

Abstract
Thyroid hormone receptors (T3Rs) are hormone-regulated transcription factors. Different T3R isoforms are expressed in a tissue-specific and developmentally regulated manner. The T3Rα-1, β-0, and β-1 isoforms typically repress target gene expression in the absence of hormone and activate transcription in the presence of hormone. Intriguingly, however, the T3Rβ-2 isoform fails to repress, and instead is able to activate transcription in both the absence and presence of hormone. We investigated the molecular mechanism behind this absence of repression by T3Rβ-2. Repression by T3Rα-1, β-0, and β-1 is mediated by the ability of these isoforms to physically recruit a SMRT/N-CoR corepressor complex. We determined that the unliganded T3Rβ-2 also recruits the SMRT corepressor; in contrast to the α-1, β-0, and β-1 isoforms, however, the T3Rβ-2 protein interacts not only with the C-terminal “receptor-interaction domain” of SMRT, but also makes additional contacts with the N-terminal “silencing domain” of the SMRT corepressor. These additional, T3Rβ-2-specific contacts interfere with the subsequent association of SMRT with mSin3, a crucial second subunit of the corepressor holo-complex. Our results suggest that T3Rβ-2 regulates transcription through a novel anti-repression mechanism, recruiting SMRT, but preventing the subsequent formation of a functional corepressor complex.
Many key aspects of vertebrate physiology, reproduction, and development are regulated through the actions of small, lipophilic hormones. These hormones include the steroids, retinoids, and thyroid hormones, and mediate their effects through the actions of a corresponding family of nuclear hormone receptors (1-7). The nuclear receptors function as hormone-regulated transcription factors, binding to specific DNA sequences, denoted hormone response elements, and regulating the transcription of adjacent target genes (1-7). Reflecting this common mode of action, nuclear hormone receptors also share a common structural architecture composed of a central DNA binding domain and a C-terminal hormone binding domain, flanked in turn by more divergent N-terminal and “hinge” regions (Fig. 1). Embedded within this global domain structure are additional amino acid motifs responsible for receptor nuclear localization, for transcription regulation, and for interactions of the receptor with a variety of accessory and modulatory proteins (1-7).
Fig. 1. Schematic representations of the different isoforms of thyroid hormone receptor and of the SMRT and mSin3 corepressor proteins.
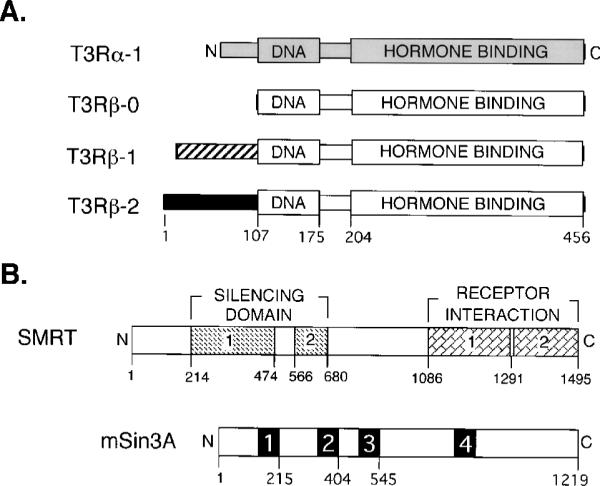
A, schematic representations of the different T3R isoforms are depicted from N to C terminus. The locations of the DNA binding (DNA) and hormone binding (HORMONE BINDING) domains are illustrated and, for the T3Rβ-2 isoform, are defined relative to their positions in the amino acid sequence. Differences in the amino acid sequences of the different isoforms are depicted by alterations in shading. B, schematic representations of the SMRT and mSin3A corepressor proteins are depicted from N to C terminus. The positions of the SMRT domains involved in transcriptional silencing (SILENCING DOMAIN) and of the C-terminal SMRT domains representing the principal sites of interaction with nuclear hormone receptors (RECEPTOR INTERACTION) are shown relative to the amino acid sequence. Both the silencing and receptor interaction domains have been further dissected into two subdomains each, as indicated (25, 27, 35, 37). The locations of presumed paired-amphipathic helices within the mSin3A polypeptide are numbered 1 to 4.
Notably, many nuclear hormone receptors are expressed from more than one locus, and/or are subject to alternative mRNA splicing so as to generate a series of distinct, although interrelated receptor “isoforms.” Thyroid hormone receptors (T3Rs),1 for example, are encoded by two distinct loci, denoted α and β, and each locus can in turn be expressed as alternatively spliced isoforms, α-1 and α-2, or β-0 (in birds), β-1, and β-2 (Fig. 1). Different receptor isoforms are synthesized in a developmentally regulated and cell-specific manner; T3Rβ-2 expression, for example, is restricted principally to the pituitary, hypothalamus, and a few other tissues, whereas the T3Rα-1 and T3Rβ-0/1 isoforms are more broadly expressed (8-14). The specific expression patterns and evolutionary conservation of these multiple isoforms, together with the results of gene ablation experiments, strongly suggest that distinct isoforms perform physiologically distinct functions (1-7).
Many nuclear hormone receptors have bimodal transcriptional properties, and can either repress or activate expression of a given target gene depending on the hormone status, nature of the promoter, and the cell context. Particularly well characterized is the ability of the T3Rα-1, β-0, and β-1 isoforms to repress transcription of many of their target genes in the absence of hormone, and to activate transcription of these same target genes in the presence of hormone (see, e.g., Refs. 15-21). These bipolar regulatory properties are closely linked to the ability of these receptors to recruit accessory proteins, denoted corepressors and coactivators, that help mediate the actual transcriptional response (reviewed in Ref. 22). The molecular mechanisms by which corepressors and coactivators modulate transcription remain to be fully elucidated but appear to involve, at least in part, modifications of the chromatin template (reviewed in Refs. 23 and 24). Thus, in the absence of hormone T3Rs physically bind to corepressor polypeptides denoted SMRT and N-CoR (25-29); SMRT and N-CoR recruit, in turn, a larger protein complex containing mSin3 and histone deacetylase that, by hypoacetylating chromatin, is believed to restrict accessibility of the DNA template to the transcriptional machinery (30-36). Conversely, binding of cognate hormone leads to a conformational change in the T3R that releases the corepressor complex, and recruits instead coactivator complexes, many of which possess histone acetyltransferase activity (16, 17, 20, 22, 25-27, 37-39); histone acetylation is believed to create a more accessible, and therefore more readily transcribed, chromatin structure (22-24).
