wild-type, 250 μM for the mutant) in the absence and presence of 2-50 μM pregnenolone sulfate. In the wild-type receptor, the major effect of pregnenolone sulfate was a dose-dependent enhancement in the apparent rate of desensitization. In control recordings from cells exposed to 1 mM GABA, the desensitization time constant was 6.3 ± 2.8 s (n = 5 cells). When 50 μM pregnenolone sulfate was coapplied with GABA, the decay time constant was 656 ± 172 ms (n = 5 cells). In cells expressing α1Q241W mutant receptors, the mode of action of pregnenolone sulfate was analogous. The decay time constant was 4.1 ± 1.8 s (n = 4 cells) in the presence of GABA, and 413 ± 78 ms (n = 4 cells) in the presence of GABA + 50 μM pregnenolone sulfate. Additionally, there was a slight (<10 %) decrease in peak response in some cells exposed to pregnenolone sulfate. Sample current traces are shown in Figure 6A-B.
Figure 6. The α1Q241W mutation does not affect channel inhibition by pregnenolone sulfate.

(A) Sample macroscopic recordings from a cell expressing wild-type α1β2γ2L receptors. The receptors were activated by 1 mM GABA (a saturating concentration) in the absence and presence of 2, 10 or 50 μM pregnenolone sulfate (PS). The current decay phases were fitted by a single exponential to a constant level yielding 6794 ms (GABA), 4214 ms (GABA + 2 μM PS), 840 ms (GABA + 10 μM PS), and 464 ms (GABA + 50 μM PS). (B) Sample macroscopic recordings from a cell expressing α1Q241W mutant receptors. The receptors were activated by 250 μM GABA (a saturating concentration) in the absence and presence of 2, 10 or 50 μM PS. The current decay phases were fitted by a single exponential to a constant level yielding 3345 ms (GABA), 1268 ms (GABA + 2 μM PS), 864 ms (GABA + 10 μM PS), and 465 ms (GABA + 50 μM PS). (C) The relative area (total charge carried) of the macroscopic response as a function of PS concentration. The data points show mean ± SEM from 4-5 cells. The curves were fitted to: Y([steroid])=Y0 + (Ymax − Y0) [steroid]n/([steroid] + EC50)n. The best-fit parameters for the wild-type receptor were: Y0=100 % (constrained), Ymax = 15 ± 2 %, EC50 = 7.4 ± 0.4 μM, nH = 0.8 ± 0.1. The best-fit parameters for the α1Q241W mutant receptor were: Y0 = 100 % (constrained), Ymax = −12 ± 8 %, EC50 = 7.7 ± 2.2 μM, nH = 0.6 ± 0.1.
The rate of development of block in the presence of 50 μM pregnenolone sulfate can be estimated from the relationship k+PS = {1/τdecay(PS) − 1/τdes}/50 μM, where τdes is the macroscopic current desensitization time constant in the presence of GABA, and τdecay(PS) is the decay time constant in the presence of GABA + pregnenolone sulfate. We estimate that the k+PS was 0.03 ± 0.01 μM−1s−1 in the wild-type receptor, and 0.04 ± 0.01 μM−1s−1 in the α1Q241W mutant receptor.
We examined the concentration-dependence of the effect of pregnenolone sulfate by comparing the area of the response (total charge transfer) during a 4 s application of GABA and 2-50 μM steroid (Figure 6C). The data show that pregnenolone sulfate had a half-maximal effect at 7.4 ± 0.4 μM and 7.7 ± 2.2 μM, on wild-type and mutant receptors, respectively. In sum, we infer that the α1Q241W mutation is without effect on channel modulation by the inhibitory steroid pregnenolone sulfate indicating that distinct sites underlie channel modulation by potentiating and inhibitory steroids.
Properties of activation, and lack of modulation by 3α5αP of the α1Q241L mutant receptor
We next examined the activation properties of the α1Q241L mutant receptor. This mutation also blocks potentiation of macroscopic currents by 3α5αP (Hosie et al., 2006; Figure 2B), and we were curious to see whether the leucine substitution similarly to the more bulky tryptophan substitution mimics the presence of the steroid.
