

Abstract
The one-step reaction of salicylaldehydes with amines and alkenyl boronic acids or alkenyl trifluoroborates to form 2H-chromenes (2H-1-benzopyrans) has been investigated in more detail and new suitable conditions have been identified, including the use of tertiary amines and protic solvents including water. This process was applied to a concise synthesis of a tocopherol analog. The analogous condensation reaction between 2-sulfamidobenzaldehydes and alkenyl trifluoroborates provides an efficient synthesis of 1,2-dihydroquinoline derivatives.
Keywords: alkenyl boronic acid, alkenyl trifluoroborate, salicylaldehyde, chromene (benzopyran), tocopherol, dihydroquinoline, quinoline
1. Introduction
The 2H-chromene (2H-1-benzopyran) moiety is a common structural feature of numerous biologically active molecules [1, 2] and is widely occurring in the structures of many natural flavonoids and anthocyanins [3], as well as the members of the vitamin E family (tocopherols and tocotrienes) [4–10]. A variety of methods are known for the synthesis of this class of compounds [2, 4–14], typically starting with 2-hydroxyacetophenone derivatives or directly from phenols via cyclization methods, electrophilic aromatic substitution, or by relying on palladium-catalyzed processes.
The 1,2-dihydroquinoline moiety is another widely occurring structural fragment in a large number of molecules with a range of biological properties, and has also been synthesized with a variety of methods, including additions to activated quinoline derivatives [15–19], and various domino-type condensation processes [20–23].
Herein, we report our studies on a common methodology for the synthesis of 2H-chromenes as well as 1,2-dihydroquinoline derivatives by utilizing aryl aldehydes and organoboron compounds [24]. This approach is based on our one-step three-component reaction among an amine, an aldehyde and an organoboron compound, that we introduced some time ago and has evolved into a versatile and synthetically useful process [25, 26]. Many variations and applications of this chemistry have been developed, including the synthesis of amino acids [27], amino alcohols [28], and various heterocycles [25].
We have earlier reported that the one-step reaction among salicylaldehydes (1), amines (2), and alkenyl or aryl boronic acids (3), at room temperature, forms aminophenol derivatives (5) [29]. When alkenyl boronic acids are used, and the reaction is performed at higher temperature, it leads to the formation of 2H-chromenes (2H-1-benzopyrans) (6), a process shown by Finn to proceed efficiently under catalytic amounts of the amine component [30]. Subsequent reports by several groups have shown that this chemistry works well in ionic liquids [31, 32], under microwave conditions [33], and by employing potassium trifluoroborates [34].
The above approach to the synthesis of 2H-chromenes (6) was postulated to involve an intramolecular nucleophilic displacement of an ammonium leaving group, as shown in intermediate 7 (Scheme 1), to form directly the product [30]. However, it is also possible that an alternative fragmentation takes place, as shown in intermediate 8, to form initially intermediate 9, which can undergo electrocyclic ring closure to form product 6. Based on this hypothesis we investigated additional aspects of this process, which are reported herein. We also extended this chemistry to the synthesis of 1,2-dihydroquinolines, by starting with 2-sulfamido-benzaldehydes.
Scheme 1.

2. Results and Discussion
The overall unified strategy for the synthesis of both 2H-chromenes (18) and 1,2-dihydroquinolines (19) is shown in Scheme 2. Thus, reaction of salicylaldehydes (11) with alkenyl boronic acids (13) or alkenyl trifluoroborates (14) in the presence of an amine (15), leads to the initial formation of an amino phenol intermediate (16), which upon heating undergoes cyclization to form the 2H-chromene product (18). A similar sequence starting with the mesyl derivatives of 2-amino benzaldehydes (12) and alkenyl trifluoroborates (14) proceeds through intermediate (17) to form 1,2-dihydroquinolines (19).
Scheme 2.
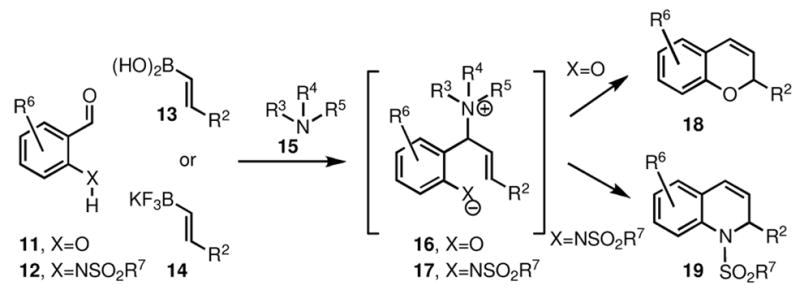
2.1 Synthesis of 2H-chromenes from salicylaldehydes
The one-step synthesis of 2H-chromenes, outlined in Scheme 1, has the advantage of being short and efficient, and it allows the direct incorporation of substituents onto the molecule. In order to further explore the scope of this process, we have carried out a more detailed investigation, which indicated that the outcome of this process depends heavily on the reaction conditions.
