

Abstract
Lacosamide has been submitted for regulatory approval in the United States and Europe for the treatment of epilepsy. Previous synthetic methods did not permit the elaboration of the structure–activity relationship (SAR) for the 3-oxy site in lacosamide. We report an expedient five-step stereospecific synthesis for N-benzyl (2R)-2-acetamido-3-oxysubstituted propionamide analogs beginning with d-serine methyl ester. The procedure incorporated alkyl (e.g. methyl, primary, secondary, and tertiary) and aryl groups at this position. The SAR for the 3-oxy site showed maximal activity in animal seizure models for small 3-alkoxy substituents.
Keywords: Amino acid N-benzylamides, Aziridine ring opening, Anticonvulsants, Diethoxytriphenylphosphorane, Structure–activity relationship
1. Introduction
Lacosamide (Vimpat®, (R)-1) is an emerging neurological agent that has shown therapeutic efficacy for the treatment of partial seizures. It has been submitted for regulatory approval in the United States and Europe under the sponsorship of UCB Pharma.1,2 Evaluation of 1 in animal seizure models at the National Institutes of Neurological Disorders and Stroke's (NINDS) Anticonvulsant Screening Program (ASP) showed that it exhibited potent anticonvulsant activity in the maximal electroshock seizure (MES) test in mice (ip) and rats (po).3
In 1996, we reported an expeditious three-step synthesis of (R)-1 from d-serine (Scheme 1).3 The 3-methoxy unit was installed by a Williamson-type ether synthesis using MeI and Ag2O. Using race-mic 4 and either EtI or allyl iodide in place of MeI gave the corresponding O-substituted derivatives 5 and 6, respectively.3 Efforts to extend the synthesis to other O-substituted analogs led to appreciably lower yields of the desired ethers.4 This synthetic impediment prevented our determining the structure–activity relationship (SAR) for the 3-oxy substituent. We report herein an expedient stereospecific synthesis of (R)-N-benzyl-2-acetamido-3-oxysubstituted propionamides (7) and document the importance of the 3-oxy substituent for anticonvulsant activity within this novel class of agents.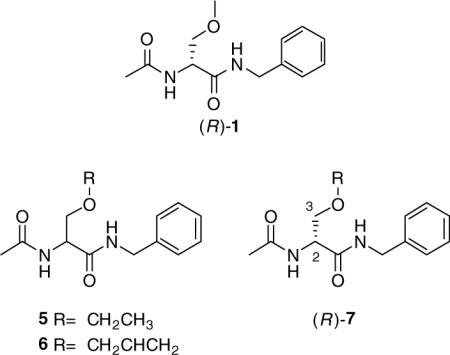
Scheme 1.
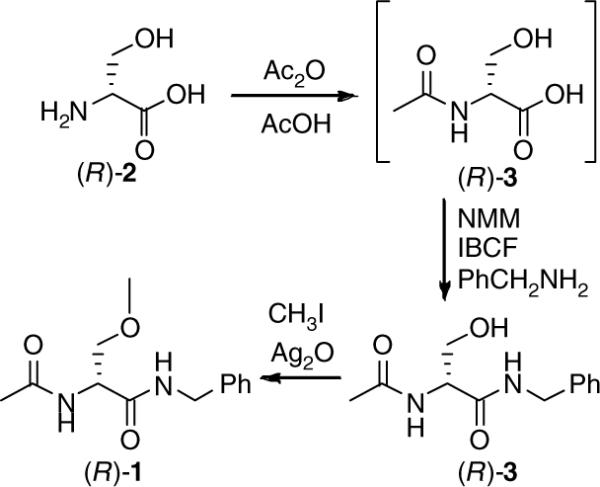
Three-step synthesis of (R)-lacosamide (1).
2. Results and discussion
2.1. Synthesis
We sought a general and efficient procedure to prepare 7 that would permit different substituents at the 3-oxy site. Okawa and coworkers demonstrated that N-Cbz-substituted aziridines 8 underwent ring opening with alcohols and thiols using catalytic amounts of BF3·Et2O to give the corresponding 3-substituted derivatives 9.5,6Similar procedures have been used by Larsson and Carlson,7 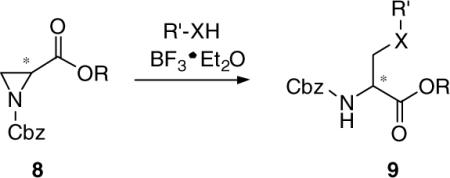 and Zavada and coworkers.8 The synthetic utility of the Okawa procedure was hampered by the time required by the five-step synthesis of 8 from the desired serine ester hydrochloride.5 In 1989, van Boom reported a solution to this problem.9 Utilizing Evans’ one-step cyclodehydration conversion of 2-amino alcohols to aziridines with diethoxytriphenylphosphorane (DTPP),10 treatment of 10 with DTPP provided the unsubstituted (2S)-aziridine-2-carboxylates 11.
and Zavada and coworkers.8 The synthetic utility of the Okawa procedure was hampered by the time required by the five-step synthesis of 8 from the desired serine ester hydrochloride.5 In 1989, van Boom reported a solution to this problem.9 Utilizing Evans’ one-step cyclodehydration conversion of 2-amino alcohols to aziridines with diethoxytriphenylphosphorane (DTPP),10 treatment of 10 with DTPP provided the unsubstituted (2S)-aziridine-2-carboxylates 11.  Thus, we treated d-serine methyl ester (12) with DTTP to give the desired methyl (2R)-aziridine-2-carboxylate (13a) along with the corresponding ethyl ester 13b in 50−60% total yield (Scheme 2). The production of the ethyl ester 13b was surprising. The amount of ethyl ester varied with each reaction and appeared to increase with increasing reaction time, and increasing amounts of DTPP.11 Since the ester group was hydrolyzed to the acid in a subsequent step to permit amide coupling, we used the binary mixture of esters without separation. With an expedient route to 13, we evaluated the generality of the BF3·Et2O catalyzed alcohol ring-opening reaction for this set of aziridines. We restricted our studies to the (R)-stereoisomer since we have shown that the pharmacological activity for this class of substituted amino acids principally resided in the d-serine stereochemical series.3 Accordingly, we converted 13a and 13b to methyl and ethyl (2R)-N-(acetyl)aziridine-2-carboxylates (14a and 14b) in 94% yield with acetic anhydride, Et3N, and a catalytic amount of DMAP. Treatment of 14a and 14b with MeOH, the primary alcohols, EtOH and diethylene glycol monomethyl ether (DEGME), the secondary alcohol, iPrOH, the tertiary alcohol, tert-BuOH, and phenol with one equivalent of BF3·Et2O gave the corresponding aziridine ring-opening products (15a,b–20a,b) in 50−60% isolated yield. The alcohols produced little differences in yields. Conversion of esters 15a,b–20a,b to the N-benzyl amides 1, 27−31, respectively, followed established procedures. First, we hydrolyzed methyl (ethyl) esters 15−20 with stoichiometric amounts of LiOH (1.0−1.1 equiv) at 0 °C to provide the free acids 21−26 upon workup. Under these conditions we detected little or no racemization. Coupling acids 21−26 with benzylamine and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM)12 provided the amides 1, 27−31, respectively. After column purification and subsequent recrystallization we saw that the spectral and analytical data for 1, 27−31 were in agreement with their proposed structures. Adding the chiral resolving agent (R)-(−)-mandelic acid to a CDCl3 solution of each amide showed only one signal for the acetamide methyl resonance consistent with the expected enantiopurity of the products.3,13
Thus, we treated d-serine methyl ester (12) with DTTP to give the desired methyl (2R)-aziridine-2-carboxylate (13a) along with the corresponding ethyl ester 13b in 50−60% total yield (Scheme 2). The production of the ethyl ester 13b was surprising. The amount of ethyl ester varied with each reaction and appeared to increase with increasing reaction time, and increasing amounts of DTPP.11 Since the ester group was hydrolyzed to the acid in a subsequent step to permit amide coupling, we used the binary mixture of esters without separation. With an expedient route to 13, we evaluated the generality of the BF3·Et2O catalyzed alcohol ring-opening reaction for this set of aziridines. We restricted our studies to the (R)-stereoisomer since we have shown that the pharmacological activity for this class of substituted amino acids principally resided in the d-serine stereochemical series.3 Accordingly, we converted 13a and 13b to methyl and ethyl (2R)-N-(acetyl)aziridine-2-carboxylates (14a and 14b) in 94% yield with acetic anhydride, Et3N, and a catalytic amount of DMAP. Treatment of 14a and 14b with MeOH, the primary alcohols, EtOH and diethylene glycol monomethyl ether (DEGME), the secondary alcohol, iPrOH, the tertiary alcohol, tert-BuOH, and phenol with one equivalent of BF3·Et2O gave the corresponding aziridine ring-opening products (15a,b–20a,b) in 50−60% isolated yield. The alcohols produced little differences in yields. Conversion of esters 15a,b–20a,b to the N-benzyl amides 1, 27−31, respectively, followed established procedures. First, we hydrolyzed methyl (ethyl) esters 15−20 with stoichiometric amounts of LiOH (1.0−1.1 equiv) at 0 °C to provide the free acids 21−26 upon workup. Under these conditions we detected little or no racemization. Coupling acids 21−26 with benzylamine and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM)12 provided the amides 1, 27−31, respectively. After column purification and subsequent recrystallization we saw that the spectral and analytical data for 1, 27−31 were in agreement with their proposed structures. Adding the chiral resolving agent (R)-(−)-mandelic acid to a CDCl3 solution of each amide showed only one signal for the acetamide methyl resonance consistent with the expected enantiopurity of the products.3,13
Scheme 2.