At GABA concentrations of 50 -1000 μM, clear-cut clusters were observed (Figure 7A-B). The intracluster open time distributions contained two components. In the presence of 1 mM GABA (a saturating concentration), the open times were 0.79 ± 0.03 ms (47 %) and 1.9 ± 0.3 ms (53 %) (averaged from 3 patches). The open time distributions were unchanged when the GABA concentration was reduced to 50 μM (Table 1). The intracluster closed time histograms were best-fitted to three exponentials. When the receptors were exposed to 1 mM GABA, the closed time components had mean durations and prevalence of 0.23 ± 0.07 (57 %), 1.0 ± 0.2 ms (35 %) and 13.3 ± 5.1 ms (8 %). In the presence of 50 μM GABA, the prevalence of the CT3 component was 29 % (Table 2). By applying the reasoning used above in the analysis of single-channel activity from the α1Q241W mutant receptor, we estimate that the prevalence of CTβ is about 21 % in the α1Q241L receptor. This value is comparable to the prevalence of CTβ in the wild-type receptor (27 ± 6 %; Li et al., 2007b), and we conclude that the leucine substitution, in contrast to the tryptophan substitution, does not decrease the fraction of CTβ. The intracluster open time histograms from the α1Q241L mutant lacked the long-lived OT3 component, and it is thus not possible to directly compare the open time data from the wild-type and α1Q241L receptors. Nonetheless, the α1Q241L mutation, unlike the α1Q241W, did not result in a frequent, long-lived open time component.
Figure 7. 3α5αP does not modulate single-channel activity from the α1Q241L mutant receptor.

(A) A sample single-channel cluster elicited by 50 μM GABA, and the open and closed time histograms. The open times were 0.47 ms (26 %) and 2.1 ms (74 %). The closed times were 0.20 ms (58 %), 2.9 ms (15 %) and 13.0 ms (27 %). (B) A sample single-channel cluster elicited by 1000 μM GABA, and the open and closed time histograms. The open times were 0.82 ms (38 %) and 2.0 ms (62 %). The closed times were 0.19 ms (59 %), 0.9 ms (32 %) and 18.9 ms (9 %). (C) A sample single-channel cluster elicited by 50 μM GABA in the presence of 1 μM 3α5αP, and the open and closed time histograms. The open times were 0.60 ms (22 %) and 2.2 ms (78 %). The closed times were 0.18 ms (58 %), 1.4 ms (16 %) and 19.6 ms (26 %). The open and closed time parameters apply to the specific patch. Averaged values from multiple patches are given in the text.
In the next set of experiments, we tested the effect of 1 μM 3α5αP on channel activation elicited by 50 μM GABA. Sample currents are shown in Figure 7C, and the data are summarized in Tables 1-2. The data indicate that the presence of steroid is without effect on the intracluster kinetic parameters. These findings are consistent with a model where the leucine substitution disrupts the interaction of 3α5αP with the receptor.
The α1Q241S mutation modifies channel potentiation by 3α5αP
In macroscopic recordings, the α1Q241S mutation shifts the steroid potentiation curve to higher steroid concentrations but has no effect on maximal potentiation (Hosie et al., 2006; Figure 2B). The previous results (see above) had indicated that mutations to α1Q241 produced changes in potentiation by neurosteroids and in the kinetic properties of single-channel currents elicited by GABA alone. Accordingly it was of interest to examine the consequences of a mutation of this residue which had relatively small effects on potentiation.
Single-channel clusters were recorded at 50 and 1000 μM GABA (Figure 8A-B). At 1 mM GABA, the open time distributions of currents from the α1Q241S receptor contained two components with mean durations and fractions of 0.76 ± 0.23 ms (75 %) and 2.0 ± 1.0 ms (25 %) (n = 3 patches). Lowering the GABA concentration to 50 μM did not alter the open time distributions (Table 1).
Figure 8. Modulation of single-channel currents from the α1Q241S mutant receptor by 3α5αP.

(A) A sample single-channel cluster elicited by 50 μM GABA, and the open and closed time histograms. The open times were 0.92 ms (63 %) and 2.1 ms (37 %). The closed times were 0.20 ms (72 %), 1.3 ms (9 %) and 19.2 ms (20 %). (B) A sample single-channel cluster elicited by 1000 μM GABA, and the open and closed time histograms. The open times were 0.72 ms (79 %) and 1.7 ms (21 %). The closed times were 0.21 ms (84 %), 1.0 ms (12 %) and 7.0 ms (3 %). (C) A sample single-channel cluster elicited by 50 μM GABA in the presence of 1 μM 3α5αP, and the open and closed time histograms. The open times were 0.61 ms (46 %) and 6.4 ms (54 %). The closed times were 0.18 ms (58 %), 1.9 ms (27 %) and 10.9 ms (15 %). The open and closed time parameters apply to the specific patch. Averaged values from multiple patches are given in the text.
We next tested the effect of 1 μM 3α5αP on channel activation elicited by 50 μM GABA. Sample currents are shown in Figure 8C, and the summary of the effects of the presence of steroid is given in Tables 1-2. The data indicate that 3α5αP has a complex effect on the intracluster open and closed time distributions. The mean duration of the longer-lived open time component was increased in the presence of 3α5αP. This effect is in qualitative agreement with the findings obtained from the wild-type receptor. However, we note that, in contrast to its effect on wild-type receptors, the application of 3α5αP did not lead to a statistically significant increase in the prevalence of long-lived openings. The presence of 3α5αP also did not affect the prevalence of CT3 in the α1Q241S mutant receptor. These findings indicate that the mutation modifies the mode of action of neurosteroid 3α5αP on the GABA-A receptor.