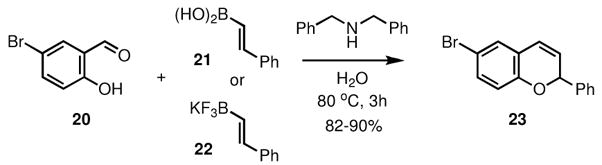
The secondary amines are generally the most reactive in this chemistry and several conditions have been reported for this process [29–31, 34]. We have also found that this transformation works particularly well in protic solvents, including ethanol and water. Among the most efficient is the use of dibenzylamine in water, and under these conditions salicylaldehyde 20 was converted to 2H-chromene 23 quite efficiently, both with alkenyl boronic acids (e.g. 21) as well as alkenyl trifluoroborates (e.g. 22), as shown in Scheme 3.
Scheme 3.
Surprisingly, however, we have also found out that tertiary amines can also mediate this process, but they are less effective (Scheme 4). Notably, highly sterically hindered secondary amines (e.g. 2,2,6,6-tetramethylpiperidine) proved less reactive than the less congested Hünig’s base, which produced the product even though is considered to be non-nucleophilic. On the other hand, tetrabutylammonium hydroxide, a relatively strong base, was found to be completely ineffective.
Scheme 4.

Taken together, these findings suggest a possible mechanism that includes a nucleophilic attack of the amine (15) to the aldehyde carbonyl, which can be aided by an intramolecular hydrogen bond with the phenolic hydroxyl group (20). The resulting intermediate (24) can then react with the boronic acid (21) to generate an ion pair (25) consisting of an electrophilic ammonium species and a nucleophilic borate species, analogously to the postulated mechanism of our three-component process [25]. Subsequent conjugate addition of the alkenyl group can lead to an ammonium phenolate intermediate (26), which subsequently can undergo fragmentation to form the 2H-chromene (23) directly, or via intermediate 27.
2.2. Synthesis of an alkyl analog of (±)-α-tocopherol
In order to illustrate the potential utility of this methodology for the synthesis of chromane derivatives related to Vitamin E, we have carried out a short synthesis of tocopherol analogs. The commonly available synthetic racemic tocopherols are produced on large industrial scale by Friedel-Crafts alkylation of trimethylhydroquinone with the tertiary allyl alcohol isophytol, or the primary allyl alcohol phytol in the presence of a Brønsted or Lewis acid, such as ZnCl2 [8]. The synthesis of individual stereoisomers of tocopherol and its analogs have also been reported. These convergent syntheses rely on palladium couplings or other reactions between the chromane derivative with a suitable side chain component [7, 9].
The use of our three-component process for tocopherol synthesis offers an alternative approach and allows the introduction of the side chain in one or two convenient steps (Scheme 5).
Scheme 5.

Thus, reaction of salicylaldehyde derivative 28 [5] with boronic acid 29 and dibenzylamine generated the chromene product 30, that was converted to the tocopherol analog 31, upon catalytic hydrogenation. The key cyclization step was relatively slow in this case (probably due to the bulkiness of the aldehyde 28) and it required prolonged heating and addition of equivalent amount of the amine, providing 57% yield of the intermediate 30. However, no side reactions were observed and the unreacted aldehyde was completely recovered. Addition of an acid catalyst (Amberlyst 15 resin) did not accelerate the reaction. At lower temperature (70 °C in ethanol), the yield dropped down to 20% (with the conversion remaining nearly quantitative), but at high temperatures (180 °C) substantial decomposition occurred. Subsequent hydrogenation of 30 under mild conditions resulted in simultaneous debenzylation and reduction of the double bond and afforded 31 in 80% yield. Unlike all-rac-α-tocopherol (32), which is a viscous air-sensitive oil and therefore is commonly used in the protected form (an acetate or other ester), compound 31 is an air-stable crystalline solid.
2.3. Synthesis of 1,2-dihydroquinolines from 2-sulfamidobenzaldehydes and alkenyl trifluoroborates
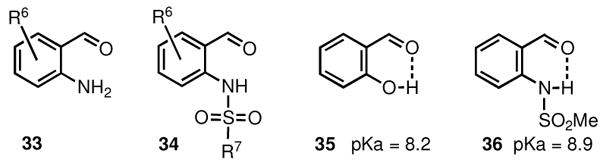
Although the three-component reaction among aryl aldehydes, amines, and organoboron compounds works very well with salicylaldehydes (1) and is quite useful for the synthesis of 2H-chromenes (Scheme 1), a similar approach to 1,2-dihydroquinolines by using 2-aminobenzaldehydes (33) [35] is not effective [30], while the limited stability of this type of aldehydes further complicates their use.
Unlike simple 2-aminobenzaldehydes (33), their N-protected analogues are quite stable at ambient temperature. Among the various possibilities, we chose to investigate the use of the readily available 2-sulfamidobenzaldehydes (34), whose sulfanilide proton (Ar-(RSO2)N-H is very similar in acidity to the phenolic proton of salicylaldehydes ArO-H. For example, the pKa of 2-(methanesulfamido)benzaldehyde (36) is calculated to be 8.9, while the the pKa of salicylaldehyde is 8.2 at 25 °C. In both cases, the acidity is further enhanced due to the intramolecular hydrogen bond (Scheme 6).