Synthesis of N-benzyl-(2R)-2-acetamido-3-oxysubstituted propionamide derivatives.
2.2. Pharmacological activity
Compounds 1 and 27−31 were tested for anticonvulsant activity by the NIH and NINDS's Anticonvulsant Screening Program (ASP) using the procedures described by Stables and Kupferberg.14 The pharmacological data are summarized in Table 1, along with similar results obtained for 1 and the established antiepileptic agents phenytoin, valproate, and phenobarbital. We observed that the N-benzyl (2R)-3-alkoxysubstituted 2-acetamidopropionamides 1, 27, and 29−31 exhibited moderate-to-excellent anticonvulsant activity in the MES-seizure test in mice (ip). Among the 3-alkoxy-substituted derivatives, activity improved with a coinciding decrease of the 3-alkoxy group's size. Thus, the MES ED50 values declined from a range between 30 mg/kg and 100 mg/kg for the tert-butoxy analog 30, down to 23 mg/kg for 29, to 7.9 mg/kg for 27 and finally to 4.5 mg/kg for the O-methoxy compound 1. The anticonvulsant activities for 1 and 27 exceeded that of phenytoin.15 Paralleling this increase, we observed a neurological toxicity increase in the rotorod test16 similar to earlier findings.3 Introduction of a 3-phenoxy unit in place of the 3-methoxy group in 1 to give 31 led to a marked activity reduction in the MES test (ED50 >100 mg/kg but still <300 mg/kg). Finally, the polyether 28 exhibited neither anticonvulsant nor neurological activity at the dose evaluated (up to 300 mg/kg). We hypothesize that the loss of neurological activity is due to the inability of this hydrophilic derivative to cross the blood brain barrier. No N-benzyl-3-oxysubstituted 2-acetamidopropionamides 1 and 27−31 showed detectable activity in the pentylenetetrazole test (scMet) in mice (data not shown), which is in agreement with similar test results observed for this class of compounds.3
Table 1.
Pharmacological data for N-benzyl (2R)-2-acetamido-3-oxysubstituted propionamides derivatives
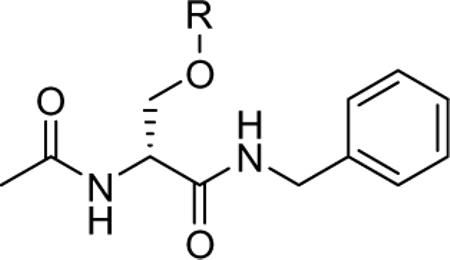 | ||||
|---|---|---|---|---|
| Compound # | R | Mice (ip)a |
||
| MES,b ED50 | Tox,c TD50 | PId | ||
| (R)-1e | CH3 | 4.5 [0.5] (3.7−5.5) | 27 [0.25] (26−28) | 6 |
| (R)-27 | CH2CH3 | 7.9 [0.25] (5.3−10.4) | 44 [0.25] (37−54) | 5.6 |
| (R)-28 | CH2CH2OCH2CH2OCH3 | >300 [0.5] | >300 [0.5] | – |
| (R)-29 | CH(CH3)2 | 23 [0.25] (20−26) | 77 [0.25] (66−96) | 3.3 |
| (R)-30 | C(CH3)3 | >30, <100 [0.5] | >100, <300 [0.5] | – |
| (R)-31 | C6H5 | >100, <300 [0.5] | >30, <100 [0.5] | – |
| Phenytoinf | – | 9.5 [2] (8.1−10) | 56 [2] (53−72) | 6.9 |
| Phenobarbitalf | – | 22 [1] (15−23) | 69 [0.5] (63−73) | 3.2 |
| Valproatef | – | 270 [0.25] (250−340) | 430 [0.25] (370−450) | 1.6 |
The compounds were administered intraperitoneally.
MES = maximal electroshock seizure test in mg/kg. Numbers in parentheses are 95% confidence intervals. The dose effect data was obtained at the ‘time of peak’ effect (indicated in hours in the brackets).
Tox = neurologic toxicity determined from rotorod test in mice. Numbers in parentheses are 95% confidence intervals. The dose effect data was obtained at the ‘time of peak’ effect (indicated in hours in the brackets).
PI = protective index (TD50/ED50).
Ref. 3.
3. Conclusions
We have established a convenient, general, stereospecific synthetic method for the preparation of N-benzyl-3-oxysubstituted 2-acetamidopropionamides (7). The route takes advantage of a rapid and short synthesis of 14 using readily available DTPP.10 Key to the preparation of these compounds is the BF3·Et2O ring opening of aziridine 14 with alcohols and phenol. Little differences in the yields for ring opening were observed when we varied the size and nucleophilicity of the alcohol or phenol.
4. Experimental
4.1. General methods
Melting points were determined with a Thomas-Hoover melting point apparatus and are uncorrected. Infrared spectra (IR) were run on a ATI Mattson Genesis Series FTIR™ spectrometer. Absorption values are expressed in wave-numbers (cm−1). Optical rotations were obtained on a Jasco P-1030 polarimeter. Proton (1H NMR, 300 MHz) and carbon (13C NMR, 75 MHz) nuclear magnetic resonance spectra were taken on a Varian Gemini 2000 spectrometer. The high-resolution mass spectrum was performed on a Bruker Apex-Q 12 Tesla FTICR by Dr. M. Crowe at the University of North Carolina—Chapel Hill. Microanalysis was provided by Atlantic Microlab, Inc. (Norcross, GA).
4.2. Synthesis of diethoxytriphenylphosphorane (DTPP)
Triphenylphosphine (110.00 g, 419 mmol) was dissolved in anhydrous toluene (500 mL) and the solvent was evaporated. The solid was redissolved in a 1:1 mixture of anhydrous CH2Cl2 (350 mL) and THF (350 mL) and cooled at −78 °C (dry ice/acetone bath). While stirring, Br2 (21.5 mL, 419 mmol) was added all at once with a syringe and the reaction was stirred at −78 °C (10 min). Commercial NaOEt (96%, 59.35 g, 838 mmol) was added portionwise over 5 min, and then anhydrous EtOH (44 mL, 838 mmol) was added dropwise to the formed suspension. The reaction was stirred at −78 °C (90 min), and allowed to warm to room temperature. The supernatant was decanted and filtered over a Celite bed. The remaining suspension was centrifuged at 1600 rpm for 15 min. The supernatant layers obtained from both fractions were combined and evaporated at 30 °C. Hexanes (500 mL) were added to the oily residue and the mixture was shaken for 5 min. The triphenylphosphine oxide (TPPO) was filtered, and the solvent removed in vacuo. Additional hexanes (500 mL) were added to the solid and the flask was let stand on ice for 20 min. Additional TPPO was filtered and the solvent evaporated to yield DTPP as a white to pale yellow solid (66.42 g, 45% yield, 90% pure by wt): 1H NMR (CDCl3) δ 0.75 (t, J = 7.0 Hz, P(OCH2CH3)2), 2.53 (app q, J = 7.0 Hz, P(OCH2CH3)2), 7.39−7.47 (m, 9 ArH), 8.04−8.12 (m, 6 ArH); 13C NMR (CDCl3) 16.4 (d, J = 5.1 Hz, P(OCH2CH3)2), 57.5 (d, J = 6.8 Hz, P(OCH2CH3)2), 127.8, 129.4, 132.7 (15 ArC), 139.2 (d, J = 173.0 Hz, 3 ArC). The DTPP obtained under these conditions was 85−95% pure by weight and was contaminated with TPPO (1H NMR analysis).