The α1S240L but not the α1Q241L mutation disrupts channel potentiation by a 3α-hydroxymethyl steroid
Lack of potentiation by 3α5αP in the α1Q241L mutant has been proposed to stem from the inability of the leucine residue to act as a H-bond acceptor to the C3-OH group of the steroid (Hosie et al., 2006). Previous studies have shown that steroid analogues with substitutions other than the hydroxyl group in the C3 position can be efficacious potentiators of the GABA-A receptor. For example, pregnane steroids with a carboxylic acid, or its amide derivative, at C3 are positive modulators of GABA-A receptor activity (Mennerick et al., 2001). The greater distance between the backbone of the steroid's A ring and the H-bonding group on C3 in such steroids as well as the rotational freedom of the C3 substituents suggests that the steroid may be interacting with a locus other than the one utilized by 3α5αP, and so we hypothesized that such steroids may remain capable of potentiating receptors containing the α1Q241L mutation.
To test this hypothesis, we compared wild-type and α1Q241L mutant channel potentiation by the steroid 3αCH2OH5βP (Figure 9A) that has a hydroxymethyl group at C3. Sample currents and steroid dose-response curves are shown in Figure 9B-D. The data demonstrate that the α1Q241L mutation has minimal effect on channel potentiation by 3αCH2OH5βP. The steroid, at 10 μM, potentiated macroscopic responses elicited by an EC20-25 concentration of GABA from the wild-type and mutant receptors by 2.7 ± 0.4-fold (n = 5 cells) and 2.2 ± 0.2-fold (n = 4 cells), respectively.
Figure 9. The α1Q241L mutation does not affect potentiation by the steroid analogue 3αCH2OH5βP.

(A) Structure of the steroid analogue 3αHOCH25βP. (B)Potentiation dose-response curves for wild-type and α1Q241L mutant receptors. The data points show mean ± SEM from 4-5 cells. Due to absence of saturation no curve fitting was attempted. (C) Sample macroscopic recordings from a cell expressing wild-type α1β2γ2L receptors. The receptors were activated by 5 μM GABA in the absence and presence of 10 μM 3αCH2OH5βP. The peak responses in this cell were 581 pA (GABA), and 1346 pA (GABA + steroid). (D) Sample macroscopic recordings from a cell expressing α1Q241L mutant receptors. The receptors were activated by 20 μM GABA in the absence and presence of 10 μM 3αCH2OH5βP. The peak responses in this cell were 559 pA (GABA), and 1480 pA (GABA + steroid).
We next examined the effects of mutations to the residues in the vicinity of α1Q241 on channel potentiation by 3α5αP and 3αCH2OH5βP. First, we tested the effect of substituting leucine for serine in position 240.
The GABA dose-response curve from the α1S240L mutant receptor was shifted to higher agonist concentrations, having an EC50 of 38 ± 4 μM (Figure 10A). The steroid effects were examined in the presence of GABA concentrations producing peak responses equal to ∼10-20 % of maximal current. We found that coapplication of 3αCH2OH5βP with GABA did not lead to potentiation of peak response in the α1S240L receptor (Figure 10B-D). The peak response was 109 ± 39 % of control (n = 8 cells) when 10 μM 3αCH2OH5βP was coapplied with 10 or 25 μM GABA.
Figure 10. Potentiation by 3αCH2OH5βP is abolished in the α1S240L and α1W245L mutant receptors.

(A) Wild-type and mutant receptors were activated by GABA, and the mean fractional peak responses (± SEM) from 5-7 cells are plotted as a function of GABA concentration. Curve fitting to the Hill equation yields the following parameters. α1S240L: EC50 = 38.1 ± 4.3 μM, nH = 1.3 ± 0.2; α1W245L: EC50 = 23.8 ± 1.6 μM, nH = 1.0 ± 0.1. The wild-type receptor data (dashed line) are replotted from Figure 1A. (B) Comparison of potentiation of wild-type, α1S240L, α1S241L, and α1W245L mutant receptors by 3α5αP or 3αCH2OH5βP. The data (mean ± SEM) show the levels of potentiation by 3 μM 3α5αP (1 μM for wild-type and α1Q241L) or 10 μM 3αCH2OH5βP of currents elicited by an EC15-25 concentration of GABA. Statistical tests (Student's t-test) were carried out with respect to control (GABA alone), and to steroid-mediated potentiation of the wild-type receptor. *, P<0.05; **, P<0.01; ***, P<0.001; ns, not significant; −, not applicable. (C) Sample macroscopic recordings from a cell expressing α1S240L mutant receptors. The receptors were activated by 10 μM GABA in the absence and presence of 10 μM 3αCH2OH5βP or 3 μM 3α5αP. The peak responses in this cell were 55 pA (10 μM GABA), 57 pA (10 μM GABA + 10 μM 3αCH2OH5βP), and 210 pA (10 μM GABA + 3 μM 3α5αP). (D) Sample macroscopic recordings from a cell expressing α1W245L mutant receptors. The receptors were activated by 5 μM GABA in the absence and presence of 10 μM 3αCH2OH5βP or 3 μM 3α5αP. The peak responses in this cell were 352 pA (10 μM GABA), 311 pA (5 μM GABA + 10 μM 3αCH2OH5βP), and 412 pA (5 μM GABA + 3 μM 3α5αP).