Scheme 6.
Indeed, following several exploratory studies, we have found suitable conditions for the one-step conversion of 2-sulfamidobenzaldehydes to 1,2-dihydroquinolines. As shown in Scheme 7, this process works best with the use of alkenyl trifluoroborates in the presence of 2 eq. of trimethylsilyl chloride and 2 eq. of triethylamine.
Scheme 7.

Following the pioneering work of Vedejs [36] on the synthesis of potassium organotrifluoroborates (RBF3K) from boronic acids (RB(OH)2), and their use in combination with trimethylsilyl chloride to generate in situ organodifluoro boranes (RBF2), the use of organo-trifluoroborates in synthesis have attracted considerable attention and new applications, recently reviewed by Genet [37,38] and Molander [39]. We have also been investigating the use of these versatile organoboron compounds in our three-component reactions, as well as other processes. In fact, the conversion of styryl fluoroborate (22) to fluorinated alcohols and amides using electrophilic fluorinating agents, was one of the earliest synthetic applications of this class of compounds [40].
Herein, we report the use of alkenyl trifluoroborates for the synthesis of 1,2-dehydroquinolines (Scheme 7). For example, under the optimal conditions, reaction of 2-(methanesulfamido)benzaldehyde (36) with styryl fluoroborate (22) gave the expected product (37) in 59% yield.
Upon varying the reaction conditions, it was found that running the reaction in anhydrous toluene and increasing the amount of TMSCl to 2 eq significantly increased the yield of the product. Use of only 1 eq of Et3N led to the formation of mixtures, while the increase in concentration of the base did not improve the reaction. The yield of 37 was 32% when the reaction is run in pure Et3N, while running the reaction in the presence of 5 eq TMSCl, along with 5 eq Et3N failed to provide any product. Other nitrogen bases (pyridine, DBU, DABCO, i-Pr2EtN, N,N-dimethylaniline) were also tested, but only i-Pr2EtN yielded 31% of the product.
Among the Lewis acids studied, only the silyl halides, silyl triflates, and boron trifluoride etherate afforded the desired product. As shown in Scheme 8, in the case of BF3·OEt2, the use of a secondary amine (e.g. dibenzylamine), along with the expected 1,2-dehydroquinoline product (37) the reaction also yielded the product of the three-component process (45).
Scheme 8.

The scope of this process was investigated with several 2-sulfamidobenzaldehyde derivatives (46a–e), provided 40–60% yields of the expected 1,2-dihydroquinoline products (47a–e), as shown in Scheme 9.
Scheme 9.

The use of various alkenyl trifluoroborates was also investigated (Scheme 10). While 2-arylvinyl (48a–c), 2-alkenylvinyl (48d) and 2-alkylalkenyl (48e) trifluoroborates provided the expected products (49a–e), the simple unsubstituted vinyltrifluoroborate (48f), under the same reaction conditions, gave only the corresponding quinoline product (49f) in low yield (18%). The more substituted alkenyl trifluoroborates (38g–h) under similar conditions did not form any product at all, presumably for steric reasons.
Scheme 10.

The 1,2-dihydroquinoline sulfamide products of this process, can be easily converted into either 2-substituted quinolines or 2-substituted 1,2,3,4-tetrahydroquinolines. As an example, we have performed both of these transformations with compound 37, which afforded both products (50 and 51) in quantitative yields, upon basic treatment or catalytic hydrogenation (Scheme 11). These additional transformations make this chemistry into a convenient method for the synthesis of these and other heterocycles.
Scheme 11.

3. Experimental
3.1. Materials and physical measurements
All reactions were performed in sealed vials with magnetic stirring, under an atmosphere of dry nitrogen. All chemicals were purchased commercially from Sigma-Aldrich (unless noted otherwise). 2-Sulfamidoaldehydes were prepared according to previously described methods [41, 42]. Potassium alkenyltrifluoroborates were prepared according to standard procedures [36, 38, 39] from the corresponding alkynes or alkenylborinc acids. Anhydrous solvents (acetonitrile and toluene, DriSolv®) were purchased from EMD Chemicals and used directly as supplied.
1H (400 MHz), 13C (100 MHz) and 19F (376 MHz) NMR spectra recorded in CDCl3 on a Varian Mercury-400 Spectrometer, and conformed to the literature data for known compounds. The purity of the products and the progress of the reactions were monitored by thin layer chromatography on Silica Gel 60 F254 glass plates (EMD) followed by visualization with potassium permanganate or vanillin stain.
