

Abstract
Nuclear hormone receptors are ligand-regulated transcription factors that modulate the expression of specific target genes in response to the binding of small, hydrophobic hormone ligands. Many nuclear hormone receptors, such as the retinoic acid receptors, can both repress and activate target gene expression; these bimodal transcription properties are mediated by the ability of these receptors to tether auxiliary factors, denoted corepressors and coactivators. Corepressors are typically bound by receptors in the absence of cognate hormone, whereas binding of an appropriate hormone agonist induces an allosteric alteration in the receptor resulting in release of the corepressor and recruitment of coactivator. Structural analysis indicates that there is a close induced fit between the hormone ligand and the receptor polypeptide chain. This observation suggests that different ligands, once bound, may confer distinct conformations on the receptor that may invoke, in turn, distinct functional consequences. We report here that different retinoids do differ in the ability to release corepressor once bound to retinoic acid receptor and suggest that these differences in corepressor release may manifest as differences in transcriptional regulation.
Nuclear hormone receptors are hormone-regulated transcription factors that mediate cellular responses to a diverse set of small lipophilic hormones; these hormones include the steroids, thyroid hormones, vitamin D3, and retinoic acid (1-7). Nuclear hormone receptors function by binding to specific DNA sequences, denoted hormone response elements, and regulating the transcription of adjacent target genes (1-7). Many nuclear hormone receptors exhibit bimodal transcriptional properties and can either repress or activate expression of their target genes, depending on the hormone status, the nature of the target promoter, and the cell context (1-7). Thyroid hormone receptors and retinoic acid receptors (RARs)1 typically repress transcription in the absence of hormone and activate gene expression in the presence of hormone (8-15).
The bimodal transcriptional properties of the nuclear hormone receptors reflect the ability of these receptors to physically recruit auxiliary proteins, denoted corepressors and coactivators, that help mediate the actual transcriptional response (15-28). In the absence of cognate hormone, thyroid hormone receptors and RARs bind to a corepressor complex composed of SMRT and/or N-CoR, mSin3A or B, histone deacetylase 1 or 2, Rb-associated proteins p46 and p48, and a number of additional proteins of as-yet unknown function (reviewed in Refs. 29 and 30). Conversely, addition of hormone leads to release of the corepressor complex by the receptor and the recruitment of a distinct set of polypeptides that serve as coactivators (reviewed in Ref. 15). Once tethered to the receptor, corepressors and coactivators appear to modulate gene transcription through multiple mechanisms that include modification of the chromatin template and direct interaction with components of the general transcriptional machinery (29-34). Many of these same corepressor and coactivator proteins also participate in transcriptional regulation by an assortment of non-receptor transcription factors (e.g. Refs. 35-40).
The ability of nuclear hormone receptors to toggle between a corepressor-bound and a coactivator-bound state is therefore a key aspect of their function. Hormone ligand plays a critical role in this switching phenomenon, with the binding of cognate hormone by receptor leading to the release of corepressor and the recruitment of coactivator (e.g. Refs. 13-17, 20, and 27). Notably, binding of hormone ligand is believed to cause a series of conformational changes in the receptor characterized by a global compacting of the polypeptide chain about the ligand and an alteration in the position of the C-terminal α-helical domain (helix 12) from an extended to a sequestered conformation (41-47).
X-ray crystallographic studies indicate that the polypeptide chain in the liganded receptor is in close approximation to the bound hormone, with the hormone providing a hydrophobic core that helps mediate the packing of the receptor about it (41-46). This intimate induced-fit between receptor and ligand suggests that different hormone ligands may confer distinct polypeptide conformations, and therefore potentially distinct properties, once bound to a given receptor (41-46). We wished to investigate if, as a consequence, different ligands differ in their ability to modulate the recruitment and release of the corepressor complex. We report here that different retinoid derivatives do indeed differ in the ability to displace SMRT corepressor from RAR; most notably, although both 9-cis-retinoic acid and all-trans-retinoic acid are bound with high affinity by RARα, only the all-trans-isomer efficiently induces release of SMRT. All-trans- and 9-cis-retinoic acid are natural products found in normal physiological contexts; our results suggest that these two retinoids may exert distinct allosteric effects on RARα and may therefore mediate distinct aspects of RAR biology.
EXPERIMENTAL PROCEDURES
In Vitro Receptor/Corepressor Binding Assays
The glutathione S-transferase (GST), GST-RARα, and GST-SMRT/TRAC-2 (codons 751−1495) constructs were created as described previously (20, 27, 37). GST fusion proteins were expressed in Escherichia coli, isolated, and immobilized by binding to glutathione agarose. [35S]Methionine-radiolabeled, full-length RARα, PML-RARα, or SMRT proteins were synthesized by a coupled in vitro transcription and translation protocol (Promega, Madison, WI). The radiolabeled proteins were incubated with the immobilized GST fusion proteins in HEMG buffer (20); the agarose matrix was then extensively washed, and proteins remaining bound to the matrix were eluted with soluble glutathione and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The electrophoretograms were visualized by autoradiography and were quantified by STORM PhosphorImager analysis (Molecular Dynamics, Sunnyvale, CA). Mutants of RARα defective for association with the SMRT corepressor (denoted RARα AHT mutants) were created by site-directed mutagenesis (17, 37).