In contrast, the mutation was essentially without effect on potentiation by 3α5αP. Application of 3 μM 3α5αP potentiated currents from the α1S240L mutant to 284 ± 103 % of control (n = 7 cells) indicating that the α1S240 residue is critical in channel modulation by 3αCH2OH5βP but not 3α5αP.
Effects of mutations to α1W245 and α1S243 residues on channel potentiation by 3αCH2OH5βP and 3α5αP
We next examined the effects of leucine mutations to residues α1W245 and α1S240 on channel activation by GABA, and modulation by 3αCH2OH5βP and 3α5αP. In the α1W245L mutant receptor, the EC50 for GABA-elicited currents was 24 ± 2 μM (Figure 10A). The effects of steroids were studied in the presence of 5 or 10 μM GABA. The data show that the steroids were ineffective at producing channel modulation (Figure 10D). Coapplication of 10 μM 3αCH2OH5βP with GABA resulted in peak current of 101 ± 15 % of control (n = 9 cells). When 3 μM 3α5αP was coapplied with GABA, the peak response was 108 ± 32 % of control (n = 11 cells).
To test the possibility that the α1W245L mutation has a more global effect on receptor function preventing channel potentiation per se, we recorded whole-cell responses in the presence of pentobarbital. This GABA-A receptor modulator potentiates current responses at micromolar concentrations. The kinetic mechanism of action of pentobarbital resembles that of neuroactive steroids (e.g., Steinbach and Akk, 2001; Akk et al., 2004) which might imply commonality of the transduction elements involved in channel modulation by the two drugs, but the effects are considered to be mediated by drug interactions with distinct sites (Akk et al., 2004; Hosie et al., 2006). We reasoned that if the α1W245L mutation allows potentiation by pentobarbital, then this serves as indication of lack of global changes in receptor function. In five cells expressing the α1W245L mutant receptor, coapplication of 100 μM pentobarbital with 5 μM GABA (EC10) increased the peak response to 906 ± 254 % of control. We interpret the findings to indicate the selective involvement of the α1W245 residue in steroid actions.
As a negative control, we examined the effect of the α1S243L mutation on channel modulation by potentiating steroids. The α-helical configuration of the M1 membrane-spanning domain places the sidechain of this residue essentially in the opposite (compared to the α1Q241 residue) surface of the domain, and we hypothesized that mutations to this residue would have minimal effect on channel modulation by either 3α5αP or 3αCH2OH5βP. The α1S243L mutation shifted the GABA dose-response curve by almost ten-fold to higher concentrations (EC50 = 80 ± 3 μM). Coapplication of 3 μM 3α5αP or 10 μM 3αCH2OH5βP with 30 μM GABA enhanced the peak response to 374 ± 41 % (n = 6 cells) or 183 ± 11 % (n = 8 cells) of control, respectively. We conclude that the α1S243L mutation has minimal effect on channel potentiation by 3α5αP and 3αCH2OH5βP.
Mutations to the M1 domain residues affect channel modulation by 3α5βP
We next examined the effects of the mutations to the M1 membrane-spanning domain on channel modulation by (3α,5β)-3-hydroxypregnan-20-one (3α5βP). Single-channel experiments have shown that 3α5αP and 3α5βP are kinetically similar as modulators of the GABA-A receptor currents (Akk et al., 2005; Li et al., 2007a). Previous work employing mutations to the α1Q241 site has indicated that 3α5αP and 3α5βP may interact with the same site to potentiate the GABA-A receptor (Hosie et al., 2006). In contrast, Mennerick et al. (2004) showed that a steroid analogue (3α,5α)-17-phenylandrost-16-en-3-ol (17-PA) selectively antagonized channel potentiation and direct activation by 3α5αP but not 3α5βP, suggesting that different sites mediate the effects of these steroids. Here, we have evaluated the effects of mutations α1S240L, α1Q241L, α1S243L and α1W245L on channel potentiation by 3α5βP in order to compare the differential effect that these mutations have on channel potentiation by 5α- and 5β-reduced steroids.