3.2. Synthesis of 6-bromo-2-phenyl-2H-chromene (23)
The mixture of either 2-phenylvinylboronic acid (148 mg, 1.0 mmol) or potassium 2-phenylvinyltrifluoroborate (210 mg, 1.0 mmol), with 5-bromosalicylaldehyde (201 mg, 1.0 mmol) and dibenzylamine (197 mg, 1.0 mmol) in 2 mL of water was stirred at 80 °C for 3 h. The reaction mixture was extracted with 3×10 mL CH2Cl2, dried over Na2SO4, filtered and evaporated. The yellow syrupy residue was purified by column chromatography (SiO2, CH2Cl2-hexane 1:5). The product, 6-bromo-2-phenyl-2H-chromene (23) (Rf ~ 0.4) was eluted, then the mixture was left on solvent-wet silica until the purple-colored band faded completely, and more product was eluted with CH2Cl2. Total yield 235 mg (82%), viscous yellowish oil
3.3. General procedure for the synthesis of 6-bromo-2-phenyl-2H-chromene (23)
The solution of 5-bromosalicylaldehyde (201 mg, 1 mmol), 2-phenylvinylboronic acid (148 mg, 1 mmol) and the appropriate amount of a secondary or tertiary amine in 2.5 mL of absolute ethanol was prepared in a 2 dram screw-cap glass vial, flushed with dry nitrogen, and stirred at 70 °C for the indicated amount of time. The reaction mixture was then evaporated to dryness and pure 6-bromo-2-phenyl-2H-chromene (23) was isolated by column chromatography on silica (eluent, dichloromethane-hexane 1:2) as light yellow oil. 1H NMR (400 MHz, CDCl3): δ 5.93 (dd, J = 9.9 Hz, 3.4 Hz, 1H), 6.00–6.04 (m, 1H), 6.55 (dd, J = 9.9 Hz, 1.1 Hz, 1H), 6.79 (d, J = 8.6 Hz, 1H), 7.24 (d, J = 2.3 Hz, 1H), 7.30 (dd, J = 8.6 Hz, 2.3 Hz, 1H), 7.42–7.58 (m, 5H). 13C NMR (100 MHz, CDCl3): δ 77.2, 113.0, 117.7, 122.9, 123.1, 125.9, 127.0, 128.5, 128.7, 129.0, 131.9, 140.1, 152.1.
3.4. Synthesis of 6-(benzyloxy)-5,7,8-trimethyl-2-tetradecyl 2H-chromene (30)
3-(Benzyloxy)-6-hydroxy-2,4,5-trimethylbenzaldehyde (28) was prepared from trimethylhydroquinone according to the procedure reported for the 3-methoxy analogue [5]. 1-Hexadecenylboronic acid was prepared in 69% yield from 1-hexadecyne (technical grade, 90%, Alfa Aesar) and catechol borane [43]. The suspension of 28 (135 mg, 0.5 mmol), 1-hexadecenylboronic acid (134 mg, 0.5 mmol) and dibenzylamine (100 mg, ~0.5 mmol) in 1 ml of 1-butanol was prepared in a 1 dram screw-cap glass vial, flushed with dry nitrogen, and stirred at 110 °C for 24 h. The reaction mixture was evaporated to dryness. The product was isolated by column chromatography on silica (eluent - ethyl acetate-hexane 1:100), followed by unreacted 28 (eluent - ethyl acetate-hexane 1:9). The fractions containing the product were evaporated to dryness, and the solid residue was recrystallized from ethanol, yielding 135 mg (57%) of crystalline air-sensitive 6-(benzyloxy)-5,7,8-trimethyl-2-tetradecyl-2H-chromene (30). 1H NMR (400 MHz, CDCl3): δ 0.92 (t, J = 6.8 Hz, 3H), 1.10–1.42 (m, 22H), 1.43–1.70 (m, 3H), 1.77–1.90 (m, 1H), 2.16 (s, 3H), 2.25 (s, 3H), 2.27 (s, 3H), 4.69–4.77 (m, 3H), 5.77 (dd, J = 10.0 Hz, 3.3 Hz, 1H), 6.60 (dd, J = 10.0 Hz, 1.7 Hz, 1H), 7.34–7.39 (m, 1H), 7.;7.46 (m, 2H), 7.49–7.54 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 11.5, 11.7, 13.1, 14.1, 22.7, 25.3, 29.4, 29.61, 29.67, 29.7, 31.9, 34.8, 74.1, 74.6, 119.0, 121.7, 122.6, 123.6, 125.4, 127.7, 127.8, 128.5, 130.4, 137.8, 147.6, 149.1.
3.5. Synthesis of 5,7,8-trimethyl-2-tetradecylchroman-6-ol (31)
Compound 30 (96 mg (0.2 mmol) was suspended in 10 mL of anhydrous ethanol in a 25 mL round-bottom flask. 10 mg of 5% palladium on carbon was added under nitrogen. The reaction mixture was stirred overnight at 60 °C under hydrogen (1 atm). Upon cooling, the product precipitated. The reaction mixture was filtered through short plug of Celite, washed with dichloromethane (100 mL). The filtrate was evaporated and dried in vacuo to give 62 mg (80%) of pure 5,7,8-trimethyl-2-tetradecylchroman-6-ol (31) as white crystals. The compound can be recrystallized from methanol. 1H NMR (400 MHz, CDCl3): δ 0.90 (t, J = 6.8 Hz, 3H), 1.18–1.80 (m, 27H), 1.94–2.03 (m, 1H), 2.11 (s, 3H), 2.14 (s, 3H), 2.17 (s, 3H), 2.61–2.73 (m, 2H), 3.78–3.88 (m, 1H), 4.25 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 11.2, 11.7, 12.1, 14.1, 22.7, 23.2, 25.6, 28.0, 29.4, 29.6, 29.66, 29.69, 31.9, 35.4, 74.8, 118.3, 118.7, 120.8, 122.3, 144.8, 147.0.