Different receptor isoforms can display distinct functions. For example, in contrast to the bipolar regulatory properties of the T3Rα-1, β-0, and β-1 isoforms described above, the T3Rβ-2 isoform does not repress, and can even activate, certain promoters in the absence of hormone; addition of thyroid hormone simply further enhances this hormone-independent activation (21, 40). We wished to better understand why the unliganded T3Rβ-2 isoform fails to repress transcription. We report here that T3Rα-1, T3Rβ-0, T3Rβ-1, and T3Rβ-2 all strongly interact with the SMRT corepressor in the absence of hormone; the T3R-β2 isoform, however, makes additional contacts with domains of the SMRT corepressor that are not contacted by the T3Rα-1, T3Rβ-0, or T3Rβ-1 forms. These additional T3Rβ-2 contacts overlap the silencing domains of the SMRT polypeptide and inhibit the ability of the SMRT protein to recruit the mSin3 subunit of the corepressor complex. Conversely, disruption of this inhibitory interaction of the T3Rβ-2 N terminus with the SMRT silencing domain, using any of a variety of experimental modalities, converts the unliganded T3Rβ-2 into a transcriptional repressor, analogous in its properties to those of the T3Rα, β-0, or β-1 isoforms. Our results therefore suggest a novel “anti-repression” model in which the T3Rβ-2 receptor recruits SMRT, but prevents SMRT-mediated repression by preventing assembly of a functional corepressor complex. This model of anti-repression may extend to other members of the nuclear hormone receptor family, or to other families of transcriptional repressors that also mediate their actions through the SMRT/N-CoR corepressor complex.
EXPERIMENTAL PROCEDURES
Protein-Protein Binding Assays in Vitro
Glutathione S-transferase (GST) fusions were constructed by using a pGEX-KG vector and inserting target DNA representing full-length TFIIB, codons 1−214 of mSin3A, codons 404−545 of mSin3A, codons 1−101 of T3Rβ-1, codons 1−107 of T3Rβ-2, or various domains of SMRT (35, 41, 42). The recombinant plasmids were transformed into Escherichia coli strain DH5α, and the resulting GST fusion proteins were purified and immobilized by binding to glutathione-conjugated agarose beads (41). [35S]Methionineradiolabeled proteins were synthesized by employing pSG5-, pCMV-, or pVZ-based plasmids in a coupled in vitro transcription/translation (TnT) system (Promega). The 35S-labeled proteins were subsequently incubated with 25 μl of the GST protein-agarose matrix for 2 h at 4 °C in 300 μl of HEMG buffer (40 mm HEPES (pH 7.8), 50 mm KCl, 0.2 mm EDTA, 5 mm MgCl2, 0.1% Triton X-100, 10% glycerol, 1.5 mm dithiothreitol, 1× Complete Proteinase Inhibitor (Roche Molecular Biochemicals), and 10 mg/ml bovine serum albumin) (27, 35, 42, 43). The agarose matrix was then washed four times with 500 μl each of HEMG buffer lacking proteinase inhibitors and bovine serum albumin. Radiolabeled proteins remaining bound to the matrix were eluted with 10 mm free glutathione and were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (27, 35, 42, 43). The electrophoretograms were visualized and quantified by PhosphorImager analysis (Molecular Dynamics Storm System).
For competition experiments codons 1−101 of T3Rβ-1, codons 1−107 of T3Rβ-2, or codons 404−545 of mSin3A were inserted into pMAL-c2. The plasmids were introduced into E. coli DH5α and the resulting maltose-binding protein (MBP) fusion polypeptides were induced and were purified using the manufacturer's protocol (New England Biolabs).
Co-immunoprecipitation Assay
COS-1 cells were transfected by a LipofectAMINE Plus (Life Technologies, Inc.) methodology. Approximately 2.5 × 105 cells were transfected with 750 ng of a pCR-SMRT plasmid and a pSG5-GAL4AD-HA plasmid containing no insert, codons 1−101 of T3Rβ-1, or codons 1−107 of T3Rβ-2. The transfected cells were incubated for 36 h and harvested into lysis buffer (1× phosphate-buffered saline, 1 mm EDTA, 1.5 mg/ml iodoacetamide, 0.5% Triton X-100, 0.2 mm phenylmethylsulfonyl fluoride, and Complete Protease Inhibitor (Roche Molecular Biochemicals)). The cell lysates were clarified by centrifugation, incubated with anti-HA-directed antibody (Santa Cruz Biotechnology) for 1 h at 4 °C, 20 μl of protein A-Sepharose was added to each sample, and the incubation was continued for 1 h. The immune complex was collected by centrifugation, extensively washed in lysis buffer, and analyzed by SDS-PAGE and Western blotting, using a rabbit polyclonal antibody directed against the SMRT C-terminal domain.
Construction of Mutants of T3Rβ-2
NotI site substitution mutants of T3Rβ-2 were constructed by a polymerase chain reaction protocol using standard recombinant DNA techniques (44). Oligonucleotides were devised to substitute four native codons, spaced every 10 amino acids within the T3Rβ-2 N terminus, with a GCT GCG GCC GCT sequence, representing the introduction of four alanines and a novel NotI restriction site. Subsequent NotI cleavage and ligation permitted the generation of defined deletions between any two of the alanine substitution mutations. Creation of the Y61F/Y65F double substitution mutation within the T3Rβ-2 N terminus was through an analogous oligonucleotide-directed, polymerase chain reaction technique (44).
Transient Transfections
Transfections of CV-1 cells were performed by a liposome/Lipofectin methodology (Life Technologies, Inc.). Approximately 5 × 104 cells were transfected with 20 ng of a pSG5-T3Rα, pSG5-T3Rβ-0, pSG5-T3Rβ-1, or pSG5-T3Rβ-2 plasmid, 50 ng of pCH110 as an internal control, and 100 ng of a pTK-DR4-luciferase reporter (43, 44). For certain experiments, a pSG5-mSin3A expression vector was also incorporated. Additional pUC18 plasmid was used to normalize the total DNA to 500 ng/transfection. The cells were placed into either hormone-depleted medium, or medium containing 100 nm T3-thyronine and incubated at 37 °C for 48 h. The cells were subsequently harvested, and the luciferase activity was determined relative to β-galactosidase activity (42-44).