4.3. General procedure to generate d-serine methyl ester ((R)-12)
d-Serine methyl ester hydrochloride (1 equiv) was suspended in acetonitrile ([C] ∼ 1 M) and Et3N (1.5 equiv) was added. After stirring at room temperature (1 h), the salts were filtered and rinsed with EtOAc and an equal volume of EtOAc was added. The mixture was stirred at 0 °C (10 min), the solids filtered, and then approximately two thirds of the solvent volume removed in vacuo. The remaining reaction mixture was stirred at 0 °C (10 min), filtered, and the solvent removed. The resulting oil was dissolved in acetonitrile and the solvent evaporated to remove excess Et3N. The preceding step was repeated until Et3N could not be detected, yielding the free amine of d-serine methyl ester as a pale yellow oil (∼90%) that solidified upon standing overnight. (R)-12 was best used as an oil since solid (R)-12 did not readily dissolve in organic solvents.
4.4. Synthesis of (R)-methyl N-acetylaziridine-carboxylate ((R)-14a) and (R)-ethyl N-acetylaziridine-carboxylate ((R)-14b)
To a solution of d-serine methyl ester (41.30 g, 347 mmol) in acetonitrile (500 mL) was added DTPP (86% by wt, 142.00 g, 346 mmol). The solution was stirred at room temperature (24 h). The solvent was removed in vacuo, the residue dissolved in minimal amount of CH2Cl2 (250 mL), and extracted with aqueous 0.1 M H2SO4 until the pH of the aqueous phase remained acidic (3× 150 mL). The combined aqueous layers were washed with EtOAc (3× 200 mL), basified (pH ∼ 10) with solid Na2CO3, saturated with NaCl until the solution became cloudy, and extracted with EtOAc (6× 200 mL). The combined organic layers were dried (Na2SO4) and evaporated to give a crude yellow liquid. Bulb-to-bulb distillation of the liquid at 80 °C under vacuum (6 mm Hg) yielded an approximately 9:1 molar mixture of (R)-13a and (R)-13b as a colorless liquid (24.60 g, 69%). The mixture was directly dissolved in CH2Cl2 (500 mL) and Et3 N (33.4 mL, 239 mmol) and DMAP (1.46 g, 12 mmol) were successively added. While stirring at room temperature (water bath), Ac2O (22.6 mL, 239 mmol) was added dropwise over 15 min and the reaction was then allowed to proceed at room temperature (45 min). The reaction was successively washed with a 10% aqueous citric acid solution (500 mL), and brine (500 mL), dried (Na2SO4), and the solvents were removed in vacuo to yield an approximately 9:1 molar mixture of a colorless residue (32.80 g, 94%) that was used without further purification: Rf = 0.38 ((R)-14a), 0.39 ((R)-14b) (2:1 hexanes/EtOAc). Spectral data for (R)-14a (∼90 mol percent based on 1H NMR integrations): 1H NMR (CDCl3) δ 2.16 (s, CH3C(O)), 2.49 (dd, J = 1.8, 7.0 Hz, NCHH′CH), 2.58 (dd, J = 3.0, 7.0 Hz, CHCOOCH3), 3.15 (dd, J = 1.8, 3.0 Hz, NCHH′CH), 3.80 (s, CO2CH3); 13C NMR (CDCl3) δ 23.8 (CH3C(O)), 31.0 (NCH2CH), 34.5 (CHCO2CH3), 52.9 (CO2CH3), 168.9 (CH3C(O)), 180.6 (COOCH3); (R)-14a was not detected by HRMS.
Spectral data for (R)-14b (∼10 mol percent based on 1H NMR integrations): 1H NMR (CDCl3) δ 1.31 (t, J = 6.9 Hz, C(O)OCH2CH3), 4.24 (q, J = 6.9 Hz, C(O)OCHH′CH3), 4.25 (q, J = 6.9 Hz, C(O)OCHH′CH3), the remaining signals were not detected and are believed to overlap with the 1H signals for (R)-14a; 13C NMR (CDCl ) no 13C signals were detected for (R)-14b; Mr (+ESI) [M+Na]+ (calcd for C7H11NO3Na+ 180.0637) 180.0631.
4.5. General procedure for the aziridine ring opening of (R)-14a/(R)-14b with alcohols. Method A
To a cooled solution (ice bath) of a binary mixture of (R)-14a and (R)-14b in the appropriate alcohol or in CH2Cl2 ([C] ∼ 0.5−1 M) was added BF3·Et2O (1 equiv) dropwise while stirring. Upon addition, the mixture was warmed to room temperature and stirred for an additional 90 min. An equal volume of saturated aqueous NaHCO3 was added to the solution and after 15 min of vigorous stirring, the organic layer separated. The aqueous layer was extracted with CH2Cl2 until no more product was detected (TLC analysis). The combined organic layers were combined, dried (Na2SO4), and removed in vacuo to yield a mixture of methyl and ethyl esters that was either used without further purification or, when needed, purified by flash column chromatography.
4.6. General procedure for the methyl/ethyl ester hydrolysis of (R)-15a/(R)-15b-(R)-20a/(R)-20b with LiOH. Method B
To a solution of methyl and ethyl esters in 2 volumes of THF ([C] ∼ 0.1 M) was added LiOH (1 equiv) in 1 volume of H2O. The reaction was stirred at room temperature (90 min), after which time the aqueous layer was washed with Et2O (2 volumes). The aqueous layer was acidified (pH 1) by the dropwise addition of aqueous concentrated HCl at 0 °C, saturated with NaCl, and extracted with EtOAc until no further product was detected (TLC analysis). The combined organic layers were combined, dried (Na2SO4), and evaporated to an oily residue that was used directly in the next step, or recrystallized when needed from EtOAc and hexanes to provide an analytical sample.
4.7. General procedure for the DMTMM amide coupling reaction. Method C
To a THF solution of acid ([C] ∼ 0.1 M) at room temperature was added benzylamine (1.2 equiv). The solution was stirred for 5−10 min until the benzylammonium carboxylate precipitated. While stirring, DMTMM (1.2 equiv) was added at once, and the resulting suspension was stirred at room temperature (3−12 h). In those cases where a salt did not precipitate, DMTMM was added after 15 min to the solution. The salts were removed by filtration, washed with THF, and the solvent was removed in vacuo. The obtained residue was purified by flash column chromatography to afford the benzylamide, and then recrystallized with EtOAc and hexanes.
4.8. Synthesis of (R)-methyl 2-acetamido-3-methoxypropionate ((R)-15a) and (R)-ethyl 2-acetamido-3-methoxypropionate ((R)-15b)
Using Method A, a ∼ 9:1 mixture of (R)-14a and (R)-14b (5.46 g, 40.6 mmol), and BF3·Et2O (5.1 mL, 40.6 mmol) in MeOH (50 mL) gave 3.95 g (56%) of (R)-15a and (R)-15b as a pale yellow residue that solidified under high vacuum and was used without further purification: Rf = 0.40 ((R)-15a), 0.42 ((R)-15b) (5:95 hexanes/EtOAc); IR (CH2Cl2 film) 3300, 3062, 2996, 2940, 1743, 1656, 1547, 1444, 1380, 1214, 1119 cm−1. Spectral data for (R)-15a (∼90 mol percent based on 1H NMR integrations): 1H NMR (CDCl3) δ 2.06 (s, CH3C(O)NH), 3.34 (s, OCH3), 3.61 (dd, J = 4.0, 9.3 Hz, CHCHH′OCH3), 3.77 (s, C(O)OCH3), 3.81 (dd, J = 4.0, 9.3 Hz, CHCHH′OCH3), 4.75 (app dt, J = 4.0, 7.8 Hz, CHCH2OCH3), 6.74 (br d, J = 7.8 Hz, CH3C(O)NH); 13C NMR (CDCl3) δ 22.9 (CH3C(O)), 52.4 (CHCH2OCH3 or C(O)OCH3), 52.5 (C(O)OCH3 or CHCH2OCH3), 59.1 (CH2OCH3), 72.2 (CHCH2OCH3), 170.1, 170.8 (CH3C(O)NH, C(O)OCH3); Mr (+ESI) [M+Na]+ (calcd for C7H13NO4Na+ 198.0742) 198.0740.