3.6. General procedure for the synthesis of 1,2-dihydroquinolines (47a–e, 49a–e) and quinolines (49f)
The mixture of solid alkenyl trifluoroborate (1 mmol), the corresponding carbonyl compound (1 mmol) and a stirring bar were sealed in a 10 mL glass vial, which was subsequently flushed with dry nitrogen. 2.5 mL of anhydrous solvent (acetonitrile or toluene) was injected followed by 0.28 mL (203 mg, 2 mmol) of triethylamine, and the resulting suspension was stirred at room temperature for 5 min. Chlorotrimethylsilane (TMSCl; 0.26 mL, 223 mg, 2.05 mmol) was then added in one portion. The mixture was stirred at 80 °C for 18 h, cooled and diluted with 30 mL of ethyl acetate and 20 mL of water. The layers were separated and the aqueous layer was extracted with 3×20 mL of ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and evaporated. The products were isolated by column chromatography on silica (eluent – ethyl acetate-hexane) and crystallized upon trituration with diethyl ether-pentane.
3.6.1. 1-(Methylsulfonyl)-2-phenyl-1,2-dihydroquinoline (47a)
1H NMR (400 MHz, CDCl3): δ 2.79 (s, 3H), 6.04 (d, J = 5.8 Hz, 1H), 6.34 (dd, J = 9.6 Hz, 5.8 Hz, 1H), 6.86 (d, J = 9.6 Hz, 1H), 6.21–6.31 (m, 6H), 7.36–7.40 (m, 1H), 7.56–7.60 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 37.7, 56.9, 126.3, 126.5, 126.7, 126.9, 127.20, 127.24, 127.97, 127.99, 128.4, 128.6, 132.9, 138.1.
3.6.2. 1-[(2-Nitrophenyl)sulfonyl]-2-phenyl-1,2-dihydroquinoline (47b)
1H NMR (400 MHz, CDCl3): δ 6.14 (d, J = 6.0 Hz, 1H), 6.21 (dd, J = 9.6 Hz, 6.0 Hz, 1H), 6.41 (d, J = 9.6 Hz, 1H), 7.09 (dd, J = 7.1 Hz, 1.7 Hz, 1H), 7.19–7.31 (m, 6H), 7.33–7.41 (m, 3H), 7.51 (d, J = 7.9 Hz, 1H), 7.63 (t, J = 7.9 Hz, 1H), 7.68 (d, J = 7.9 Hz, 1H). 13C NMR (100 MHz, CDCl3): 57.1, 123.5, 124.9, 126.4, 127.1, 127.3, 127.5, 128.0, 128.1, 128.4, 128.5, 129.0, 130.4, 130.6, 130.8, 131.8, 133.7, 137.4, 147.7.
3.6.3. 7-Bromo-1-(methylsulfonyl)-2-phenyl-1,2-dihydroquinoline (47c)
1H NMR (400 MHz, CDCl3): δ2.82 (s, 3H), 6.03 (d, J = 5.8 Hz, 1H), 6.36 (ddd, J = 9.6 Hz, 5.8 Hz, 1.2 Hz, 1H), 6.62 (d, J = 9.6 Hz, 1H), 7.10 (dd, J = 8.1 Hz, 1.2 Hz, 1H), 7.25–7.37 (m, 6H), 7.75 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 38.2, 56.9, 121.7, 125.5, 126.8, 127.2, 127.6, 127.8, 128.2, 128.6, 129.5, 129.6, 134.1, 137.7.
3.6.4. 6,7-Dimethoxy-1-(methylsulfonyl)-2-phenyl-1,2-dihydroquinoline (47d)
1H NMR (400 MHz, CDCl3): δ 2.71 (s, 3H), 3.78 (s, 3H), 3.82 (s, 3H), 5.89 (d, J = 5.8 Hz, 1H), 6.14 (dd, J = 9.6 Hz, 5.8 Hz, 1H), 6.65 (s, 1H), 6.71 (d, J = 9.6 Hz, 1H), 7.06 (s, 1H), 7.14–7.23 (m, 3H), 7.28–7.33 (m, 2H). 13C NMR (100 MHz, CDCl3): δ37.0, 55.8, 56.5, 108.7, 110.9, 121.0, 124.4, 125.87, 125.90, 127.1, 127.8, 128.2, 138.1, 147.4, 148.5.
3.6.5. 1-(Methylsulfonyl)-6-nitro-2-phenyl-1,2-dihydroquinoline (47e)
1H NMR (400 MHz, CDCl3): δ2.87 (s, 3H), 6.12 (d, J = 6.0 Hz, 1H), 6.46 (dd, J = 9.6 Hz, 6.0 Hz, 1H), 6.90 (d, J = 9.6 Hz, 1H), 7.24–7.35 (m, 5H), 7.71 (d, J = 9.1 Hz, 1H), 8.06 (dd, J = 9.1 Hz, 2.5 Hz, 1H), 8.10 (d, J = 2.5 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 39.2, 57.4, 121.8, 123.4, 124.6, 125.6, 127.0, 128.0, 128.5, 128.7, 129.7, 137.4, 138.9, 145.0.