RESULTS
The T3Rβ-2 Isoform Does Not Repress Target Gene Expression in the Absence of Hormone
It has been reported that the neither the avian nor the mammalian β-2 isoform of T3R represses target gene expression, but instead either has no effect on target gene transcription in the absence of hormone, or displays a hormone-independent transcriptional activation that is further enhanced on addition of hormone (21, 40). To confirm this phenomenon, we employed transient transfections of CV-1 cells to examine the ability of the different T3R isoforms to modulate expression of a suitable T3R-responsive luciferase reporter gene. CV-1 cells contain little or no endogenous T3Rs, and exhibited little or no hormone regulation of luciferase expression when this reporter construct was introduced alone (Fig. 2). As expected from prior work (21), introduction of the T3Rα-1, β-0, or β-1 isoforms led to repression of reporter gene expression in the absence of hormone (Fig. 2, A and B). In clear contrast, however, introduction of the T3Rβ-2 isoform did not repress reporter gene expression in the absence of hormone; instead, the unliganded T3Rβ-2 either had no effect on luciferase expression or had a slight stimulatory activity when compared with the basal levels of reporter expression observed in the absence of an introduced receptor (Fig. 2 and data not shown). Also as expected, addition of hormone led to stimulation of reporter gene expression by all the T3R isoforms tested (Fig. 2B and data not shown). Our results confirm that the T3Rβ-2 isoform exhibits unique transcriptional regulatory properties, and in contrast to other T3R isoforms, fails to repress transcription in the unliganded state.
Fig. 2. Lack of repression by T3Rβ-2.

A, T3Rβ-2 fails to repress in the absence of hormone. Either an empty pSG5 vector (vector) or a pSG5 vector expressing one of the T3R isoforms, as indicated below the panel, was introduced into CV-1 cells together with a thymidine kinase promoter-luciferase reporter containing a DR-4 thyroid hormone response element; a pCH110 lacZ plasmid was also included as an internal control. The cells were incubated in thyroid hormone-depleted medium for 48 h, harvested, and the luciferase activity was determined relative to the β-galactosidase activity. The data represent the average and standard deviation of from three to eight experiments. B, T3Rβ-2 activates in the presence of hormone. A similar experiment as in panel A was performed but either in the absence (solid fill) or presence (diagonal fill) of 100 nm T3-thyronine hormone. A horizontal line is drawn to permit comparison to the basal activity of the reporter (i.e. relative luciferase activity in the absence of a transfected T3R).
T3Rβ-2 Interacts with SMRT Corepressor, but Makes Novel Contacts with the SMRT Silencing Domain Not Observed for T3Rα-1, T3Rβ-0, or T3Rβ-1
We wished to determine the molecular basis for the failure of the T3Rβ-2 isoform to repress gene transcription. We first tested if the T3Rβ-2 isoform was able to interact with the SMRT corepressor, given that the recruitment of SMRT is crucial for repression by the other T3R isoforms (25-29, 45). We initially employed a “GST-pull-down” methodology. GST fusions, representing different domains of SMRT, were immobilized by binding to glutathione agarose. The various immobilized SMRT derivatives were then incubated with the radiolabeled T3R, and the T3R protein bound to the SMRT matrix was subsequently eluted, detected by SDS-PAGE, and quantified by PhosphorImager analysis (Fig. 3).
Fig. 3. Binding of different isoforms of T3R to different domains of SMRT.

Each T3R isoform, indicated to the left of the panels, was synthesized as a full-length radiolabeled protein by in vitro transcription and translation and was tested for the ability to bind to different GST-SMRT fusion constructs, immobilized on glutathioneagarose. Non-recombinant GST was employed as a negative control (top panel). Radiolabeled receptor that bound to each GST construct in the absence (solid fill) or presence (diagonal fill) of 100 nm T3 thyronine hormone was subsequently eluted, was resolved by SDS-PAGE, and was quantified by PhosphorImager analysis. The results of a representative experiment are presented. Note the change in scale between the top three panels and the bottom panel.
Although unable to repress in vivo, the T3Rβ-2 isoform nonetheless interacted strongly with the C terminus of SMRT (codons 1086−1495) (Fig. 3); this region of SMRT has previously been identified as the principal site of interaction of corepressor with the nuclear hormone receptors (Fig. 1). The SMRT/T3Rβ-2 interaction was comparable in intensity to those observed between SMRT and T3Rα-1, T3Rβ-0, or T3Rβ-1 (Fig. 3). Also in common with these other isoforms, the interaction of the T3Rβ-2 with the SMRT (codons 1055−1495) domain was inhibited by the addition of thyroid hormone (Fig. 3; compare solid to diagonal fill bars). None of the T3R isoforms tested interacted with a non-recombinant GST construct employed as a negative control, or with a GST fusion representing an internal domain (codons 751−1074) of SMRT (Fig. 3). Intriguingly, however, the T3Rβ-2 isoform did bind to a domain within the N-terminal domain of SMRT that was not contacted by the other T3R isoforms. This novel, T3Rβ-2-specific contact mapped to amino acids 566−680 of SMRT (Fig. 3) and overlaps a region in the N terminus of SMRT crucial for SMRT-mediated transcriptional silencing (Fig. 1).
The Novel SMRT Contacts Observed for T3Rβ-2 Are Mediated by the Unique N Terminus of This Receptor Isoform
It would appear likely that the ability of T3Rβ-2, but not β-0 or β-1, to interact with the SMRT silencing domain reflects the divergent nature of the N termini of the different isoforms, given that the DNA-binding and hormone-binding domains of these isoforms are identical (Fig. 1). Notably, the N terminus of T3Rβ-0 is highly truncated relative to that of β-2 (Fig. 1), suggesting that the T3Rβ-2 N terminus might serve to stabilize the interaction with the SMRT silencing domain, rather than the N terminus of the other isoforms acting to inhibit this interaction. Supporting this conclusion, an artificially truncated T3Rβ2 product, lacking an N-terminal domain (Fig. 4, denoted T3Rβ2-t), failed to bind to the SMRT (codons 566−680) silencing domain, whereas the full-length T3Rβ-2 translation product, employed in the same assay, did bind to the SMRT silencing domain (Fig. 4). As expected, both the full-length and truncated T3Rβ2 forms bound to the SMRT C-terminal (codons 1086−1495) domain (Fig. 4); this interaction is mediated by the C-terminal receptor domain that is retained in both T3Rβ-2 and T3Rβ2-t.
Fig. 4. Differential SMRT binding by full-length versus artificially truncated T3Rβ-2.

The in vitro translation system was employed to produce both full-length receptor (T3Rβ2) and an internally initiated truncated derivative (T3Rβ2-t; Ref. 46) lacking the native N terminus (note 10% input lane). The ability of the full-length and truncated receptor derivatives to bind to non-recombinant GST, or to the various GST-SMRT fusions was determined by the protocol described in Fig. 3. A reproduction of the resulting PhosphorImager scan is depicted above, and a quantification of the results, expressed as the percentage of radiolabeled receptor bound to each GST fusion relative to the amount of the same polypeptide in the original input, is presented below. A representative experiment is presented.