Spectral data for (R)-15b (∼10 mol percent based on 1 HNMR integrations): 1H NMR (CDCl3) δ 1.29 (t, J = 7.2 Hz, C(O)OCH2CH3) 1.99 (s, CH3C(O)NH), 3.44 (s, OCH3), 3.92 (dd, J = 4.0, 9.3 Hz, CHCHH′OCH3), 4.19−4.28 (m, C(O)OCH2CH3), 6.40−6.50 (br d, CH3C(O)NH), the remaining peaks were not detected and are believed to overlap with (R)-15a signals; 13C NMR signals were not detected for (R)-15b; Mr (+ESI) 212.0896 [M+Na]+ (calcd for C8H15NO4Na+ 212.0899 [M+Na]+).
4.9. Synthesis of (R)-methyl 2-acetamido-3-ethoxypropionate ((R)-16a) and (R)-ethyl 2-acetamido-3-ethoxypropionate ((R)-16b)
Using Method A, a mixture of (R)-14a and (R)-14b (∼90% (R)-14a and ∼10% (R)-14b) (1.88 g, 13.0 mmol), and BF3·Et2O (1.63 mL, 13.0 mmol) in EtOH (25 mL) gave 1.34 g (54%) of (R)-16a and (R)-16b as a pale yellow oil that was used without further purification: Rf = 0.43 ((R)-16a), 0.45 ((R)-16b) (5:95 hexanes/EtOAc); IR (neat) 3287, 3064, 2977, 2876, 1745, 1661, 1542, 1442, 1375, 1212, 1119 cm−1. Spectral data for (R)-16a (∼90 mol percent based on 1H NMR integrations): 1H NMR (CDCl3) δ 1.16 (t, J = 7.2 Hz, CH2OCH2CH3), 2.06 (s, CH3C(O)NH), 3.50 (q, J = 7.2 Hz, CH2OCHH′CH3), 3.51 (q, J = 7.2 Hz, CH2OCHH′CH3), 3.65 (dd, J = 4.0, 8.7 Hz, CHCHH′OCH2CH3), 3.76 (s, C(O)OCH3), 3.84 (dd, J = 4.0, 8.7 Hz, CHCHH′OCH2CH3), 4.75 (app dt, J = 4.0, 7.5 Hz, CHCH2OCH2CH3), 6.54 (br d, J = 7.5 Hz, CH3C(O)NH); 13C NMR (CDCl3) δ 15.0 (OCH2CH3), 23.3 (CH3C(O)), 52.7 (CHCH2OCH3 or C(O)OCH3), 52.8 (C(O)OCH3 or CHCH2OCH2H3), 67.1 (CHCH2OCH2CH3), 70.2 (CHCH2OCH2CH3), 170.1, 171.1 (CH3C(O)NH, C(O)OCH3); Mr (+ESI) 212.0897 [M+Na]+ (calcd for C8H15NO4Na+ 212.0899 [M+Na]+).
Spectral data for (R)-16b (∼10 mol percent based on 1 HNMR integrations): 1H NMR (CDCl3) δ 1.24 (t, J = 7.2 Hz, CH2OCH2CH3 or C(O)OCH2CH3), 1.29 (t, J = 7.2 Hz, C(O)OCH2CH3 or CH2OCH2CH3), 1.99 (s, CH3C(O)NH), 3.96−4.04 (m, CH2OCHH′CH3), 4.19−4.30 (m, C(O)OCH2CH3), 6.10−6.22 (br d, J = 6.8 Hz, CH3C(O)NH), the remaining peaks were not detected and are believed to overlap with (R)-16a signals; 13C NMR signals were not detected for (R)-16b; Mr (+ESI) 226.1054 [M+Na]+ (calcd for C9H17NO4Na+ 226.1055 [M+Na]+).
4.10. Synthesis of (R)-methyl 2-acetamido-3-(2-(2-methoxyethoxy)ethoxy)propionate ((R)-17a) and (R)-ethyl 2-acetamido-3-(2-(2-methoxyethoxy)ethoxy)propionate ((R)-17b)
Using Method A, a ∼1:1 mixture of (R)-14a and (R)-14b (4.50 g, 30.0 mmol), DEGME (11.3 g, 94.5 mmol), and BF3·Et2O (3.8 mL, 30.0 mmol) in CH2Cl2 (30 mL) gave 2.84 g (35%) of (R)-17a and (R)-17b as a colorless viscous oil after purification by flash chromatography column (2:1 hexanes/EtOAc to EtOAc): Rf = 0.31 ((R)-17a), 0.33 ((R)-17b) (EtOAc); IR (neat) 3300, 3063, 2940, 2940, 1744, 1659, 1553, 1446, 1216, 1120 cm−1. Spectral data for (R)-17a and (R)-17b (1:1): 1H NMR (CDCl3) δ 1.28 ((R)-17b, t, J = 7.2 Hz, C(O)OCH2CH3), 2.06 ((R)-17a,b, s, CH3C(O)NH), 3.39 ((R)-17a,b, s, CH2CH2OCH3), 3.52−3.58 ((R)-17a,b, m, CH2CH2OCH3), 3.60−3.64 ((R)-17a,b, m, OCH2CH2OCH2), 3.68−3.74 ((R)-17a,b, m, CHCHH′OCH2), 3.76 ((R)-17a, s, C(O)OCH3), 3.94, 3.97 ((R)-17a,b, app t, J = 4.2 Hz CHCHH′OCH2), 4.22 ((R)-17b, t, J = 7.2 Hz, C(O)OCH2CH3), 4.68−4.76 ((R)-17a,b, m, CHCH2OCH2), 6.52−6.70 ((R)-17a,b, m, CH3C(O)NH); 13C NMR (CDCl3) δ 14.3 ((R)-17a,b, C(O)OCH2CH3), 23.1 ((R)-17a,b, CH3C(O)), 52.6, 52.9, 53.0 ((R)-17a,b, CHCH2OCH2, (R)-17a, C(O)OCH3), 61.7 ((R)-17b, C(O)OCH2CH3), 70.5, 70.6, 71.1, 71.2, 71.3 ((R)-17a,b, OCH2-CH2OCH2CH2OCH3), 170.1, 170.4, 170.9 ((R)-17a,b, CH3C(O)NH, C(O)OCH3), the remaining resonances were not detected and are believed to overlap with nearby signals; Compound (R)-17a was not detected by HRMS; (R)-17b: Mr (+ESI) 300.1421 [M+Na]+ (calcd for C12H23NO6Na+ 300.1423 [M+Na]+).
4.11. Synthesis of (R)-methyl 2-acetamido-3-isopropoxypropionate ((R)-18a) and (R)-ethyl 2-acetamido-3-isopropoxypropionate ((R)-18b)
Using Method A, a ∼9:1 mixture of (R)-14a and (R)-14b (3.40 g, 23.5 mmol), and BF3·Et2O (2.95 mL, 23.5 mmol) in iPrOH (30 mL) gave 2.98 g (62%) of (R)-18a and (R)-18b as a pale yellow oil: Rf = 0.46 ((R)-18a), 0.48 ((R)-18b) (5:95 hexanes/EtOAc); IR (neat) 3295, 3062, 2972, 2877, 1748, 1663, 1538, 1442, 1375, 1212, 1147 cm−1. Spectral data for (R)-18a (∼90 mol percent based on 1H NMR integrations): 1H NMR (CDCl3) δ 1.10 (d, J = 6.0 Hz, CH2OCHCH3(C′H3)), 1.12 (d, J = 6.0 Hz, CH2OCHCH3(C′H3)), 2.06 (s, CH3C(O)NH), 3.55 (hept, J = 6.0 Hz, CH2OCH(CH3)2), 3.64 (dd, J = 3.7, 9.3 Hz, CHCHH′OCH(CH3)2), 3.76 (s, C(O)OCH3), 3.84 (dd, J = 3.7, 9.3 Hz, CHCHH′OCH(CH3)2), 4.72 (app dt, J = 3.7, 7.2 Hz, CHCH2OCH(CH3)2), 6.41 (br d, J = 7.2 Hz, CH3C(O)NH); 13C NMR (CDCl3) δ 21.5 (OCHCH3(C′H3)), 21.6 (OCHCH3(C′H3)), 22.9 (CH3C(O)), 52.1 (CHCH2OCH(CH3)2 or C(O)OCH3), 52.6 (C(O)OCH3 or CHCH2OCH(CH3)2), 67.5 (CHCH2OCH(CH3)2), 70.2 (CHCH2OCH(CH3)2), 169.6, 170.4 (CH3C(O)NH, C(O)OCH3); Mr (+ESI) 226.1054 [M+Na]+ (calcd for C9H17NO4Na+ 226.1055 [M+Na]+).