The summary of results is given in Figure 11. Coapplication of 3 μM 3α5βP with GABA resulted in potentiation of peak current in α1β2γ2L wild-type receptors and α1S243L mutant receptors, but not when the receptor contained the α1S240L, α1Q241L, or α1W245L mutation. Potentiation of receptors containing the α1S243L mutation is in agreement with the data suggesting that the orientation of the α1S243 residue is such that substitutions here have little effect on modulation by steroids (see above). The lack of potentiation in the α1W245L mutant is similar to the effect of this mutation on modulation by 3α5αP, confirming our earlier conclusion that this residue is critical to channel potentiation by neuroactive steroids. The lack of potentiation by 3α5βP in the α1Q241L mutant is also similar to the effect of the mutation on modulation by 3α5αP. But the α1S240L mutation had different effects on potentiation by 3α5αP and 3α5βP. The mutation fully abolishes potentiation by 3α5βP (107 ± 19 % of control, n = 5 cells) but is without effect on potentiation by 3α5αP (Figure 10B). Thus, mutations to the α1 subunit M1 domain differentially affect channel modulation by 3α5αP and 3α5βP suggesting that the two steroids are oriented differently in the binding pocket.
Figure 11. Effects of mutations to the M1 domain on potentiation by 3α5αP.

Comparison of potentiation of wild-type, and α1S240L, α1Q241L, α1S243L and α1W245L mutant receptors by 3α5βP. The data (mean ± SEM) from 3-6 cells show the levels of potentiation by 3 μM 3α5βP of currents elicited by an EC15-25 concentration of GABA. Statistical tests (Student's t-test) were carried out with respect to control (GABA alone), and to 3α5βP-mediated potentiation of the wild-type receptor. *, P<0.05; **, P<0.01; ***, P<0.001; ns, not significant; −, not applicable.
Potentiation of the GABA-A receptor by 5α-pregnan-20-one
The data presented above on the effects of the mutations to the α1S240 and α1Q241 residues on channel potentiation by 3α5αP and 3αCH2OH5βP could be interpreted as indicating that the hydroxyl group of 3α5αP interacts, possibly via H-bonding, with the α1Q241 residue, and the hydroxymethyl group of 3αCH2OH5βP interacts with the α1S240 residue. On the other hand, the finding that potentiation by 3α5βP is sensitive to the nature of the residue in both the α240 and α241 positions, and the single-channel data on the α1Q241S mutation that retains, but modifies, the mechanism of potentiation by 3α5αP, are not fully compatible with this simple model.
A possible explanation is that the α1Q241 residue is not directly interacting with the C3-OH group of the steroid molecule but is rather a necessary component to appropriately shape the binding surface to accommodate the steroids 3α5αP and 3α5βP. To explore this hypothesis, we examined receptor modulation by 5α-pregnan-20-one (3deoxy5αP). This steroid (Figure 12A) is devoid of H-bonding groups on C3 and is therefore predicted to not interact, at least via H-bonding, with the residues in the α1 subunit M1 domain. The interaction of 3deoxy5αP with the wild-type receptor could be considered functionally analogous to 3α5αP interaction with the α1Q241L mutant receptor.
Figure 12. Steroid 3deoxy5αP potentiates the wild-type but not the α1Q241L mutant receptor.

(A) Structure of the steroid analogue 3deoxy5αP. (B) Comparison of potentiation of the wild-type receptor and the α1Q241L mutant receptor by 1 μM 3deoxy5αP. The data show mean ± SEM from 5-7 cells. (C) Sample macroscopic recordings from a cell expressing wild-type receptors. The receptors were activated by 5 μM GABA in the absence and presence of 1 μM 3deoxy5αP. The peak responses in this cell were 114 pA (GABA), and 281 pA (GABA + 3deoxy5αP). (D) Sample macroscopic recordings from a cell expressing α1Q241L mutant receptors. The receptors were activated by 20 μM GABA in the absence and presence of 1 μM 3deoxy5αP. The peak responses in this cell were 317 pA (GABA), and 305 pA (GABA + 3deoxy5αP).
We examined macroscopic currents from the wild-type receptor exposed to 5 μM GABA in the absence and presence of 0.2-5 μM 3deoxy5αP. Channel potentiation was observed at steroid concentrations 0.5 μM and higher, reaching maximal potentiation at 1 μM 3deoxy5αP. Exposure to 1 μM 3deoxy5αP increased the peak response to 202 ± 16 % of control (n = 5 cells; Figure 12B-C). We suspect that poor solubility of the steroid in aqueous solutions prevented stronger potentiation when nominally higher steroid concentrations were used. In any case, channel potentiation by a steroid that is devoid of groups on C3 that can form a H-bond indicates that a H-bond between the A ring of the steroid and the receptor is not required to produce channel potentiation.