Is the T3Rβ-2 N terminus alone sufficient for this interaction with the SMRT silencing domain? Consistent with this proposal, radiolabeled constructs limited to either the first 70 or first 107 amino acids of T3Rβ-2 were able to bind to the GST-SMRT silencing domain, but not to a non-recombinant GST construct employed as a negative control (Fig. 5). The T3Rβ-2 N terminus also exhibited a weaker interaction with the C terminus of SMRT, but not with GST alone or with an internally derived SMRT domain (Fig. 5). In contrast, the N terminus of the β-1 T3R isoform did not interact with any region of SMRT (Fig. 5). The ability of the N terminus of T3Rβ-2, but not of T3Rβ-1, to interact strongly with SMRT was also observed in a reciprocal experiment using SMRT as the radiolabeled protein and GST fusions representing the T3R N termini (Fig. 6A); a construct representing the N-terminal silencing domains of SMRT was sufficient for this interaction (Fig. 6B). The ability of the T3Rβ-2 N terminus to interact with SMRT extended further to co-transfection experiments in mammalian cells; immunoprecipitation of an HA-tagged construct representing the T3Rβ-2 N terminus efficiently co-precipitated the SMRT protein from lysates of transfected COS-1 cells, whereas an HA-tagged T3Rβ-1 N terminus construct did not (Fig. 6C). We conclude that the receptor N terminus mediates the contacts that the T3Rβ-2 isoform makes with the SMRT silencing domain, whereas the N termini of the other T3Rβ isoforms do not interact with SMRT.
Fig. 5. Binding of the abstracted N terminus of T3Rβ-2 to the SMRT corepressor.

The same type of in vitro binding experiment as in Fig. 3 was repeated, but employing radiolabeled receptor derivatives restricted to the N-terminal domain of each T3R isoform. The receptor derivatives tested included: amino acids 1−107 of T3Rβ-2, amino acids 1−70 of T3Rβ-2, or amino acids 1−101 of T3Rβ-1, as indicated to the left of the panels. The radiolabeled protein bound to each GST-fusion was eluted, was resolved by SDS-PAGE, and was visualized by PhosphorImager analysis. Quantification of protein binding, relative to input, was as in Fig. 4.
Fig. 6. Binding of radiolabeled SMRT and N-CoR proteins to the T3Rβ-2 N terminus.

A, the similar experiment as in Fig. 5 was repeated but in a reciprocal fashion, assaying the ability of full-length radiolabeled SMRT or N-CoR corepressor proteins to bind to GST fusions representing the N-terminal domain of T3Rβ-1 (amino acids 1−101) or T3Rβ2 (amino acids 1−107), as indicated above the panel. Non-recombinant GST was employed as a negative control. The radiolabeled corepressor proteins bound to each GST-fusion were eluted, resolved by SDS-PAGE, and visualized by PhosphorImager analysis. Adjacent lanes represent duplicate experiments. Quantification of protein binding, relative to input, was as in Fig. 4. B, the experiment in panel A was repeated, except using in vitro translation products restricted to the N-terminal silencing domains of SMRT (amino acids 1−680) or N-CoR (amino acids 1−2129, plus six C-terminal amino acids retained as a result of the construction), as indicated. C, an expression vector, either empty (−) or containing the N termini of T3Rβ-1 or β-2 linked to an HA-epitope tag as indicated above the panel, were introduced by transfection into COS-1 cells, together with a SMRT expression vector. The cells were subsequently lysed, the lysates were immunoprecipitated using anti-HA antibodies, and the immunoprecipitates were analyzed by a Western blot method using anti-SMRT antibodies (upper panel). A Western blot analysis of the total lysates, using anti-SMRT antibodies, is presented below.
We next extended these experiments to N-CoR; N-CoR is a second corepressor polypeptide that is related to SMRT both structurally and functionally (26, 28). The T3Rβ-2 N terminus interacted with full-length N-CoR in a similar fashion as it interacted with SMRT, i.e. radiolabeled N-CoR bound in vitro to GST fusions representing the T3Rβ-2 N terminus, but not to GST fusions representing the T3Rβ-1 N terminus, or to non-recombinant GST used as a negative control (Fig. 6A). As with SMRT, the N-terminal “silencing region” of N-CoR was sufficient for this interaction with the T3Rβ-2 N terminus (Fig. 6B).
The Interaction of T3Rβ-2 with the SMRT Silencing Domain Inhibits the Ability of SMRT to Recruit mSin3A
An important component of SMRT repression is mediated through the ability of the SMRT silencing domain to physically interact with mSin3, which in turn helps tether the histone deacetylases and additional corepressor components that serve as the actual effectors of transcriptional silencing (reviewed in Refs. 23 and 24). Might the interaction of the T3Rβ-2 N terminus with the SMRT silencing domain interfere with these downstream aspects of SMRT-mediated repression? We first tested if the T3R-β2 N terminus inhibited the ability of SMRT to recruit the mSin3 corepressor subunit. The primary in vitro interaction site between SMRT and mSin3A overlaps the PAH-3 domain of the latter (Fig. 1); as previously reported, radiolabeled SMRT was strongly bound by a GST-mSin3A construct representing this mSin3A domain, but not by a non-recombinant GST construct or by other mSin3A domains employed as negative controls (Fig. 7A and Ref. 35). Notably, addition of increasing amounts of the N-terminal domain of T3Rβ-2, synthesized as a MBP fusion, strongly inhibited this interaction between SMRT and mSin3A (Fig. 7A, solid fill); in fact, the MBP-T3Rβ-2 N terminus polypeptide more effectively interfered with the mSin3A/SMRT interaction than did a MBP-mSin3A polypeptide used in a homologous competition (Fig. 7A). In contrast, equivalent amounts of the non-recombinant MBP or an MBP fused to the N terminus of T3Rβ-1 had no effect on the SMRT/mSin3A interaction (Fig. 7A).
Fig. 7. Inhibition of mSin3A binding to SMRT and N-CoR by the T3Rβ-2 N-terminal domain.

A, the binding of mSin3A to SMRT is inhibited by the T3Rβ-2 N-terminal domain. Radiolabeled SMRT, synthesized in vitro, was incubated with a GST-mSin3A (codons 404−545) fusion protein immobilized on glutathione agarose. Included in the incubation was 0 (none), 10, 100, or 1000 ng of a MBP fusion polypeptide containing the N-terminal 107 amino acids of T3Rβ-2 (T3Rβ-2), a non-recombinant MBP polypeptide (MBP-only), a MBP fusion polypeptide containing the N-terminal 101 amino acids of T3Rβ-1, or a MBP-mSin3A (amino acids 404−545) construct, as indicated. Radiolabeled SMRT bound to the GST-mSin3A was subsequently eluted, resolved by SDS-PAGE, and quantified by PhosphorImager analysis. The background binding of SMRT to a non-recombinant GST matrix was subtracted from each value presented. B, the binding of mSin3A to N-CoR is inhibited by the T3Rβ-2 N-terminal domain. A similar experiment as in panel A was performed, but employing radiolabeled N-CoR and a mSin3A (codons 1−215) construct. C, the binding of SMRT to TFIIB is not inhibited by the T3Rβ-2 N-terminal domain. A similar experiment as in panel A was performed, but employing radiolabeled TFIIB together with the GST-SMRT (amino acids 95−680) construct.
