

Abstract
We report here a class of thiazolidine-2,4-diones and 2-thioxothiazolidin-4-ones as potent inhibitors of the lymphoid specific tyrosine phosphatase (Lyp) identified from high throughput screens. Chemical modification by incorporating known phosphotyrosine (pTyr) mimics led to discovery of a salicylate-based inhibitor with submicromolar potency.
Keywords: Thiazolidine, Lyp, pTyr mimics, salicylate
The lymphoid specific phosphatase (Lyp), encoded by the PTPN22 gene, plays an important role in T cell signaling. A single-nucleotide polymorphism in PTPN22 is associated with autoimmune disorders such as type 1 diabetes, rheumatoid arthritis, systemic lupus erythematosus and Grave's disease. The autoimmunity-predisposing allele corresponds to a gain-of-function mutation, producing a variant phosphatase that is more active than the wild type enzyme in inhibiting T cell signaling.1,2 Inhibition of Lyp therefore provides a novel approach to the treatment of a broad spectrum of autoimmune diseases.
To identify small molecule inhibitors of Lyp, high-throughput screens were performed at Columbia University as part of the Molecular Library Screening Center Network (MLSCN) of the NIH Roadmap for Medical Research. The Lyp assay3 was established using a bacterially expressed fully active 62 KDa N-terminal catalytic domain of Lyp. Compounds were assayed for their capacity to inhibit Lyp-catalyzed conversion of DiFMUP(6,8-difluoro-4-methylumbeliferyl phosphate) to a fluorescent product. Fluorescence with excitation at 360nm and emission at 465 nm was recorded at the endpoint of the reaction. From the compound libraries provided by NIH, a variety of active hits were identified through the high throughput screens and confirmed manually by the dose-response assays. Intriguingly, a number of these compounds share a thiazolidine core structure and thus form an active cluster, providing a starting point for the assessment of structure-activity relationships and medicinal chemistry efforts (Figure 1).
Figure 1.

An active cluster of Lyp inhibitors
Protein tyrosine phosphatases (PTPases) share a highly conserved active site, the phosphotyrosine (pTyr) binding pocket that is the main target for PTPase inhibitor design. Thus, most inhibitors share a pharmacophore structurally similar to the pTyr substrate. Effective pTyr mimics are often charged bidentate anions that competitively bind to the highly polarized pTyr pocket. Several classes of mimics have been reported,4 including the difluoromethylenephosphonates (DFMP) and benzoic acids such as 2-(oxalylamino)-benzoic acids (OBA), salicylic acids (SA) and its derivatives (Figure 2).
Figure 2.

The phosphotyrosine(pTyr) mimic
We noticed that several of the thiazolidine hits contained benzoic acid moiety, and thus could be replaced with a pTyr-mimicking fragment. Fragment-based drug design is a new approach that has been successfully applied to challenging targets.5 This strategy allows hits to be optimized by combining and linking different fragments. We reasoned that combining a known pTyr surrogate with the thiazolidine core structure we identified from high throughput screens might result in synergistically improved potency. Salicylic acid and its derivatives, which are potent pTyr surrogates, were chosen as our initial building blocks for novel Lyp inhibitors.
As shown in Scheme 1, a series of thiazolidine-dione and 2-thioxothiazolidin-4-one compounds with an appended salicylic moiety were designed and synthesized.6 Dioxothiazolidinyl esters 3a-3d were synthesized from thiazolidine-2, 4-dione 1 which was first converted to potassium salt 2 by potassium hydroxide and subsequently alkylated with methyl (tert-butyl) bromoacetate in acetone at 50°C, or methyl (tert-butyl) bromopropionate in dimethylformamide at 90°C in the presence of potassium iodide. The acids 4a-4b were obtained by the treatment of the tert-butyl esters 3c-3d with trifluoroacetic acid. Suzuki coupling of boronic acid 5a-5b with 5-iodo-2-hydroxybenzoate 6a-6b conveniently yielded salicylic acid derived aldehydes 7a-7d, which were condensed with thiazolidine-diones 3a,3c, 4a, 4b and commercially available 2-thioxothiazolidin-4-ones 4c-4d in toluene to afford the final product 8a-8s in yields of 72-85%.
Scheme 1.

The synthesis of Lyp inhibitors 8a-8s
A triacid analog 11 was synthesized according to Scheme 2.6 Alkylation of compound 7a with 2-bromoacetate gave aldehyde 9, followed by deprotection of methyl ester with lithium hydroxide to yield aldehyde 10. Similar condensation of aldehyde 10 and 4d readily afforded the desired product 11 in a total yield of 62% over three steps.
Scheme 2.