We also examined the effect of the α1Q241L mutation on channel potentiation by 3deoxy5αP. The summary of the data and sample currents are shown in Figure 12B-D. The data demonstrate that the steroid is unable to potentiate currents from the α1Q241L mutant receptor. Coapplication of 1 μM 3deoxy5αP with 20 μM GABA (∼EC15) led to peak response of 97 ± 1 % of control (n = 7 cells). This finding is not consistent with a model where the lack of steroid potentiation in a receptor containing the α1Q241L mutation is due to the inability of the leucine residue to form a H-bond with the C3-OH group.
DISCUSSION
We have examined the effects of mutations to residues in the GABA-A receptor α1 subunit M1 membrane-spanning region on channel modulation by neuroactive steroids. A previous report (Hosie et al., 2006) had implicated the α1Q241 residue in the actions of neurosteroids by showing that when the glutamine residue was replaced with a tryptophan or leucine residue, potentiation by 3α5αP and 3α5βP was greatly diminished. It was concluded that the α1Q241 interacts with the steroid 3α-hydroxyl group (Hosie et al., 2006). The goal of the present work was to provide a more detailed kinetic and pharmacological characterization of the effects of mutations on channel activation, and modulation by neurosteroids. Overall, our data are consistent with the α1Q241 residue acting as a critical site for channel modulation by 3α5αP. However, our data are most consistent with a model where the α1Q241 residue forms a crucial intraprotein contact rather than participates in direct interaction with the C3-OH group. We have identified an additional residue in the α1 subunit M1 domain (α1S240) that is required for modulation by the steroid analogue 3αCH2OH5βP but not 3α5αP, and a residue (α1W245) that may participate as a transduction element in channel potentiation by steroids but not by pentobarbital.
The α1Q241W and α1Q241L mutations affected the kinetic properties of channels activated by GABA. The tryptophan substitution enhanced channel opening efficacy while the leucine substitution had a slightly deleterious effect on channel function. Our data confirm that the α1Q241W and α1Q241L mutations drastically diminish receptor potentiation by 3α5αP, but we show that different mechanisms underlie the absence of potentiation in the mutant receptors. The α1Q241W mutation functionally mimics the presence of steroid, and receptors activated by GABA (or P4S) demonstrate single-channel kinetic properties found in wild-type receptors exposed to GABA (or P4S) in the presence of high concentrations of potentiating steroids. The coapplication of 3α5αP with GABA had no further effect on mutant receptor activation.
The major effect of the α1Q241L mutation on channel activity was the loss of the longest-lived open time component. The application of 3α5αP did not modify single-channel or whole-cell currents elicited by GABA, consistent with the previous hypothesis (Hosie et al., 2006) that the leucine substitution interferes with the ability of 3α5αP to interact with the steroid binding site.
As with all mutational studies, the possibility exists that the amino acid substitutions have a deleterious structural effect. Figure 13 shows a DOPE plot (Shen and Sali, 2006) of the M1 region, illustrating that there are no major energetic changes caused by the α1Q241W or α1Q241L mutations. DOPE provides a statistical potential for assessing the deviation of a particular structure from an idealized reference state, and the plots shown in Figure 13 show the results of a sliding window of 13 residues through the models. Examining the M1 region, we see that the mutations studied produced small changes in this parameter, supporting the idea that no major structural consequences resulted from the mutations.
Figure 13.

The DOPE plots of the M1 domains of the wild-type (solid line), α1Q241W (dashed line), and α1Q241L mutant receptor (dotted) over a 13 residue evaluation window. Negative scores show a favorable trend to the idealized reference state, while positive scores indicate unfavorable deviations from the reference state. There were only minimal changes in the scores arising from the mutations.
We also examined the effect of the α1Q241S mutation on channel activation and 3α5αP-induced potentiation. Hosie and coworkers (Hosie et al., 2006) had found that the α1Q241S mutation had little effect on steroid potentiation, and concluded that the serine side-chain was able to replace glutamine as a H-bond acceptor and so retain potentiation. We confirm that 3α5αP elicits potentiation of macroscopic currents (Figure 2B), but find that the single-channel potentiation profile is distinct in the mutant and wild-type receptors. In the wild-type receptor, the presence of 3α5αP results in increases in the duration and prevalence of the longest open time component and a decrease in the prevalence of the activation-related closed time component. In the α1Q241S mutant receptor, 3α5αP has a strong effect on the mean duration of the longest-lived open time component but is largely without effect on the prevalence of long openings. The presence of steroid has a complex effect on the intracluster closed time distributions. However, the signature feature of steroid modulation in the wild-type receptor - a decrease in the prevalence of CT3 - is absent in the mutant receptor.
Additionally, we have shown that the α1Q241W mutation does not interfere with the actions of the inhibitory neurosteroid pregnenolone sulfate. This is in agreement with previous studies that have concluded that the sites of action for potentiating and inhibitory neuroactive steroids are distinct (Park-Chung et al., 1999; Akk et al., 2001).