A analogous inhibitory effect of the T3Rβ-2 N terminus, but not that of β-1 or of MBP alone, was observed in reciprocal experiments using radiolabeled full-length mSin3A and a GST-SMRT construct (data not shown). Consistent with the inhibition being mediated by a direct interaction between the T3Rβ-2 N terminus and the SMRT silencing domain, inhibition of the SMRT/mSin3A interaction by the T3Rβ-2 N terminus was not observed with deleted versions of SMRT that are unable to bind to the T3Rβ-2 N terminus (data not shown).
The SMRT-related N-CoR corepressor displays interactions with two regions of mSin3A in vitro: the PAH-3 domain interaction also observed for SMRT, and an additional interaction with the mSin3A N-terminal PAH-1 domain (Fig. 1). We therefore tested the effect of the T3Rβ-2 N terminus on the ability of N-CoR to tether mSin3A. The T3Rβ-2 N terminus strongly interfered with the ability of N-CoR to interact with the PAH-1 domain (Fig. 7B) and somewhat more modestly with the ability of N-CoR to interact with the PAH-3 (data not shown). Neither the T3Rβ-1 N terminus nor the MBP-alone constructs demonstrated this interference (Fig. 7B).
Although the ability of SMRT to recruit mSin3 plays an important role in repression, inhibitory interactions of SMRT with the general transcriptional machinery, including TFIIB, may also contribute to transcriptional silencing under certain conditions (35, 47). This SMRT/TFIIB interaction is mediated by the same silencing region of SMRT as implicated in the mSin3 interaction (35). We therefore next examined if the T3Rβ-2 N terminus also interferes with the SMRT/TFIIB interaction. Radiolabeled TFIIB bound readily to a GST-SMRT construct under our GST-pull down conditions (Fig. 7C and Ref. 35). In contrast to our results with mSin3A, co-introduction of the T3Rβ-2 N terminus had no demonstrable effect on the SMRT/TFIIB interaction (Fig. 7C). We conclude that the effects of the T3Rβ-2 N terminus are specific, in that the β-2 N terminus interferes with the ability of SMRT to recruit mSin3, but not TFIIB.
Disruption of the Ability of T3Rβ-2 to Interact with the SMRT Silencing Region Converts T3Rβ-2 into a Transcriptional Repressor
Our results suggest that the N terminus of T3Rβ-2 competes with mSin3A for occupancy of the SMRT silencing domain. If so, then overexpression of mSin3A in transfected cells might outcompete the effects of the β-2 N terminus and confer on T3Rβ-2 the ability to repress transcription. Consistent with this hypothesis, the introduction of increasing amounts of an mSin3A expression vector into CV-1 cells led to conversion of the unliganded T3Rβ-2 from an activator to a strong repressor of reporter gene transcription (Fig. 8A). In contrast, overexpression of mSin3A had little or no effect on the repression observed for the unliganded T3Rβ-0 or T3Rβ-1 isoforms (Fig. 8A). Similar results were observed with a different reporter based on the “negative response element” in the TSH-α promoter; overexpression of mSin3A interfered with reporter activation in the absence of hormone by T3Rβ-2, but not by β-0 (data not shown).
Fig. 8. Effects of disrupting the interaction of the T3Rβ-2 N terminus with SMRT.

A, overexpression of mSin3A converts unliganded T3Rβ-2 into a repressor. The transfection experiment in Fig. 2A was repeated, except introducing different quantities of a pSG-5 mSin3A expression vector, as indicated below the panel. The average and range of two experiments are presented. B, deletions in the receptor N terminus inhibit binding of SMRT by T3Rβ-2. A series of in-frame deletions were created in the N terminus of T3Rβ-2; the codons encompassed by each deletion are indicated below the panel. Each mutant receptor, synthesized by in vitro transcription and translation, was tested for the ability to bind to a GST-SMRT(aino acids 566−680) fusion protein as in Fig. 3. Binding of wild-type T3Rβ-2 was defined as 100. The average and range of duplicate experiments are presented. C, deletions in the receptor N terminus convert T3Rβ-2 into a repressor. The T3Rβ-2 mutants described in panel B were tested for the ability to repress reporter gene transcription in the absence of hormone, as in Fig. 2. Results are presented for the various T3Rβ-2 deletions and the wild-type T3Rβ-2, as indicated below the panel. Parallel transfections using an empty vector or wild-type T3β-0 are included for comparison. The average and standard deviation of multiple experiments are shown.
To approach the same question by genetic means, we mapped the SMRT interaction domain within the N terminus of T3Rβ-2 by creating a series of deletion mutations within the receptor. We first examined the effect of these mutations on the ability of the encoded protein to bind to the SMRT silencing domain (Fig. 8B); a Δ(6−60) or Δ(6−70) deletion abolished virtually all interaction of the receptor with the SMRT silencing domain (Fig. 8B). We next employed transient transfections to establish if mutations that interfered with the T3Rβ-2/SMRT silencing domain interaction also altered the transcriptional properties of the receptor. Notably, the T3Rβ-2 N-terminal deletions that severely inhibited the interaction with the SMRT silencing domain in vitro, i.e. Δ(6−60) or Δ(6−70), also converted the β-2 isoform into a repressor in the absence of hormone, resulting in a phenotype indistinguishable from that observed with the wild-type T3Rα, β-0, and β-1 isoforms (Fig. 8C). We conclude that there is a close correspondence between the ability of the T3Rβ-2 N terminus to contact the silencing domain of SMRT and the absence of repression by T3Rβ-2.
A short region in the N terminus of T3Rβ-2, centered on tyrosines 61 and 65, can act as a transcriptional activator when abstracted from the receptor and fused to a GAL4-DNA binding domain; the integrity of these tyrosines, in particular, is crucial for this transcriptional activation function (21). We therefore examined if these two tyrosines contributed to the SMRT interaction/anti-repression phenomenon we report here. In contrast to their contribution to transcriptional activation in the GAL4DBD fusion, conversion of these tyrosines to phenylalanines had no observable effect either on the ability of the β-2 receptor to interact with SMRT in vitro or on the absence of transcriptional repression by T3Rβ-2 in transient transfections (Table I). We conclude that, although mapping to an overlapping region, the lack of repression observed for the T3Rβ-2 isoform can be distinguished from the transcriptional activation domain previously mapped to this region of the receptor.