3.6.6. 2-(4-Chlorophenyl)-1-(methylsulfonyl)-1,2-dihydroquinoline (49a)
1H NMR (400 MHz, CDCl3): δ2.79 (s, 3H), 5.99 (d, J = 6.0 Hz, 1H), 6.31 (dd, J = 9.8 Hz, 6.0 Hz, 1H), 6.87 (d, J = 9.8 Hz, 1H), 7.21–7.33 (m, 7H), 7.54–7.59 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 37.8, 56.2, 126.6, 126.75, 126.79, 127.0, 127.9, 128.6, 128.7, 128.9, 132.6, 133.9, 136.6.
3.6.7. 2-(4-Methoxyphenyl)-1-(methylsulfonyl)-1,2-dihydroquinoline (49b)
1H NMR (400 MHz, CDCl3): δ 2.78 (s, 3H), 3.77 (s, 3H), 5.98 (d, J = 5.8 Hz, 1H), 6.29 (dd, J = 9.6 Hz, 5.8 Hz, 1H), 6.80 (d, J = 8.7 Hz, 2H), 6.84 (d, J = 9.6 Hz, 1H), 7.20–7.30 (m, 5H), 7.52–7.56 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 37.8, 55.2, 56.6, 113.8, 126.1, 126.5, 126.6, 127.0, 127.5, 128.1, 128.6, 128.7, 129.9, 132.9, 159.4.
3.6.8. 1-(Methylsulfonyl)-2-[4-(trifluoromethyl)phenyl]-1,2-dihydroquinoline (49c)
1H NMR (400 MHz, CDCl3): δ2.81 (s, 3H), 6.08 (d, J = 6.0 Hz, 1H), 6.37 (dd, J = 9.6 Hz, 6.0 Hz, 1H), 6.90 (d, J = 9.6 Hz, 1H), 7.22–7.31 (m, 3H), 7.48–7.57 (m, 4H), 7.61 (d, J = 7.5 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 37.8, 56.3, 121.2 (q, 1JC,F = 271 Hz), 125.4 (q, 3JC,F = 4 Hz), 126.2, 126.88, 126.91, 126.98, 127.1, 127.6, 127.8, 129.0, 130.1 (q, 2JC,F = 32 Hz), 132.6, 142.3. 19F NMR (376 MHz, CDCl3): δ −62.59.
3.6.9. 2-Cyclohex-1-en-1-yl-1-(methylsulfonyl)-1,2-dihydroquinoline (49d)
1H NMR (400 MHz, CDCl3): δ 1.35–1.62 (m, 4H), 1.74–1.95 (m, 3H), 2.09–2.20 (m, 1H), 5.15 (d, J = 5.6 Hz, 1H), 5.53–5.58 (m, 1H), 6.00 (dd, J = 9.5 Hz, 5.6 Hz, 1H), 6.64 (d, J = 9.5 Hz, 1H), 7.09–7.23 (m, 3H), 7.53 (d, J = 7.9 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 21.9, 22.4, 24.9, 25.1, 37.3, 59.0, 125.4, 125.7, 126.2, 126.4, 126.7, 127.1, 128.1, 128.2, 133.2, 133.8.
3.6.10. 7-bromo-2-butyl-1-(methylsulfonyl)-1,2-dihydroquinoline (49e)
1H NMR (400 MHz, CDCl3): δ 0.84 (t, J = 7.0 Hz, 1H), 1.17–1.47 (m, 6H), 2.66 (s, 3H), 4.65–4.73 (m, 1H), 6.11 (dd, J = 9.6 Hz, 5.8 Hz, 1H), 6.49 (d, J = 9.6 Hz, 1H), 7.01 (d, J = 8.3 Hz, 1H), 7.33 (dd, J = 8.3 Hz, 1.7 Hz, 1H), 7.77 (d, J = 1.7 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 13.9, 22.0, 27.2, 32.7, 37.8, 55.1, 121.2, 124.0, 127.0, 127.6, 129.7, 130.1, 130.2, 133.9.
3.6.11. 7-Bromoquinoline (49f)
1H NMR (400 MHz, CDCl3): δ 7.37–7.42 (m, 1H), 7.59–7.64 (m, 1H), 7.67 (d, J = 8.7 Hz, 1H), 8.08–8.13 (m, 1H), 8.26–8.29 (m, 1H), 8.88–8.91 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 121.4, 123.5, 126.8, 129.0, 130.1, 131.7, 135.9, 148.7, 151.2.