Spectral data for (R)-18b (∼10 mol percent based on 1H NMR integrations): 1H NMR (CDCl3) δ 1.16 (d, J = 6.0 Hz, CH2OCHCH3(C′H3)), 1.21 (d, J = 6.0 Hz, CH2OCHCH3(C′H3)), 1.28 (t, J = 6.0 Hz, C(O)OCH2CH3), 1.99 (s, CH3C(O)NH), 4.06−4.12 (m, CHCHH′OCH(CH3)2), 4.18−4.26 (m, C(O)OCH2CH3), 5.95−6.10 (br m, CH3C(O)NH), the remaining signals were not detected and are believed to overlap with (R)-18a signals; 13C NMR signals were not detected for (R)-18b; Mr (+ESI) 240.1211 [M+Na]+ (calcd for C10H19NO4Na+ 240.1212 [M+Na]+).
4.12. Synthesis of (R)-methyl 2-acetamido-3-tert-butoxypropionate ((R)-19a) and (R)-ethyl 2-acetamido-3-tert-butoxypropionate ((R)-19b)
Using Method A, a ∼9:1 mixture of (R)-14a and (R)-14b (3.50 g, 24.2 mmol), and BF3·Et2O (3.05 mL, 24.2 mmol) in tert-BuOH (30 mL) gave 2.75 g (52%) of (R)-19a and (R)-19b as a pale yellow oil: Rf = 0.52 ((R)-19a), 0.54 ((R)-19b) (5:95 hexanes/EtOAc); IR (neat) 3298, 3062, 2974, 1749, 1663, 1537, 1370, 1204, 1098 cm−1. Spectral data for (R)-19a (∼90 mol percent based on 1H NMR integrations): 1H NMR (CDCl3) δ 1.14 (s, CH2OC(CH3)3), 2.06 (s, CH3C(O)NH), 3.56 (dd, J = 3.0, 9.0 Hz, CHCHH′OC(CH3)3), 3.76 (s, C(O)OCH3), 3.81 (dd, J = 3.0, 9.0 Hz, CHCHH′OC(CH3)3), 4.72 (app dt, J = 3.0, 7.2 Hz, CHCH2OC(CH3)3), 6.41 (br d, J = 7.2 Hz, CH3C(O)NH); 13C NMR (CDCl3) δ 22.9 (CH3C(O)), 27.7 (OC(CH3)3), 52.1 (CHCH2OC(CH3)3 or C(O)OCH3), 53.3 (C(O)OCH3 or CHCH2OCH(CH3)2), 62.4 (CHCH2OC(CH3)3), 73.8 (CHCH2OC(CH3)3), 170.3, 171.5 (CH3C(O)NH, C(O)OCH3); Mr (+ESI) 240.1211 [M+Na]+ (calcd for C10H19NO4Na+ 240.1212 [M+Na]+).
Spectral data for (R)-19b (∼10 mol percent based on 1H NMR integrations): 1H NMR (CDCl3) δ 1.21 (s, CH2OC(CH3)3), 1.99 (s, CH3C(O)NH), 4.18−4.24 (m, C(O)OCH2CH3), 5.95−6.15 (br m, CH3C(O)NH), the remaining signals were not detected and are believed to overlap with (R)-19a signals or are too small to be detected; 13C NMR signals were not detected for (R)-19b; Mr (+ESI) 254.1368 [M+Na]+ (calcd for C11H21NO4Na+ 254.1368 [M+Na]+).
4.13. Synthesis of (R)-methyl 2-acetamido-3-phenoxypropionate ((R)-20a) and (R)-ethyl 2-acetamido-3-phenoxypropionate ((R)-20b)
Using Method A, a ∼3:7 mixture of (R)-14a and (R)-14b (2.00 g, 13.1 mmol), phenol (3.95 g, 42.0 mmol), and BF3·Et2O (1.6 mL, 13.1 mmol) in CH2Cl2 (20 mL) gave 1.40 g (43%) of (R)-20a and (R)-20b as a slight yellow residue: Rf = 0.50 ((R)-20a), 0.52 ((R)-20b) (5:95 hexanes/EtOAc); IR (neat) 3067, 2984, 1743, 1660, 1596, 1541, 1498, 1379, 1296, 1238, 1159 cm−1; Spectral data for (R)-20a and (R)-20b (∼3:7): 1H NMR (CDCl3) δ 1.25 ((R)-20b, t, J = 7.2 Hz, C(O)OCH2CH3), 2.06 ((R)-20a,b, s, CH3C(O)NH), 3.76 ((R)-20a, s, C(O)OCH3), 4.20−4.23 ((R)-20a,b, m, CHCHH′OPh), 4.24 ((R)-20b, q, J = 7.2 Hz, C(O)OCH2CH3), 4.36−4.43 ((R)-20a,b, m, CHCHH′OPh), 4.68−5.06 ((R)-20a,b, m, CHCH2OPh), 6.52 ((R)-20a,b, d, J = 7.2 Hz, CH3C(O)NH), 6.84−6.90 ((R)-20a,b, m, 2 ArH (o)), 6.95−7.00 ((R)-20a,b, m, ArH (p)), 7.24−7.32 ((R)-20a,b, m, 2 ArH (m)); 13C NMR (CDCl3) δ 14.3 ((R)-20b, C(O)OCH2CH3), 23.1 ((R)-20a,b, CH3C(O)), 52.4, 52.5, 52.9 ((R)-20a,b, CHCH2OPh, (R)-20a, C(O)OCH3), 62.1 ((R)-20b, C(O)OCH2CH3), 68.1, 68.2 ((R)-20a,b, CHCH2OPh), 114.8, 121.7, 129.7, 158.3, 158.4 ((R)-20a,b, CH2OPh), 170.0, 170.1, 170.5 ((R)-20a,b, CH3C(O)NH, (R)-20a C(O)OCH3), the remaining signals were not detected and are believed to overlap with nearby peaks; Compound (R)-20a, Mr (+ESI) 268.0895 [M+Na]+ (calcd for C12H15NO4Na+ 260.0899 [M+Na]+); Compound (R)-20b, Mr (+ESI) [M+Na]+ (calcd for C12H15NO4Na+ 274.1055 [M+Na]+) 274.1052.
4.14. Synthesis of (R)-2-acetamido-3-methoxypropionic acid ((R)-21)
Using Method B, a mixture of (R)-15a and (R)-15b (3.79 g, 21.5 mmol) in THF (210 mL), and LiOH (515 mg, 21.5 mmol) in H2O (100 mL) gave 1.31 g (38%) of (R)-21 as a white solid after work-up and recrystallization from EtOAc: mp 108−109 °C; −20.9° (c 0.65, MeOH) (lit.17 −16.9° (c 1.2; MeOH)) for a partially racemized sample (∼4:1, (R)to (S))); Rf = 0−0.1 (EtOAc); IR (nujol mull) 3352, 3100−2200, 1746, 1631, 1549, 1459, 1375 cm−1; 1H NMR (DMSO-d6) δ 1.86 (s, CH3C(O)), 3.25 (s, CH2OCH3), 3.49 (dd, J = 3.9, 10.0 Hz, CHH′OCH3), 3.63 (dd, J = 6.0, 10.0 Hz, CHH′OCH3), 4.36−4.45 (m, CHCH2O), 8.20 (d, J = 7.2 Hz, CH3C(O)NH), 12.7 (s, CO2H); 13C NMR (DMSO-d6) δ 22.3 (CH3C(O)), 52.1 (CHCH2OCH3), 58.3 (OCH3), 71.8 (CHCH2OCH3), 169.4, 171.7 (CHCO2H, CH3C(O)NH). (R)-21 was not detected by HRMS. Anal. Calcd for C6H11NO4: C, 44.72; H, 6.88; N, 8.69. Found: C, 44.75; H, 6.82; N, 8.77.