Table I. Comparison of SMRT binding and transcriptional repression properties of wild-type and mutant T3Rβ-2 proteins.
Binding to the SMRT (codons 566−680) domain was determined by a GST “pull-down” protocol as in Fig. 3. Transcriptional repression was determined by transient transfections, as in Fig. 2. -Fold repression is defined as the relative luciferase activity observed with the empty pSG5 vector, divided by the relative luciferase activity observed in the presence of the T3R allele indicated; values below 1.0 represent activation.
| T3R allele | Binding to codons 566−680 of SMRT | Repression in absence of T3 |
|---|---|---|
| -fold | ||
| T3Rβ-0 | <1.0 | 1.85 ± 0.43 |
| T3Rβ-2 wild-type | 6.1 ± 1.3 | 0.82 ± 0.28 |
| T3Rβ-2 (Y61F/Y65F) mutant | 6.5 ± 0.4 | 0.86 ± 0.19 |
DISCUSSION
The T3Rβ-2 Isoform Recruits SMRT Corepressor, but Makes Additional Corepressor Contacts Not Mediated by the Other T3R Isoforms
The T3Rα-1, β-0, and β-1 isoforms can repress transcription in the absence of hormone, reflecting the ability of these receptors to recruit a SMRT/N-CoR corepressor complex to the target gene promoter (23-29, 48, 49). The goal of the experiments reported here was to determine why the T3Rβ-2 isoform, in contrast to these other T3R isoforms, does not repress transcription (21). The simplest hypothesis, that the T3Rβ-2 isoform is unable to recruit the SMRT corepressor, proved not to be true. On the contrary, T3Rβ-2 bound to SMRT with the same or greater overall avidity as did the T3Rα-1, β-0, and β-1 isoforms. Also in common with these other isoforms, the primary T3Rβ-2 interaction with SMRT was hormone-labile and was mediated by contacts between the receptor hormone binding domain and an interaction domain previously identified within the C terminus of SMRT. However, the T3Rβ-2 isoform made additional strong contacts with the N-terminal region of SMRT that were not observed for the T3R isoforms that are able to confer repression.
These novel contacts of T3Rβ-2 with SMRT were mediated by the N-terminal domain of the receptor, a region that, although highly conserved between the avian and mammalian T3Rβ-2 isoforms, diverges extensively in sequence from the N termini of T3Rα-1, β-0, and β-1 (3, 8, 50). This novel SMRT interaction domain of T3Rβ-2 mapped to a region spanning the first 60−70 amino acids of this receptor. Sequence analysis failed to elucidate any identifiable amino acid motif within this N-terminal domain, nor was the sequence of the β-2 N terminus recapitulated in any other protein currently in the data base.2 There was also no detectable sequence relatedness between the SMRT interaction domain within the T3Rβ-2 N terminus and the previously identified SMRT interaction domains located in the C-terminal domains of the nuclear hormone receptors. We conclude that the N terminus of the T3Rβ-2 isoform possesses a novel SMRT interaction motif that is not found in receptors that are capable of mediating repression.
The N Terminus of T3Rβ-2 Interacts with a Region of SMRT Essential for Transcriptional Repression and Can Interfere with Assembly of a Corepressor Complex
Two adjacent domains within SMRT, denoted silencing domain (SD)-1 and SD-2, play important roles in corepressor-mediated transcriptional repression (25-27, 31, 33, 35, 45). Mutations in either of these silencing domains impair the ability of SMRT to function as a corepressor and deletion of both SD-1 and SD-2 results in virtually all loss of SMRT-mediated transcriptional repression (25-27, 31, 33, 35, 45). SD-1 is known to participate in at least two interactions that are believed to contribute to the silencing phenotype. The first, and more extensively characterized, is the ability of SD-1 to bind to mSin3, which in turn recruits histone deacetylase and other critical components of the corepressor complex (31, 33, 35, 45). The ability to assemble this SMRT/mSin3/HDAC complex is closely linked to the ability of SMRT to repress, and abridging the assembly or actions of this complex can impair or prevent transcriptional repression (31, 33, 35, 45). It was therefore provocative that the site of interaction of the T3Rβ-2 N terminus on SMRT overlaps the SD-2 region, and maps close to the SD-1 region. Might the T3Rβ-2 N terminus be able to inhibit SMRT function by blocking the actions of these SMRT silencing domains? In fact, introduction of the T3Rβ-2 N terminus strongly interferes with the ability of SMRT to recruit the mSin3 polypeptide in vitro. A similar phenomenon is also observed with N-CoR. In contrast, introduction of the T3Rβ-1 N terminus, or of other non-receptor proteins, has no effect on the SMRT/mSin3 or N-CoR/mSin3 interaction.
In addition to recruitment of mSin3, the SMRT SD-1 domain is also able to interact with TFIIB. Although less well characterized than the mSin3 interaction, this and other interactions with the general transcriptional machinery may also contribute to SMRT-mediated silencing, at least in certain contexts, by interfering with assembly of the transcriptional preinitiation complex (35, 47). This interaction of SMRT with TFIIB may contribute to transcriptional repression by functioning in conjunction with the mSin3/HDAC complex, or conceivably may mediate aspects of SMRT repression in contexts where the mSin3/HDAC complex is inoperative. Although the T3Rβ-2 N terminus inhibited the ability of the SMRT SD-1 to recruit mSin3, it did not detectably inhibit the ability of the same SMRT SD-1 to interact with TFIIB. These results raise the intriguing possibility that SMRT tethered to T3Rβ-2 may still be able to interact with, and repress the function of, TFIIB. Therefore, T3Rβ-2 may potentially be capable of repression in certain promoter or cell contexts where the SMRT-TFIIB interaction, rather than the SMRT/mSin3/HDAC interaction, plays the predominant role.
We Propose an Anti-repression Model for the T3Rβ-2 Isoform, by Which This Receptor Tethers, but Prevents the Function of, the SMRT Corepressor Complex
In summary, we suggest that the unliganded T3Rβ-2 fails to repress transcription not due to a failure of this isoform to recruit SMRT, but due to the ability of the T3Rβ-2 N terminus to make additional, inhibitory contacts with the silencing domain of SMRT. These inhibitory contacts are proposed to block, in all or in part, the proper association of SMRT with mSin3, and thereby abort the formation of a functional corepressor complex. Consistent with this hypothesis, interfering with the ability of the T3Rβ-2 N terminus to interact with the SMRT silencing domain converts the unliganded T3Rβ-2 into a transcriptional repressor. For example, overexpression of mSin3A in transfected cells converts the unliganded T3Rβ-2 from a modest transcriptional activator into a strong repressor, presumably by competing with the T3Rβ-2 N terminus for SMRT occupancy. In contrast, mSin3A overexpression has no effect on T3R isoforms with N termini that fail to interact with SMRT and that already function as repressors. Similarly, N-terminal mutations that disrupt the ability of the T3Rβ-2 to interact with the SMRT silencing domain also convert the T3Rβ-2 phenotype to that of T3Rα, β-0, or β-1: repression in the absence, and activation in the presence, of hormone.