3.7. Reaction of potassium 2-phenylvinyltrifluoroborate with N-(2-formylphenyl)methanesulfonamide and dibenzylamine
The mixture of potassium 2-phenylvinyltrifluoroborate (210 mg, 1 mmol), N-(2-formylphenyl)methanesulfonamide (199 mg, 1 mmol) and dibenzylamine (394 mg, 2 mmol) was sealed in a 10 mL glass vial equipped with a stirring bar and flushed with dry nitrogen. 2.5 mL of anhydrous toluene was injected and the resulting suspension was stirred at room temperature for 5 min. Boron trifluoride-diethyl ether complex (BF3·Et2O; 0.25 mL, 281 mg, ~2 mmol) was then added in one portion. The mixture was stirred at 80 °C for 18 h, cooled and worked up as described above in the general procedure. Rf values (25% ethyl acetate in hexane): for compound 37 Rf=0.4, for compound 45 Rf=0.6; both stained grey upon heating with vanillin stain. The products were isolated by column chromatography on silica (eluent – ethyl acetate-hexane 1:9, changed to 1:5 after elution of the compound 45), yielding 124 mg (26%) of 45 and 86 mg (30%) of 37.
3.8. Synthesis of 2-phenylquinoline (50)
The solution of 285 mg (1 mmol) of compound 37 and 400 mg (10 mmol) of NaOH in 10 mL of absolute ethanol was stirred at 50 °C for 12 h. The reaction mixture was then poured into 30 mL of water, extracted with 3×20 mL of ethyl acetate, and the combined organic extracts were washed with 25 mL of water. The organic layer was dried over Na2SO4, filtered and evaporated to give yellowish crystals of 2-phenylquinoline (50) (205 mg, >99%). 1H NMR (400 MHz, CDCl3): δ 7.45–7.50 (m, 1H), 7.51–7.57 (m, 3H), 7.71–7.77 (m, 1H), 7.83 (d, J = 8.3 Hz, 1H), 7.88 (d, J = 8.3 Hz, 1H), 8.15–8.24 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 119.0, 126.2, 127.1, 127.4, 127.5, 128.8, 129.3, 129.6, 129.7, 136.7, 139.7, 148.3, 157.3.
3.9. 1-(Methylsulfonyl)-2-phenyl-1,2,3,4-tetrahydroquinoline (51)
To the solution of 285 mg (1 mmol) of compound 37 in 10 mL of absolute ethanol 20 mg of 5% palladium on carbon was added under nitrogen. The reaction mixture was stirred overnight at room temperature under hydrogen (1 atm), filtered through a short plug of silica gel and washed with 200 mL of ethyl acetate. The filtrate was evaporated and dried in vacuo to yield 1-(methylsulfonyl)-2-phenyl-1,2,3,4-tetrahydroquinoline (51) (290 mg, >99%) as viscous colorless oil. 1H NMR (400 MHz, CDCl3): δ 2.05–2.15 (m, 1H), 2.48–2.57 (m, 1H), 2.64–2.81 (m, 2H), 2.89 (s, 3H), 5.61 (t, J = 6.8 Hz, 1H), 7.12–7.20 (m, 2H), 7.23–7.37 (m, 6H), 7.82 (d, J = 7.9 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 25.1, 31.4, 39.0, 59.0, 123.4, 124.5, 125.9, 126.9, 127.0, 128.4, 128.6, 131.4, 136.2, 141.7.
Acknowledgments
Financial support by the National Institutes of Health and the Loker Hydrocarbon Research Institute of the University of Southern California is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ellis GP, editor. Chromenes, Chromanones, and Chromones, The Chemistry of Heterocyclic Compounds. Vol. 31. Wiley-Interscience; New York: 1977. [Google Scholar]
- 2.Nicolaou KC, Pfefferkorn JA, Roecker AJ, Cao GQ, Barluenga S, Mitchell HJ. J Am Chem Soc. 2000;122:9939–9953. [Google Scholar]
- 3.Veitch NC, Grayer RJ. Natural Product Reports. 2008;25:555–611. doi: 10.1039/b718040n. [DOI] [PubMed] [Google Scholar]
- 4.Wehrli PA, Fryer RI, Metlesics W. J Org Chem. 1971;36:2910–2912. doi: 10.1021/jo00818a050. [DOI] [PubMed] [Google Scholar]
- 5.Hussain HH, Babic G, Durst T, Wright JS, Flueraru M, Chichirau A, Chepelev LL. J Org Chem. 2003;68:7023–7032. doi: 10.1021/jo0301090. [DOI] [PubMed] [Google Scholar]
- 6.Trost BM, Toste FD. J Am Chem Soc. 1998;120:9074–9075. [Google Scholar]
- 7.Trost0 BM, Shen HC, Dong L, Surivet JP, Sylvain C. J Am Chem Soc. 2004;126:11966–11983. doi: 10.1021/ja048078t. [DOI] [PubMed] [Google Scholar]