The synthesis of the triacid analog 11
To further modify the salicyclic compounds, Schiff base analogs with the thiazolidinedione head group replaced with the hydantoin ring were synthesized.6 As outlined in Scheme 3, simply mixing aldehydes with the hydrochloride salt of 1-aminohydantoin in ethanol readily gave the corresponding Schiff bases 12a-12d in quantitative yields. The synthesis of 17a and 17b started from protection of 1-aminohydantoin with benzaldehyde by forming the imine 13. Deprotonation of 13 followed by alkylation with bromoacetate provided the imidyl esters 15a and 15b. Simultaneous deprotection of ester and imine by refluxing in hydrochloric acid afforded the corresponding amino acid 16. Condensation between the hydantoin-derived amino acid and aldehydes 7c and 7d furnished the Schiff base analogs 17a and 17b in good yields.
Scheme 3.

The synthesis of Schiff base analogs
A total of 25 salicylic compounds were synthesized, and their ability to inhibit Lyp was tested in vitro with IC50s determined from the dose response assays. Results for thiazolidine compounds are summarized in Table 1, while the results for Schiff base analogs are shown in Table 2. 17 compounds showed reasonable activities against Lyp. Among them, compound 8p was shown to be a submicromolar inhibitor with an impressive IC50 of 0.39 μM. Several structure-activity relationships could be observed from these data. The presence of a free carboxyl group in the molecule is essential for the activity. For example, diester compounds 8b, 8c, 8d and 8k are basically inactive, consistent with the expectation that negative charge is important for the activity of phosphatase inhibitors. As demonstrated by compounds 8j, 8r, 8m and 8q, the thiophene ring is generally more potent than the furan ring. Interestingly, 2-thione also seems preferable to 2-one in the thiozolidine ring that is best illustrated by increased potency of compound 8a and 8j. The length of the side chain (n=0, 1 and 2), depending on the overall structure of the compound, has a varying impact on the activities. 2-(carboxymethoxy) benzoic acid (CMBA), a derivative of salicyclic acid, is also a potent pTyr mimic. However, our attempt to replace salicyclic acid with CMBA resulted in a great loss of activity of compound 11 compared with 8e, indicating that the free hydroxyl group is necessary for retaining the activity.
Table 1.
The Lyp inhibitory activities of compound 8a-8s and 11
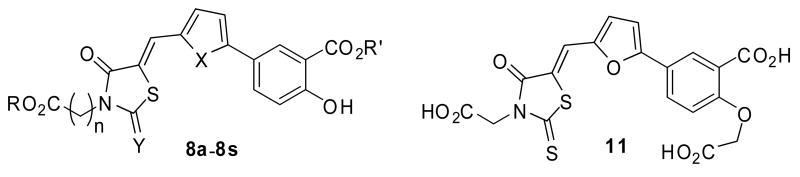 | ||||||
|---|---|---|---|---|---|---|
| Comp | R | R' | n | X | Y | IC50(μM)a |
| 8a | H | CH3 | 1 | O | S | 21.0 |
| 8b | CH3 | CH3 | 2 | O | O | 44.0 |
| 8c | CH3 | CH3 | 2 | S | O | >44 |
| 8d | CH3 | CH3 | 1 | S | O | >44 |
| 8e | H | H | 1 | O | S | 8.7 |
| 8f | H | H | 2 | O | O | 5.1 |
| 8g | H | H | 2 | S | O | 5.9 |
| 8h | H | H | 1 | S | O | 2.8 |
| 8i | H | H | 1 | S | S | 5.4 |
| 8j | H | CH3 | 1 | S | S | 4.3 |
| 8k | CH3 | CH3 | 1 | O | O | >44 |
| 8l | H | CH3 | 1 | S | O | 18 |
| 8m | H | CH3 | 1 | O | O | >44 |
| 8n | H | H | 1 | O | O | 5.6 |
| 8o | H | CH3 | 2 | O | O | 4.3 |
| 8p | H | CH3 | 2 | S | O | 0.39 |