It should be noted that the unliganded T3Rβ-2 protein not only fails to repress, but can actually stimulate target gene expression in certain contexts, and ligand-independent activation functions have been previously mapped within the N terminus of the T3Rβ-2 isoform (21, 51). Our own experiments demonstrate that these hormone-independent activation domains exist, but are distinct in their location and/or their properties from the anti-repression domains we report here. In particular, tyrosine 61 and 65, previously identified as required for transcriptional activation by the T3Rβ-2 N terminus (21), neither played a role in the SMRT interaction described here nor were necessary for anti-repression. Similarly, a second activation function within the T3Rβ-2 N terminus, important in regulation of negative response elements, maps to amino acids 89−116 and therefore lies outside of the anti-repression determinants analyzed here (51).3 Perhaps the anti-repression properties of the T3Rβ-2 N terminus contribute to receptor physiology by permitting these hormone-independent activation functions to be manifested by the unliganded T3Rβ-2 despite the concomitant presence of the tethered SMRT protein. Therefore, we suggest that the unique transcriptional properties of the T3Rβ-2 isoform are actually a sum of the combined functions of the anti-repression, hormone-independent activation, and hormone-dependent activation domains of the receptor; the combinatorial outcome of these distinct functions may be different in different cell types and on different target promoters.
Our anti-repression model is consistent with previous reports establishing a role for the T3Rβ-2 N terminus in the unique transcriptional properties of this isoform (51). Furthermore, by proposing that corepressor tethered to T3Rβ-2 is rendered functionally inert by the β-2 N terminus, our work helps explain the previous, and apparently paradoxical observation that N-CoR corepressor physically associates with T3Rβ-2, but does not detectably influence the transcriptional properties of this isoform (40). It is also of note that T3Rβ-2 exhibits unique regulatory properties on “negative” hormone response elements, a distinct subclass of DNA binding sites that are turned off, rather than on, by hormone (40, 52). It will be interesting to determine if the contributions of the T3Rβ-2/SMRT interactions described here also influence these negative thyroid response elements.
The T3Rβ-2 isoform exhibits a highly tissue-specific expression pattern and possesses unique regulatory properties (21, 40, 51, 52). It appears that the lack of repression observed for this isoform, mediated in part by the anti-repression model proposed here, reflects the unique physiological role of this isoform. The anti-repression model also suggests a possible mechanism by which other transcription factors that operate through the SMRT corepressor complex may conceivably modulate their own transcriptional silencing properties. The PLZF and BCL-6 transcriptional repressors, for example, all display multiple points of contact with SMRT/N-CoR that include interactions with the SMRT silencing domain (see, e.g, Refs. 43 and 53). More work will be necessary to establish if anti-repression plays a role in the functions of these non-receptor transcription factors, and in the actions of other members of the nuclear hormone receptor family.
Acknowledgments
We sincerely thank R. M. Evans and B. Vennstrom for providing the mammalian and avian T3R molecular clones adapted for use in this research, C. Glass and M. G. Rosenfeld for providing the N-CoR molecular clone, and M. A. Lazar for providing the GAL4 expression and reporter vectors.
Footnotes
This work was supported in part by Public Health Services/National Institutes of Health Grants R37 CA-53394 and R01 DK-53528. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: T3R, thyroid hormone receptor; GST, glutathione S-transferase; SD, silencing domain; MBP, maltose-binding protein; HA, hemagglutinin; PAGE, polyacrylamide gel electrophoresis; TFIIB, transcription factor IIB.
Z. Yang, S.-H. Hong, and M. L. Privalsky, unpublished observations.
Z. Yang, unpublished observations.
REFERENCES
- 1.Beato M, Herrlich P, Schutz G. Cell. 1995;83:851–858. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 2.Kastner P, Mark M, Chambon P. Cell. 1995;83:859–870. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- 3.Lazar MA. Endocrinol. Rev. 1993;14:184–193. doi: 10.1210/edrv-14-2-184. [DOI] [PubMed] [Google Scholar]
- 4.Mangelsdorf DJ, Evans RM. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 5.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. Cell. 1995;83:835–840. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribeiro RC, Apriletti JW, West BL, Wagner RL, Fletterick RJ, Schaufele F, Baxter JD. Ann. N. Y. Acad. Sci. 1993;758:366–389. doi: 10.1111/j.1749-6632.1995.tb24843.x. [DOI] [PubMed] [Google Scholar]
- 7.Tsai MJ, O'Malley BW. Annu. Rev. Biochem. 1994;63:451–483. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 8.Forrest D, Sjoberg M, Vennstrom B. EMBO J. 1990;9:1519–1528. doi: 10.1002/j.1460-2075.1990.tb08270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodin RA, Lazar MA, Wintman BI, Darling DS, Koenig RJ, Larsen PR, Moore DD, Chin WW. Science. 1989;244:76–78. doi: 10.1126/science.2539642. [DOI] [PubMed] [Google Scholar]
- 10.Lechan RM, Qi Y, Berrodin TJ, Davis KD, Schwartz HL, Strait KA, Oppenheimer JH, Lazar MA. Endocrinology. 1993;132:2461–24698. doi: 10.1210/endo.132.6.7684976. [DOI] [PubMed] [Google Scholar]
- 11.Lechan RM, Qi Y, Jackson IMD, Mahdavi V. Endocrinology. 1994;135:92–100. doi: 10.1210/endo.135.1.7516871. [DOI] [PubMed] [Google Scholar]
- 12.Li M, Boyages SC. Brain Res. 1997;773:125–131. doi: 10.1016/s0006-8993(97)00917-7. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz HL, Lazar MA, Oppenheimer JH. J. Biol. Chem. 1994;269:24777–24782. [PubMed] [Google Scholar]