Interestingly, the α1Q241L mutation did not affect channel potentiation by the steroid analogue 3αCH2OH5βP. Site-directed mutagenesis of residues in the vicinity of α1Q241 led to the identification of two additional residues adjacent to (or within) the steroid binding pocket: α1S240 and α1W245. Mutation of the α1S240 residue to leucine disrupted the potentiating effect of 3αCH2OH5βP but not 3α5αP. The α1W245L mutation abolished potentiation by both steroids, but left intact channel potentiation by pentobarbital and the marine cembranoid eupalmerin acetate (data not shown). In addition, the α1W245L mutation did not affect channel modulation by the inhibitory steroid pregnenolone sulfate (data not shown). We propose that the α1W245 residue participates as a transduction element in the actions of potentiating neuroactive steroids.
In single-channel recordings, some neuroactive steroids have three kinetically distinct effects on channel open and closed time distributions (Akk et al., 2004; Li et al., 2007a). Many of our previous findings could be interpreted as the effects being produced by steroid interactions with multiple non-overlapping sites. The dose-response relationships for the effects on open and closed times can be different for a given steroid. For example, (3α,5β,17β)-3-hydroxy-18-norandrostance-17-carbonitrile elicits an increase in the prevalence of OT3 with an EC50 of <100 nM while the EC50 for the increase of the duration of OT3 is >10 μM (Akk et al., 2004). We have also found that the steroid etiocholanolone, that has a single kinetic effect (to increase the prevalence of OT3), does not compete with a steroid having three kinetic effects (3α5βP) suggesting that etiocholanolone is unable to bind to the sites that mediate the increase in the duration of OT3 and the decrease in the prevalence of CTβ (Li et al., 2007a). Additional evidence for multiple binding sites for steroids comes from the finding that potentiation by 5α- but not 5β-reduced steroids is inhibited by a steroid analogue 17-PA (Mennerick et al., 2004), and that the dose-response curves for steroid enhancement of muscimol-elicited 36Cl− uptake are biphasic (Morrow et al., 1990).
In contrast, the finding that the α1Q241L mutation abolishes all kinetically distinguishable effects that 3α5αP exerts in the wild-type receptor suggests that the steroid interacts with a single site on the receptor to produce potentiation. The presence of all three types of effects in the receptor containing the α1Q241W mutation, that we believe mimics the presence of steroid, is similarly in favor of a single binding site for steroid.
We hypothesize that the steroid binding pocket presents a hydrophobic surface capable of accommodating steroid molecules of different structure. Different steroids, through interactions with non-overlapping loci, elicit a particular combination of kinetic effects that lead to channel potentiation. The lack of competition between etiocholanolone and 3α5βP could be explained if the α1Q241 residue was not a docking site for the steroid per se, but rather one of the necessary components to maintain the structure of the surface to which steroids bind. Mutations to the M1 domain differently affect the structure of the binding surface so that 3αCH2OH5βP remains capable of interacting with its binding site following the α1Q241L but not the α1S240L mutation while 3α5βP requires both α1S240 and α1Q241 intact in order to modulate receptor activity. In this model, differential sensitivity of 3α5αP and 3α5βP to mutations in the M1 domain suggests that the two steroids bind to different loci, accounting for their different sensitivity to the steroid antagonist 17-PA.
Support for this hypothesis comes from the finding that the α1Q241S mutation alters the kinetic mode of action of 3α5αP. The data showing that the α1Q241L mutation disrupts potentiation by 3deoxy5αP is further indication that the glutamine-to-leucine mutation does not act by simply preventing H-bonding between the A ring of the steroid and the receptor. Instead, the mutation may alter the structure of the protein so that the binding site can no longer accommodate 3deoxy5αP. By extension, the data suggest that the α1Q241 residue does not constitute the docking site for neurosteroids.
In sum, the present study confirms the crucial role of the amino acid residues in the α1 subunit M1 domain in GABA-A receptor modulation by potentiating neurosteroids. The data demonstrate a critical role of the α1Q241 residue in channel potentiation by 3α-hydroxysteroids and 3deoxy5αP. We identified two additional residues in the α1 subunit M1 membrane-spanning domain, α1S240 and α1W245, whose mutation interferes with the ability of potentiating neuroactive steroids to modulate the GABA-A receptor. Our data are most consistent with a model where mutations to residues in the M1 membrane-spanning domain shape the binding surface on the GABA-A receptor to which multiple steroid molecules can bind.
ACKNOWLEDGMENTS
We thank Drs. C.F. Zorumski and S. Mennerick for comments on the manuscript. JHS is the Russell and Mary Shelden Professor of Anesthesiology.
This work was supported by National Institutes of Health grants AA14707 (to GA), and GM47969 (to DFC and JHS).