4.15. Synthesis of (R)-2-acetamido-3-ethoxypropionic acid ((R)-22)
Using Method B, a mixture of (R)-16a and (R)-16b (2.48 g, 13.0 mmol) in THF (130 mL), and LiOH (312 mg, 13.0 mmol) in H2O (65 mL) gave 1.57 g (69%) of (R)-22 as a white solid after work-up and recrystallization from EtOAc: mp 149−151 °C; −31.5° (c 0.70, MeOH); Rf = 0−0.15 (5:95 hexanes/EtOAc); IR (nujol mull) 3355, 3300−2100 (br), 1951, 1747, 1630, 1545, 1457, 1374, 1204, 1107 cm−1; 1H NMR (DMSO-d6) δ 1.09 (t, J = 6.9 Hz, OCH2CH3), 1.86 (s, CH3C(O)), 3.39−3.49 (m, CH2OCH2CH3), 3.53 (dd, J = 4.2, 10.0 Hz, CHH′OCH2CH3), 3.63 (dd, J = 6.0, 10.0 Hz, CHH′OCH2CH3), 4.36−4.42 (m, CHCH2O), 8.15 (d, J = 7.2 Hz, CH3C(O)NH), the carboxylic acid proton could not be detected; 13C NMR (DMSO-d6) δ 14.9 (OCH2CH3), 22.3 (CH3C(O)), 52.4 (CHCH2OCH3), 65.8 (OCH2CH3), 69.6 (CHCH2OCH2CH3), 169.4, 171.7 (CHCO2H, CH3C(O)NH); Mr (+ESI) 214.0480 [M+K]+ (calcd for C7H13NO4K+ 214.0482 [M+K]+). Anal. Calcd for C7H13NO4: C, 47.99; H, 7.48; N, 8.00. Found: C, 48.21; H, 7.56; N, 7.95.
4.16. Synthesis of (R)-2-acetamido-3-(2-(2-methoxyethoxy)ethoxy)propionic acid ((R)-23)
Using Method B, a mixture of (R)-17a and (R)-17b (3.85 g, 14.2 mmol) in THF (160 mL), and LiOH (376 mg, 15.6 mmol) in H2O (80 mL) gave 2.44 g (68%) of (R)-23 as a colorless viscous oil after work-up: −38.5° (c 1.15, CHCl3); Rf = 0−0.11 (10:90 MeOH/CHCl3); IR (neat) 3500−2500 (br), 1974, 1731, 1654, 1547, 1103 cm−1; 1H NMR (CDCl3) δ 2.08 (s, CH3C(O)), 3.40 (s, OCH3), 3.54−3.70 (m, OCH2CH2OCH2CH2OCH3), 3.75 (dd, J = 3.3, 9.7 Hz, CHH′OCH2), 3.98 (dd J = 3.3, 9.7 Hz, CHH′OCH2), 4.68−4.78 (m, CHCH2O), 6.98 (d, J = 7.8 Hz, C(O)NHCH), 9.10−9.50 (br s, C(O)OH); 13C NMR (CDCl3) δ 22.9 (CH3C(O)), 53.0 (CHCH2OCH2CH2), 59.0 (OCH3), 70.3, 70.4, 70.7, 70.8, 72.1 (CH2OCH2CH2OCH2CH2O), 171.4, 172.3 (C(O)OH, CH3C(O)NH); Mr (+ESI) 272.1107 [M+Na]+ (calcd for C18H20N2O3Na+ 272.1110 [M+Na]+). Anal. Calcd for C10H9NO6·0.33H2O: C, 46.50; H, 7.81; N, 5.42. Found: C, 46.34; H, 7.74; N, 5.46.
4.17. Synthesis of (R)-2-acetamido-3-isopropoxypropionic acid ((R)-24)
Using Method B, a mixture of (R)-18a and (R)-18b (2.47 g, 12.0 mmol) in THF (120 mL), and LiOH (288 mg, 12.0 mmol) in H2O (60 mL) gave 2.15 g (95%) of (R)-24 as a white solid after work-up. Recrystallization from EtOAc and hexanes afforded an analytical sample: mp 128−130 °C; −36.5° (c 0.60, MeOH); Rf = 0.05−0.18 (5:95 hexanes/EtOAc); IR (nujol mull) 3366, 3300−2100 (br), 1751, 1636, 1548, 1457, 1376, 1328 cm−1; 1H NMR (DMSO-d6) δ 1.06 (d, J = 6.0 Hz, OCHCH3(C′H3)), 1.07 (d, J = 6.0 Hz, OCHCH3(C′H3), 1.87 (s, CH3C(O)), 3.49−3.50 (m, CHH′OCH(CH3)2), 3.64 (dd, J = 5.7, 9.9 Hz, CHH′OCH(CH3)2), 4.33−4.40 (m, CHCH2O), 8.10 (d, J = 7.2 Hz, CH3C(O)NH), 12.70 (CO2H); 13C NMR (DMSO-d6) δ 21.8 (OCHCH3(C′H3)), 21.9 (OCHCH3(C′H3), 22.4 (s, CH3C(O)), 52.6 (CHCH2OCH(CH3)2), 67.4 (CH2OCH(CH3)2), 71.3 (CH2OCH(CH3)2), 169.4, 171.8 (CH3C(O)NH, CO2H); Mr (+ESI) 212.0897 [M+Na]+ (calcd for C8H15NO4Na+ 212.0899 [M+Na]+). Anal. Calcd for C8H15NO4: C, 50.78; H, 7.99; N, 7.40. Found: C, 50.87; H, 8.02; N, 7.34.
4.18. Synthesis of (R)-2-acetamido-3-tert-butoxypropionic acid ((R)-25)
Using Method B, a mixture of (R)-19a and (R)-19b (2.17 g, 9.90 mmol) in THF (100 mL), and LiOH (237 mg, 9.90 mmol) in H2O (50 mL) gave 1.10 g (55%) of (R)-25 as a white solid after work-up. Recrystallization from EtOAc and hexanes afforded an analytical sample: mp 154−156 °C; −46.7° (c 0.8, MeOH); Rf = 0.48 (5:95 hexanes/EtOAc); IR (nujol mull) 3370, 3300−2100 (br), 1875, 1708, 1613, 1542, 1459, 1371, 1229, 1103 cm−1; 1H NMR (DMSO-d6) δ 1.11 (s, OC(CH3)3), 1.87 (s, CH3C(O)), 3.47 (dd, J = 4.2, 9.3 Hz, CHH′OC(CH3)3), 3.60 (dd, J = 5.1, 9.3 Hz, CHH′OC(CH3)3), 4.30−4.38 (m, CHCH2O), 8.01 (d, J = 7.2 Hz, CH3C(O)NH), 12.6 (CO2H); 13C NMR (DMSO-d6) δ 22.4 (s, CH3C(O)), 27.2 (OC(CH3)3), 52.8 (CHCH2OC(CH3)3), 61.7 (CH2OC(CH3)3), 72.8 (CHCH2OC(CH3)3), 169.4, 171.9 (CH3C(O)NH, CO2H); Mr (+ESI) 242.0794 [M+K]+ (calcd for C9H17NO4K+ 242.0795 [M+K]+). Anal. Calcd for C9H17NO4: C, 53.19; H, 8.43; N, 6.89. Found: C, 53.04; H, 8.49; N, 6.84.
4.19. Synthesis of (R)-2-acetamido-3-phenoxypropionic acid ((R)-26)
Using Method B, a mixture of (R)-20a and (R)-20b (1.4 g, 5.7 mmol) in THF (60 mL), LiOH (142 mg, 5.9 mmol) in H2O (30 mL) gave 950 mg (74%) of (R)-26 after recrystallization: mp 168−169.5 °C; −91.2° (c 0.5, MeOH); Rf = 0−0.15 (10:90 MeOH/CHCl3); IR (nujol mull) 3362, 2300−2800 (br), 1950, 1746, 1607, 1551, 1461 cm−1; 1H NMR (DMSO-d6) δ 1.90 (s, CH3C(O)), 4.13 (dd, J = 3.9, 9.6 Hz, CHH′OPh), 4.37 (dd J = 5.1, 9.6 Hz, CHH′OPh), 4.60−4.68 (m, CHCH2O), 6.88−6.98 (m, OC6H5, (o) and (p)), 7.24−7.32 (m, OC6H5, (m)), 8.42 (d, J = 7.5 Hz, NHCHCH2), 12.50−13.00 (br, C(O)OH); 13C NMR (DMSO-d6) δ 22.3 (CH3C(O)), 51.8 (CHCH2OPh), 67.6 (CH2OPh), 114.6 (OC6H5, (o)), 121.0 (OC6H5,(p)), 129.8 (OC6H5, (m)), 158.1 (OC6H5, (i)), 169.5, 171.3 (CH3C(O)NH, CH3C(O)OH); Mr (+ESI) 246.0739 [M+Na]+ (calcd for C11H13NO4Na+ 246.0742 [M+Na]+). Anal. Calcd for C11H13NO4·0.25H2O: C, 58.02; H, 5.98; N, 6.15. Found: C, 58.00; H, 5.98; N, 5.91.