- 14.Sjoberg M, Vennstrom B, Forrest D. Development. 1992;114:39–47. doi: 10.1242/dev.114.1.39. [DOI] [PubMed] [Google Scholar]
- 15.Baniahmad A, Kohne AC, Renkawitz R. EMBO J. 1992;11:1015–1023. doi: 10.1002/j.1460-2075.1992.tb05140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baniahmad A, Leng X, Burris TP, Tsai SY, Tsai MJ, O'Malley BW. Mol. Cell. Biol. 1995;15:76–86. doi: 10.1128/mcb.15.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casanova J, Helmer E, Selmi-Ruby S, Qi JS, Au-Flieger M, Desai-Yajnik V, Koudinova N, Yarm F, Raaka BM, Samuels HH. Mol. Cell. Biol. 1994;14:5756–5765. doi: 10.1128/mcb.14.9.5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Damm K, Thompson CC, Evans RM. Nature. 1989;339:593–597. doi: 10.1038/339593a0. [DOI] [PubMed] [Google Scholar]
- 19.Sap J, Munoz A, Schmitt H, Stunnenberg H, Vennstrom B. Nature. 1989;340:242–244. doi: 10.1038/340242a0. [DOI] [PubMed] [Google Scholar]
- 20.Schulman IG, Juguilon H, Evans RM. Mol. Cell. Biol. 1996;16:3807–3813. doi: 10.1128/mcb.16.7.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sjoberg M, Vennstrom B. Mol. Cell. Biol. 1995;15:4718–4726. doi: 10.1128/mcb.15.9.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung L. Mol. Endocrinol. 1996;10:1167–1177. doi: 10.1210/mend.10.10.9121485. [DOI] [PubMed] [Google Scholar]
- 23.Pazin MJ, Kadonaga JT. Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- 24.Wolffe AP. Nature. 1997;387:16–17. doi: 10.1038/387016a0. [DOI] [PubMed] [Google Scholar]
- 25.Chen JD, Evans RM. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 26.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamel Y, Soderstrom M, Glass CK, Rosenfeld MG. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 27.Sande S, Privalsky ML. Mol. Endocrinol. 1996;10:813–825. doi: 10.1210/mend.10.7.8813722. [DOI] [PubMed] [Google Scholar]
- 28.Seol W, Mahon MJ, Lee YK, Moore DD. Mol. Endocrinol. 1996;10:1646–1655. doi: 10.1210/mend.10.12.8961273. [DOI] [PubMed] [Google Scholar]
- 29.Zamir I, Harding HP, Atkins GB, Horlein A, Glass CK, Rosenfeld MG, Lazar MA. Mol. Cell. Biol. 1996;16:5458–5465. doi: 10.1128/mcb.16.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alland L, Muhle R, Hou H, Potes J, Chin L, Schreiber-Agus N, DePinho RA. Nature. 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- 31.Bagy L, Kao H-Y, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 32.Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE. Cell. 1997;89:341–347. doi: 10.1016/s0092-8674(00)80214-7. [DOI] [PubMed] [Google Scholar]
- 33.Heinzel T, Lavinsky RM, Mullen T-M, Soderstrom M, Laherty CD, Torchia J, Yang W-M, Brard G, Ngo SG, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 34.Laherty CD, Yang W-M, Sun J-M, Davie JR, Seto E, Eisenman RN. Cell. 1997;89:349–356. doi: 10.1016/s0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- 35.Wong C-W, Privalsky ML. Mol. Cell. Biol. 1998;18:5500–5510. doi: 10.1128/mcb.18.9.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Iratni R, Erdjument-Bromage H, Tempst P, Reinberg D. Cell. 1997;89:357–364. doi: 10.1016/s0092-8674(00)80216-0. [DOI] [PubMed] [Google Scholar]
- 37.Chen JD, Umesono K, Evans RM. Proc. Natl. Acad. Sci. U. S. A. 1996;93:7567–7571. doi: 10.1073/pnas.93.15.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin BC, Hong S-H, Krig S, Yoh SM, Privalsky ML. Mol. Cell. Biol. 1997;17:6131–6138. doi: 10.1128/mcb.17.10.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner RL, Apriletti JW, McGrath ME, West BL, Baxter JD, Fletterick RJ. Nature. 1995;378:690–697. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- 40.Hollenberg AN, Monden T, Madura JP, Lee K, Wondisford FE. J. Biol. Chem. 1996;271:28516–28520. doi: 10.1074/jbc.271.45.28516. [DOI] [PubMed] [Google Scholar]
- 41.Guan KL, Dixon JE. Anal. Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 42.Wong C-W, Privalsky ML. Mol. Cell. Biol. 1998;18:5724–5733. doi: 10.1128/mcb.18.10.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong S-H, David G, Wong C-W, Dejean A, Privalsky ML. Proc. Natl. Acad. Sci. U. S. A. 1997;94:9028–9033. doi: 10.1073/pnas.94.17.9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoh SM, Chatterjee VKK, Privalsky ML. Mol. Endocrinol. 1997;11:470–480. doi: 10.1210/mend.11.4.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Leo C, Schroen DJ, Chen JD. Mol. Endocrinol. 1997;11:2025–2037. doi: 10.1210/mend.11.13.0028. [DOI] [PubMed] [Google Scholar]
- 46.Bigler J, Eisenman RN. Mol. Cell. Biol. 1993;8:4155–4161. doi: 10.1128/mcb.8.10.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muscat GE, Burke LJ, Downes M. Nucleic Acids Res. 1998;26:2899–2907. doi: 10.1093/nar/26.12.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zamir I, Zhang J, Lazar MA. Genes Dev. 1997;11:835–846. doi: 10.1101/gad.11.7.835. [DOI] [PubMed] [Google Scholar]
- 49.Zhang J, Zamir I, Lazar MA. Mol. Cell. Biol. 1997;17:6887–6897. doi: 10.1128/mcb.17.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Showers MO, Darling DS, Keifer GD, Chin WW. DNA Cell Biol. 1991;10:211–221. doi: 10.1089/dna.1991.10.211. [DOI] [PubMed] [Google Scholar]
- 51.Langlois M-F, Zanger K, Monden T, Safer JD, Hollenberg AN, Wondisford FE. J. Biol. Chem. 1997;272:24927–24933. doi: 10.1074/jbc.272.40.24927. [DOI] [PubMed] [Google Scholar]
- 52.Safer JD, Langlois MF, Cohen R, Monden T, John-Hope D, Maduare J, Hollenberg AN, Wondisford FE. Mol. Endocrinol. 1997;11:16–26. doi: 10.1210/mend.11.1.9867. [DOI] [PubMed] [Google Scholar]
- 53.Dhordain PO, Albagli O, Lin RJ, Ansieau S, Quief S, Leutz A, Kerckaert JP, Evans RM, Leprince D. Proc. Natl. Acad. Sci. U. S. A. 1997;94:10762–10767. doi: 10.1073/pnas.94.20.10762. [DOI] [PMC free article] [PubMed] [Google Scholar]