| 8q | CH3 | H | 1 | O | O | 4.8 |
| 8r | CH3 | H | 1 | S | O | 1.8 |
| 8s | H | 0 | O | S | 1.4 | |
| 11 | >44 | |||||
IC50 values are determined from a single experiment performed in triplicate
Table 2.
The inhibitory activities of the Schiff base analogs
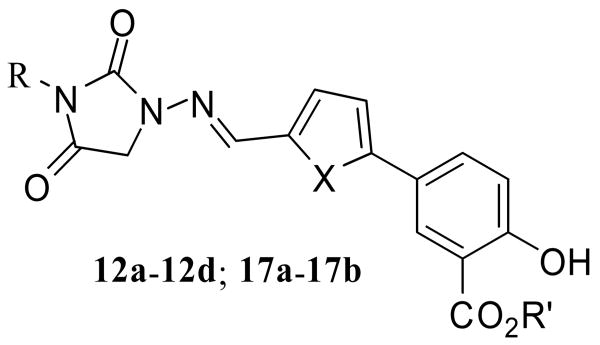 | ||||
|---|---|---|---|---|
| Comp | R | R' | X | IC50(μM)a |
| 12a | H | CH3 | O | >44 |
| 12b | H | H | O | >44 |
| 12c | H | CH3 | S | >44 |
| 12d | H | H | S | 34.0 |
| 17a | CH2CO2H | H | O | 36.0 |
| 17b | CH2CO2H | H | S | >44 |
IC50 values are determined from a single experiment performed in triplicate
Coincidently, when this manuscript was being prepared, the crystal structure of Lyp in complex with a salicyclic acid-based inhibitor was published.7 It was shown that the hydroxyl group of salicyclic acid makes critical polar interactions in the active site of Lyp.
As can be seen from Table 2, the Schiff base analogs are poor inhibitors in general. Only compound 12d and 17a showed moderate activities with the IC50s of 34.0 μM and 36.0 μM, respectively.
In summary, we have successfully identified a cluster of thiozolidine compounds with good Lyp inhititory activities via high throughput screening. Improved potency (<1 μM) was achieved by rational design and chemical synthesis. PTPase selectivity and cell studies of the promising compounds are currently being pursued.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, et al. Nat Genet. 2004;36:337. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 2.Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F, et al. Nat Genet. 2005;37:1317. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 3.See the assay protocol published in PubChem (AID: 606).
- 4.Burke TR, Lee K. Acc Chem Res. 2003;36:426. doi: 10.1021/ar020127o. [DOI] [PubMed] [Google Scholar]
- 5.Hajduk PJ, Greer J. Nat Rev Drug Discov. 2007;6(3):211. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 6.Analytical data for representative compounds: 8p, 1H NMR (300 MHz, CD3OD) δ 10.65 (br, 1H), 8.12 (s, 1H), 7.99 (s, 1H), 7.91(d, 1H), 7.68(d, 1H), 7.62 (d, 1H), 7.06(d, 1H), 3.90 (s, 3H), 3.83-3.80 (m, 2H), 2.60-2.55 (m, 2H); 13C NMR (75 MHz, CD3OD) δ 172.5, 168.8, 166.9, 165.7, 160.6, 149.8, 137.5, 136.2, 133.2, 127.7, 126.8, 125.5, 124.8, 119.3, 118.6, 115.1, 53.4, 38.4, 32.4; ESI-MS(M+ + H): 434.3. 11, 1H NMR (300 MHz, CD3OD) 8.11 (s, 1H), 7.93 (d, 1H), 7.73(s, 1H), 7.39(d, 1H), 7.31 (d, 1H), 7.22(d, 1H), 4.86 (s, 2H), 4.72 (s, 2H); 13C NMR (75 MHz, CD3OD) δ 194.2, 170.3, 167.9, 167.3, 166.6, 158.2, 157.7, 149.5, 129.4, 127.6, 124.7, 123.3, 122.0, 120.0, 118.0, 115.2, 110.4, 66.0, 45.8; ESI-MS(M+ + H): 464.0. 12d, 1H NMR (300 MHz, CD3OD) δ 11.2 (s, 1H), 8.01 (s, 1H), 7.98 (s, 1H), 7.85(d, 1H), 7.43 (d, 1H), 7.35 (d, 1H), 7.01 (d, 1H), 4.32 (s, 2H); 13C NMR (75 MHz, CD3OD) δ 171.9, 169.5, 161.5, 153.8, 144.6, 138.8, 138.2, 133.4, 132.4, 127.5, 125.3, 124.2, 118.9, 114.3, 49.9; ESI-MS(M+ + H): 346.3.
- 7.Yu Xiao, Sun JP, He Y, Guo X, Liu S, Zhou B, Hudmon A, Zhang ZY. Proc Natl Acad Sci U S A. 2007;104(50):19767. doi: 10.1073/pnas.0706233104. [DOI] [PMC free article] [PubMed] [Google Scholar]