4.20. Synthesis of (R)-2-acetamido-N-benzyl-3-methoxypropionamide ((R)-1)
Using Method C, (R)-21 (100 mg, 0.62 mmol), benzylamine (81 μL, 0.74 mmol), and DMTMM (205 mg, 0.74 mmol) in anhydrous THF (10 mL) gave 95 mg (61%) of (R)-1 after flash column chromatography (10:90 MeOH/CHCl3) and recrystallization from EtOAc: mp 142−143 °C (lit.17 mp 142−143 °C); +16.1° (c 0.9, MeOH) (lit.17 +16.0° (c 1.0; MeOH)); Rf = 0.47 (10:90 MeOH/CHCl3); 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 3.37 (s, CH2OCH3), 3.46 (dd, J = 7.2, 8.4 Hz, CHH′OCH3), 3.79 (dd J = 4.2, 8.4 Hz, CHH′OCH3), 4.40−4.52 (m, NHCH2C6H5), 4.52−4.60 (m, CHCH2O), 6.40−6.60 (br m, CH3C(O)NH), 6.78−6.92 (br m, C(O)NHCH2Ph), 7.18−7.38 (m, NHCH2C6H5), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-1 gave only one signal for the acetyl methyl protons and the methoxy protons, addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of authentic (S)-1 and (R)-1 (1:2 ratio) gave two signals for the acetyl methyl protons (δ 2.023 (S) and 2.010 (R)(Δppm = 0.013)), and two signals the methoxy protons (δ 3.311 (S) and 3.350 (R)(Δppm = 0.039)); 13C NMR (CDCl3) δ 23.4 (CH3C(O)), 43.7 (NHCH2Ph), 52.6 (CHCH2OCH3), 59.3 (CH2OCH3), 71.9 (CH2OCH3), 127.6, 127.7, 138.1 (NHCH2C6H5), 170.2, 170.5 (CHC(O)NH, CH3C(O)NH), the remaining aromatic resonance was not detected and is believed to overlap with nearby signals.
4.21. Synthesis of (R)-2-acetamido-N-benzyl-3-ethoxypropionamide ((R)-27)
Using Method C, (R)-22 (1.34 g, 7.7 mmol), benzylamine (1.00 mL, 9.2 mmol), and DMTMM (2.54 g, 9.2 mmol) in anhydrous THF (80 mL) gave 1.11 g (46%) of (R)-27 as a white solid after flash column chromatography (8:92 MeOH/CHCl3) and two recrystallizations from EtOAc: mp 129−130 °C; −34.1° (c 0.64, CHCl3); Rf = 0.35 (5:95 MeOH/CHCl3); IR (nujol mull) 3283, 1634, 1555, 1456, 1375, 1114 cm−1; 1H NMR (CDCl3) δ 1.15 (t, J = 7.2 Hz, OCH2CH3), 2.04 (s, CH3C(O)), 3.44 (dd, J = 8.4, 9.3 Hz, CHH′OCH2CH3), 3.48−3.62 (m, OCH2CH3), 3.85 (dd J = 4.2, 9.3 Hz, CHH′OCH2CH3), 4.40−4.58 (m, CHCH2OCH2, NHCH2C6H5), 6.40−6.50 (br d, CH3C(O)NH), 6.78−6.90 (br t, C(O)NHCH2Ph), 7.22−7.38 (m, NHCH2C6H5), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-27 gave only one signal for the acetyl methyl protons (δ 2.017); 13C NMR (CDCl3) δ 15.1 (OCH2CH3), 23.3 (CH3C(O)), 43.6 (NHCH2Ph), 52.7 (CHCH2OCH2CH3), 67.0 (CHCH2OCH2CH3), 69.9 (CHCH2OCH2CH3), 127.6, 127.7, 128.7, 138.1 (NHCH2C6H5), 170.3, 170.5 (CHC(O)NH, CH3C(O)NH); Mr (+ESI) 287.1374 [M+Na]+ (calcd for C14H20N2O3Na+ 287.1372 [M+Na]+). Anal. Calcd for C14H20N2O3: C, 63.62; H, 7.63; N, 10.60. Found: C, 63.62; H, 7.56; N, 10.47.
4.22. Synthesis of (R)-2-acetamido-N-benzyl-3-(2-(2-methoxyethoxy)ethoxy)propionamide ((R)-28)
Using Method C, (R)-23 (1.67 g, 6.70 mmol), benzylamine (876 μL, 8.04 mmol), and DMTMM (2.22 g, 8.04 mmol) in THF (70 mL) gave a residue that was purified twice by flash chromatography (5:95 MeOH/CHCl3) to yield (R)-28 (1.20 g, 53%) as a yellow oil that progressively turned to an amorphous solid after 3 d under high vacuum: mp 48−52 °C; +7.7° (c 1.18, MeOH); Rf = 0.51 (10:90 MeOH/CHCl3); IR (neat) 3313, 3072, 2921, 2358, 2245, 1657, 1538, 1103 cm−1; 1H NMR (CDCl3) δ 2.02 (s, CH3C(O)), 3.26 (s, OCH3), 3.39−3.80 (m, CHH′OCH2CH2OCH2CH2OCH3), 4.05 (dd, J = 3.9 Hz, 9.9 Hz, CHH′OCH2CH2O), 4.48 (d, J = 6.0 Hz, NHCH2C6H5), 4.54−4.62 (m, CHCH2O), 6.77 (d, J = 6.0 Hz, NHCH2C6H5), 7.20−7.39 (m, C6H5), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-28 gave only one signal for the acetyl peak protons; 13C NMR (CDCl3) δ 23.4 (CH3C(O)), 43.7 (NH2CH2C6H5), 52.5 (CHCH2OCH2CH2), 59.0 (OCH3), 70.4, 70.5, 70.6, 71.9 (OCH2-CH2OCH2CH2O), 127.5, 127.7, 128.8, 137.8 (C6H5), 170.3, 170.4 (CHC(O)NH, CH3C(O)), the remaining signal was not detected and is believed to overlap with nearby peaks; Mr (+ESI) 361.1743 [M+Na]+ (calcd for C17H26N2O5Na+ 361.1739 [M+Na]+). Anal. Calcd for C17H26N2O5·0.33H2O: C, 58.77; H, 7.83; N, 8.06. Found: C, 58.59; H, 7.88; N, 8.10.
4.23. Synthesis of (R)-2-acetamido-N-benzyl-3-isopropoxypropionamide ((R)-29)
Using Method C, (R)-24 (1.90 g, 10.0 mmol), benzylamine (1.31 mL, 12.0 mmol), and DMTMM (3.32 g, 12.0 mmol) in anhydrous THF (100 mL) gave 2.01 g (72%) of (R)-29 as a white solid after flash column chromatography (5:95 MeOH/CHCl3) and recrystallization from EtOAc: mp 151−153 °C; −23.4° (c 0.50, CHCl3); Rf = 0.37 (5:95 MeOH/CHCl3); IR (nujol mull) 3280, 3098, 1642, 1555, 1458, 1377, 1298, 1258, 1147 cm−1; 1H NMR (CDCl3) δ 1.09 (d, J = 6.0 Hz, OCHCH3(C′H3)), 1.13 (d, J = 6.0 Hz, OCHCH3(C′H3)), 2.04 (s, CH3C(O)), 3.40 (app t, J = 8.7 Hz, CHH′OCH(CH3)2), 3.63 (hept, J = 6.0 Hz, OCH(CH3)2), 3.84 (dd J = 3.9, 8.7 Hz, CHH′OCH(CH3)2), 4.38−4.57 (m, CHCH2OCH, NHCH2C6H5), 6.42−6.50 (br d, CH3C(O)NH), 6.82−6.94 (br t, C(O)NHCH2Ph), 7.24−7.38 (m, NHCH2C6H5), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-29 gave only one signal for the acetyl methyl protons (δ 2.014); 13C NMR (CDCl3) δ 22.0 (OCHCH3(C′H3)), 22.2 (OCHCH3(C′H3)), 23.4 (CH3C(O)), 43.7 (NHCH2Ph), 52.9 (CHCH2OCH(CH3)2), 67.5 (CH2OCH(CH3)2), 72.7 (CH2OCH(CH3)2), 127.7, 128.8, 138.1 (NHCH2C6H5), 170.5 (CHC(O)NH or CH3C(O)NH), the remaining aromatic signal was not detected and is believed to overlap with nearby signals, the second C(O) peak was not observed; Mr (+ESI) 301.1530 [M+Na]+ (calcd for C15H22N2O3Na+ 301.1528 [M+Na]+). Anal. Calcd for C15H22N2O3: C, 64.73; H, 7.97; N, 10.06. Found: C, 64.56; H, 8.00; N, 10.12.