- 8.Kokubo Y, Hasegawa A, Kuwata S, Ishihara K, Yamamoto H, Ikariya T. Adv Synth Catal. 2005;347:220–224. [Google Scholar]
- 9.Tietze LF, Sommer KM, Zinngrebe J, Stecker F. Angew Chem Int Ed. 2005;44:257–259. doi: 10.1002/anie.200461629. [DOI] [PubMed] [Google Scholar]
- 10.Chenevert R, Courchesne G, Pelchat N. Bioorganic & Medicinal Chemistry. 2006;14:5389–5396. doi: 10.1016/j.bmc.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 11.Trost BM, Shen HC, Dong L, Surivet JP. J Am Chem Soc. 2003;125:9276–9277. doi: 10.1021/ja036052g. [DOI] [PubMed] [Google Scholar]
- 12.Palucki M, Yasuda N. Tetrahedron Lett. 2005;46:987–990. [Google Scholar]
- 13.Lee YR, Choi JH, Yoon SH. Tetrahedron Lett. 2005;46:7539–7543. [Google Scholar]
- 14.Shi YL, Shi M. Org Biomol Chem. 2007;5:1499–1504. doi: 10.1039/b618984a. [DOI] [PubMed] [Google Scholar]
- 15.Takamura M, Funabashi K, Kanai M, Shibasaki M. J Am Chem Soc. 2000;122:6327–6328. doi: 10.1021/ja010654n. [DOI] [PubMed] [Google Scholar]
- 16.Chang YM, Park YS, Lee SH, Yoon CM. Tetrahedron Lett. 2004;45:9049–9052. [Google Scholar]
- 17.Yamaoka Y, Miyabe H, Takemoto Y. J Am Chem Soc. 2007;129:6686–6687. doi: 10.1021/ja071470x. [DOI] [PubMed] [Google Scholar]
- 18.Amiot F, Cointeaux L, Jan Silve E, Alexakis A. Tetrahedron. 2004;60:8221–8231. [Google Scholar]
- 19.Yadav JS, Reddy BVS, Yadav NN, Gupta MK, Sridhar B. J Org Chem. 2008;73:6857–6859. doi: 10.1021/jo8007034. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi H, Bekkali Y, Capolino AJ, Gilmore T, Goldrick SE, Nelson RM, Terenzio D, Wang J, Zuvela-Jelaska L, Proudfoot J, Nabozny G, Thomson D. Bioorganic & Medicinal Chemistry Letters. 2006;16:1549–1552. doi: 10.1016/j.bmcl.2005.12.043. [DOI] [PubMed] [Google Scholar]
- 21.Ranu BC, Hajra A, Dey SS, Jana U. Tetrahedron. 2003;59:813–819. [Google Scholar]
- 22.Luo Y, Li Z, Li CJ. Org Lett. 2005;7:2675–2678. doi: 10.1021/ol050826b. [DOI] [PubMed] [Google Scholar]
- 23.Xiao F, Chen Y, Liu Y, Wang J. Tetrahedron. 2008;64:2755–2761. [Google Scholar]
- 24.Presented at the XIII IMEBORON Meeting in Platja d’Aro, Spain, September 21–25, 2008.
- 25.Petasis NA. In: Multicomponent Reactions; Zhu J, Bienayme H, editors. Wiley-VCH; 2005. pp. 199–223. [Google Scholar]
- 26.Petasis NA. Aust J Chem. 2007;60:795–798. [Google Scholar]
- 27.Petasis NA, Zavialov IA. J Am Chem Soc. 1997;119:445–446. [Google Scholar]
- 28.Petasis NA, Zavialov IA. J Am Chem Soc. 1998;120:11798–11799. [Google Scholar]
- 29.Petasis NA, Boral S. Tetrahedron Lett. 2001;42:539–542. [Google Scholar]
- 30.Wang Q, Finn MG. Org Lett. 2000;2:4063–4065. doi: 10.1021/ol006710r. [DOI] [PubMed] [Google Scholar]
- 31.Kabalka G, Venkataiah B, Das BC. Synlett. 2004:2194–2196. [Google Scholar]
- 32.Kabalka GW, Venkataiah B, Dong G. Tetrahedron Lett. 2004;45:729–731. [Google Scholar]
- 33.McLean NJ, Tye H, Whittaker M. Tetrahedron Lett. 2004;45:993–995. [Google Scholar]
- 34.Liu F, Evans T, Das BC. Tetrahedron Lett. 2008;49:1578–1581. doi: 10.1016/j.tetlet.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith LI, Opie JW. Organic Syntheses, Coll. 1955;3:56. [Google Scholar]
- 36.Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J Org Chem. 1995;60:3020–3027. [Google Scholar]
- 37.Darses S, Genet JP. Eur J Org Chem. 2003;2003:4313–4327. [Google Scholar]
- 38.Darses S, Genet JP. Chem Rev. 2008;108:288–325. doi: 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- 39.Molander GA, Ellis N. Acc Chem Res. 2007;40:275–286. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- 40.Petasis NA, Yudin AK, Zavialov IA, Prakash GKS, Olah GA. Synlett. 1997:606–608. [Google Scholar]
- 41.Hechavarria Fonseca M, Eibler E, Zabel M, Konig B. Tetrahedron: Asymmetry. 2003;14:1989–1994. [Google Scholar]
- 42.Nishiguchi A, Ikemoto T, Ito T, Miura S, Tomimatsu K. Heterocycles. 2007;71:1183–1192. [Google Scholar]
- 43.Brown HC, Gupta SK. J Am Chem Soc. 1975;97:5249–5255. [Google Scholar]