Abbreviations
- 3α5αP
(3α,5α)-3-hydroxypregnan-20-one
- 3α5βP
(3α,5β)-3-hydroxypregnan-20-one
- P4S
piperidine-4-sulfonic acid
- 3αCH2OH5βP
(3α,5β)-3-hydroxymethylpregnan-20-one
- 17-PA
(3α,5α)-17-phenylandrost-16-en-3-ol
- 3deoxy5αP
5α-pregnan-20-one
REFERENCES
- Akk G, Bracamontes J, Steinbach JH. Pregnenolone sulfate block of GABAA receptors: mechanism and involvement of a residue in the M2 region of the α subunit. J Physiol. 2001;532:673–684. doi: 10.1111/j.1469-7793.2001.0673e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Bracamontes JR, Covey DF, Evers A, Dao T, Steinbach JH. Neuroactive steroids have multiple actions to potentiate GABAA receptors. J Physiol. 2004;558:59–74. doi: 10.1113/jphysiol.2004.066571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF, Mennerick S. Neurosteroid access to the GABAA receptor. J Neurosci. 2005;25:11605–11613. doi: 10.1523/JNEUROSCI.4173-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Li T, Benkwitz C, Czajkowski C, Pearce RA. Effects of the γ2S subunit incorporation on GABAA receptor macroscopic kinetics. Neuropharmacol. 2003;44:1003–1012. doi: 10.1016/s0028-3908(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HMA, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Li P, Covey DF, Steinbach JH, Akk G. Dual potentiating and inhibitory actions of a benz[e]indene neurosteroid analog on recombinant α1β2γ2 GABAA receptors. Mol Pharmacol. 2006;69:2015–2026. doi: 10.1124/mol.106.022590. [DOI] [PubMed] [Google Scholar]
- Li P, Bracamontes J, Katona BW, Covey DF, Steinbach JH, Akk G. Natural and enantiomeric etiocholanolone interact with distinct sites on the rat α1β2γ2L GABAA receptor. Mol Pharmacol. 2007a;71:1582–1590. doi: 10.1124/mol.106.033407. [DOI] [PubMed] [Google Scholar]
- Li P, Shu HJ, Wang C, Mennerick S, Zorumski CF, Covey DF, Steinbach JH, Akk G. Neurosteroid migration to intracellular compartments reduces steroid concentration in the membrane and diminishes GABA-A receptor potentiation. J Physiol. 2007b;584:789–800. doi: 10.1113/jphysiol.2007.142794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Reichert DE, Rodriguez AD, Manion BD, Evers AS, Eterovic VA, Steinbach JH, Akk G. Mechanisms of potentiation of the mammalian GABAA receptor by the marine cembranoid eupalmerin acetate. Br J Pharmacol. 2008;153:598–608. doi: 10.1038/sj.bjp.0707597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HL, Shu YC, Wu YH. Molecular dynamics simulations to determine the optimal loop length in the helix-loop-helix motif. J Biomol Struct Dyn. 2003;20:741–745. doi: 10.1080/07391102.2003.10506890. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Mienville JM, Vicini S. Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neurosci Lett. 1988;90:279–284. doi: 10.1016/0304-3940(88)90202-9. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zeng CM, Benz A, Shen W, Izumi Y, Evers AS, Covey DF, Zorumski CF. Effects on γ-aminobutyric acid (GABA)A receptors of a neuroactive steroid that negatively modulates glutamate neurotransmission and augments GABA neurotransmission. Mol Pharmacol. 2001;60:732–741. [PubMed] [Google Scholar]
- Mennerick S, He Y, Jiang X, Manion BD, Wang M, Shute A, Benz A, Evers AS, Covey DF, Zorumski CF. Selective antagonism of 5α-reduced neurosteroid effects at GABAA receptors. Mol Pharmacol. 2004;65:1191–1197. doi: 10.1124/mol.65.5.1191. [DOI] [PubMed] [Google Scholar]
- Morrow AL, Pace JR, Purdy RH, Paul SM. Characterization of steroid interactions with γ-aminobutyric acid receptor-gated chloride ion channels: evidence for multiple steroid recognition sites. Mol Pharmacol. 1990;37:263–270. [PubMed] [Google Scholar]
- Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH. Sulfated and unsulfated steroids modulate γ-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/s0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Qin F, Auerbach A, Sachs F. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys J. 1996;70:264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Mennerick S, Covey DF, Zorumski CF. Pregnenolone sulfate modulates inhibitory synaptic transmission by enhancing GABAA receptor desensitization. J Neurosci. 2000;20:3571–3579. doi: 10.1523/JNEUROSCI.20-10-03571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JH, Akk G. Modulation of GABAA receptor gating by pentobarbital. J Physiol. 2001;537:715–733. doi: 10.1111/j.1469-7793.2001.00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Zorumski C, Bracamontes J, Steinbach JH. Endogenous subunits can cause ambiguities in the pharmacology of exogenous γ-aminobutyric acidA receptors expressed in human embryonic kidney 293 cells. Mol Pharmacol. 1996;50:931–938. [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]