4.24. Synthesis of (R)-2-acetamido-N-benzyl-3-tert-butoxypropionamide ((R)-30)
Using Method C, (R)-25 (1.10 g, 5.4 mmol), benzylamine (0.71 mL, 6.5 mmol), and DMTMM (1.80 g, 6.5 mmol) in anhydrous THF (50 mL) gave 730 mg (46%) of (R)-30 as a white solid after flash column chromatography (5:95 MeOH/CHCl3) and recrystallization from EtOAc: mp 126−127 °C; −22.9° (c 0.85, CHCl3); Rf = 0.39 (5/95 MeOH/CHCl3); IR (nujol mull) 3280, 3091, 1641, 1550, 1459, 1372, 1246, 1194, 1090 cm−1; 1H NMR (CDCl3) δ 1.14 (s, OC(CH3)3), 2.04 (s, CH3C(O)), 3.40 (app t, J = 8.5 Hz, CHH′OC(CH3)3), 3.84 (dd, J = 4.2, 8.5 Hz, CHH′OC(CH3)3), 4.38−4.57 (m, CHCH2O, NHCH2C6H5), 6.40−6.50 (br d, CH3C(O)NH), 6.80−6.92 (br t, C(O)NHCH2Ph), 7.23−7.40 (m, NHCH2C6H5), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-30 gave only one signal for the acetyl methyl protons (δ 2.009) and the tert-butoxy methyl protons (δ 1.112); 13C NMR (CDCl3) δ 23.4 (CH3C(O)), 27.6 (OC(CH3)3), 43.8 (NHCH2Ph), 53.2 (CHCH2OC(CH3)3), 61.7 (CH2OC(CH3)3), 74.5 (CH2OC(CH3)3), 127.7, 127.8, 128.8, 138.1 (NHCH2C6H5), 170.3, 170.5 (CHC(O)NH, CH3C(O)NH); Mr (+ESI) 315.1687 [M+Na]+ (calcd for C16H24N2O3Na+ 315.1685 [M+Na]+). Anal. Calcd for C16H24N2O3: C, 65.73; H, 8.27; N, 9.58; Found: C, 65.64; H, 8.08; N, 9.57.
4.25. Synthesis of (R)-2-acetamido-N-benzyl-3-phenoxypropionamide ((R)-31)
Using Method C, (R)-26 (376 mg, 1.68 mmol), benzylamine (219 μL, 2.02 mmol), and DMTMM (557 mg, 2.02 mmol) in THF (20 mL) gave a residue that was purified by flash column chromatography (5:95 MeOH/CHCl3) and further recrystallized from EtOAc to yield (R)-31 (305 mg, 58%) as a white solid: mp 169−170 °C; −18.0° (c 0.4, MeOH); Rf = 0.52 (5/95 MeOH/CHCl3); IR (nujol mull) 3288, 3073, 1687, 1551, 1458, 1375 cm−1; 1H NMR (CDCl3) δ 2.03 (s, CH3C(O)), 4.05 (dd, J = 7.5, 9.6 Hz, CHH′OCH3), 4.37 (dd J = 4.2, 9.6 Hz, CHH′OCH3), 4.40−4.56 (m, NHCH2C6H5), 4.78−4.86 (m, CHCH2O), 6.66 (d, J = 6.0 Hz, CH3C(O)NH), 6.87 (d, J = 7.8 Hz, OC6H5 (o)), 6.98 (t, J = 7.8 Hz, OC6H5 (p)), 7.16−7.35 (m, CH2C6H5 (m) and NHCH2C6H5), addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-31 gave only one signal for the acetyl peak protons; 13C NMR (CDCl3) δ 23.4 (CH3C(O)), 43.9 (NH2CH2Ph), 52.6 (CHCH2OPh), 67.4 (CH2OPh), 114.8 (OC6H5, (o)), 121.9 (OC6H5, (p)), 127.7, 127.8, 128.9 (CH2C6H5), 129.8 (OC6H5, (m)), 137.8 (CH2C6H5), 157.9 (OC6H5, (i)), 169.5, 170.6 (CHC(O)NH, CH3C(O)); Mr (+ESI) 335.1366 [M+Na]+ (calcd for C18H20N2O3Na+ 335.1372 [M+Na]+). Anal. Calcd for C18H20N2O3: C, 69.21; H, 6.45; N, 8.97; Found: C, 69.29; H, 6.52; N, 9.05.
4.26. Pharmacology
Compounds were screened under the auspices of the National Institutes of Health's Anticonvulsant Screening Program. Experiments were performed in male rodents [albino Carworth Farms No. 1 mice (intraperitoneal route, ip), albino Sprague–Dawley rats (oral route, po)]. Housing, handling, and feeding were in accordance with recommendations contained in the ‘Guide for the Care and Use of Laboratory Animals’. Anticonvulsant activity was established using the MES test14,18 and the scMet test,14 using previously reported methods.19,20
Acknowledgments
The authors thank the NINDS and the Anticonvulsant Screening Program (ASP) at the National Institutes of Health for supporting the pharmacological studies via the ASP's contract site at the University of Utah with Drs. H. Wolfe, S. White, M. Franklin, and K. Wilcox. The project described was supported by Grants RO1NS054112 (H.K.), and F31NS060358 (P.M.) from the National Institute of Neurological Disorders and Stroke. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2008.08.055.
References and notes
- 1.Doty P, Rudd GD, Stöhr T, Thomas D. Neurotherapeutics. 2007;4:145–148. doi: 10.1016/j.nurt.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beyreuther BK, Freitag J, Heers C, Krebsfänger N, Scharfenecker U, Stöhr T. CNS Drug Rev. 2007;13:21–42. doi: 10.1111/j.1527-3458.2007.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi D, Stables JP, Kohn H. J. Med. Chem. 1996;39:1907–1916. doi: 10.1021/jm9508705. [DOI] [PubMed] [Google Scholar]
- 4.Choi D, Kohn H. unpublished results.
- 5.Nakajima K, Neya M, Yamada S, Okawa K. Bull. Chem. Soc. Jpn. 1982;55:3049–3050. [Google Scholar]
- 6.Nakajima K, Oda H, Okawa K. Bull. Chem. Soc. Jpn. 1983;56:520–522. [Google Scholar]
- 7.Larsson U, Carlson R. Acta Chem. Scand. 1994;48:511–516. [Google Scholar]
- 8.Bělohradský M, Ridvan L, Závada J. Collect. Czech Chem. Commun. 2003;68:1319–1328. [Google Scholar]
- 9.Kuyl-Yeheskiely E, Dreef-Tromp C, van der Marel G, van Boom J. Recl. Trav. Chim. Pays-Bas. 1989;108:314–316. [Google Scholar]
- 10.Mathieu-Pelta I, Evans SA. J. Org. Chem. 1994;59:2234–2237. [Google Scholar]
- 11.Salomé C, Kohn H. unpublished results.
- 12.Kunishima M, Kawachi C, Monta J, Terao K, Iwasaki F, Tani S. Tetrahedron. 1999;55:13159–13170. [Google Scholar]
- 13.Morrison J, Weisman G. Asymmetric Synthesis—Analytical Methods. Vol. 1. Academic Press; New York: 1983. pp. 153–171. [Google Scholar]
- 14.Stables JP, Kupferberg HG. In: Molecular and Cellular Targets for Antiepileptic Drugs. Avanzini G, Tanganelli P, Avoli M, editors. John Libbey; London: 1977. pp. 191–198. [Google Scholar]
- 15.Porter RJ, Cereghino JJ, Gladding GD, Hessie BJ, Kupferberg HJ, Scoville B, White BG. Cleve. Clin. Q. 1984;51:293–305. doi: 10.3949/ccjm.51.2.293. [DOI] [PubMed] [Google Scholar]
- 16.Dunham NW, Miya TS. J. Am. Pharm. Assoc. 1957;46:208–209. doi: 10.1002/jps.3030460322. [DOI] [PubMed] [Google Scholar]
- 17.Andurkar SV, Stables JP, Kohn H. Tetrahedron: Asymm. 1998;9:3841–3854. [Google Scholar]
- 18.Krall RL, Penry JK, Kupferberg HJ, Swinyard EA. Epilepsia. 1978;19:393–407. doi: 10.1111/j.1528-1157.1978.tb04506.x. [DOI] [PubMed] [Google Scholar]
- 19.LeTiran A, Stables JP, Kohn H. J. Med. Chem. 2002;45:4762–4773. doi: 10.1021/jm020225f. [DOI] [PubMed] [Google Scholar]
- 20.LeTiran A, Stables JP, Kohn H. Bioorg. Med. Chem. 2001;9:2693–2708. doi: 10.1016/s0968-0896(01)00204-8. [DOI] [PubMed] [Google Scholar]