Mammalian Two-hybrid Assays
The construction of pSG5 vectors expressing either a GAL4 DNA binding domain (DBD)-SMRT fusion or a GAL4 activation domain (AD)-RARα fusion has been previously described (37, 48). The RARα (AHT) mutation was transferred into the pSG5-GAL4AD background in the form of an EcoRV to XhoI fragment generated by polymerase chain reaction. The actual transfections were performed in 12-well tissue culture plates containing 7 × 104 CV-1 cells per well. Transient transfections were performed employing a liposome/Lipofectin methodology (Life Technologies, Inc.), using 25 ng of pSG5 GAL4DBD vector, 100 ng of pSG5 GAL4AD vector, 100 ng of pGL2-GAL4 (17-mer) luciferase reporter plasmid, and 100 ng of a pCMV-promoter-lacZ reporter plasmid (used as an internal control) per well. The cells were subsequently incubated for 48 h in the presence or absence of all-trans- or 9-cis-retinoic acid and harvested, and the luciferase and β-galactosidase activities were determined as previously detailed (27, 34, 37, 49).
Protease Resistance Assays
[35S]Methionine-radiolabeled, full-length RARα was synthesized in vitro by the coupled transcription and translation protocol. For the trypsin and elastase assays, 1 μl of radiolabeled protein was diluted to 16 μl in 50 mm Tris-Cl (pH 7.4) buffer containing various concentrations of retinoid hormone or an equivalent volume of ethanol carrier. Proteolysis was initiated by adding 4 μl of protease (either 2 μg/μl type IV elastase or 0.125 μg/μl trypsin pre-treated with tosyl-l-phenylalanine chloromethyl ketone; Sigma). After a 10-min incubation at room temperature, proteolysis was terminated by addition of 14 μl of SDS-PAGE sample buffer, and the samples were frozen rapidly by transfer to dry ice. The samples were subsequently boiled for 10 min and resolved by SDS-PAGE. The electrophoretograms were visualized by STORM PhosphorImager analysis.
Transient Transfections
CV-1 cell transfections were performed by the same liposome protocol described for the two-hybrid assay. Approximately 4 × 105 cells were transfected with 25 ng of pSG5-RARα, 100 ng of a reporter construct representing the thymidine kinase promoter joined to luciferase and driven by a β-RARE, and 100 ng of the pCMV-lacZ plasmid employed as an internal control. Alternatively, the cells were transfected with 25 ng of a pSG5-GAL4DBD-RARα construct, 100 ng of the pGL2-GAL4(17-mer)-luciferase reporter, and 100 ng of the pCMV-lacZ internal control. Retinoid hormone was added (or not) 5 h post-transfection, and the cells were harvested at 48 h, and relative luciferase activity (normalized to β-galactosidase activity) was subsequently determined.
RESULTS
Different Retinoids Differ in the Ability to Trigger Release of Corepressor by RARα
The SMRT polypeptide represents a principal site of contact between the nuclear hormone receptors and the corepressor complex (16, 17, 19, 20-22, 24, 28). We therefore investigated the effects of different retinoid derivatives on the association in vitro between SMRT and RARα by employing a “GST-pull down” assay. A glutathione S-transferase (GST) derivative of SMRT was synthesized in E. coli, purified, and immobilized on a glutathione-agarose matrix (48). Radiolabeled, full-length RARα, synthesized by in vitro transcription and translation, was then incubated with the GST-SMRT matrix (20, 27). After incubation, the matrix was extensively washed, and any radiolabeled RARα remaining bound to the matrix was subsequently eluted with soluble glutathione, resolved by SDS-PAGE, and quantified by PhosphorImager analysis (20, 27). As we and others have previously reported (16, 17, 20, 22, 27, 34), radiolabeled RARα bound efficiently to the GST-SMRT matrix in the absence of hormone but not to a non-recombinant GST construct employed as a negative control (Fig. 1). Also as previously noted (20), the ability of RARα to bind to SMRT was severely inhibited by inclusion of either 50 or 500 nm all-trans-retinoic acid in the binding assay (Fig. 1).
Fig. 1. Different retinoids have different effects on the binding of RARα to GST-SMRT.
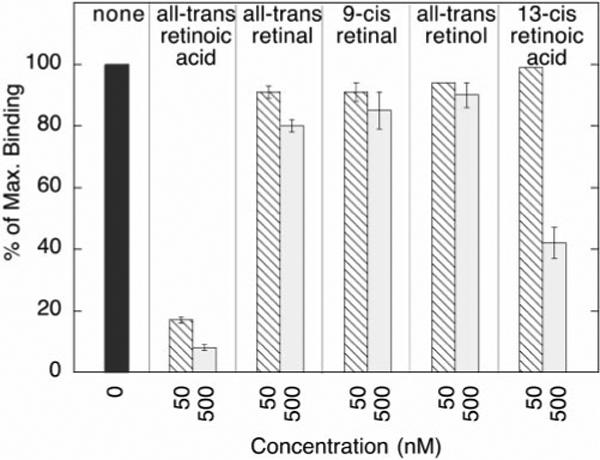
Radiolabeled, full-length RARα was synthesized by in vitro transcription and translation and was incubated with a GST-SMRT (codons 751−1495) construct immobilized on a glutathione-agarose matrix. The incubation buffer contained either hormone-free ethanol carrier (none), 50 or 500 nm of the retinoids indicated. RARα bound to the GST-SMRT matrix after incubation was extensively washed, eluted with soluble glutathione, resolved by SDS-PAGE, and quantified by PhosphorImager analysis. The amount of receptor bound to the GST-SMRT matrix in the absence of hormone was defined as 100%. RARα binding to non-recombinant GST, immobilized to glutathione-agarose and employed as a negative control, was typically 2% or less under identical conditions.
We next evaluated the ability of an assortment of other retinoids to inhibit the interaction of RARα with SMRT corepressor. In general, the ability of a given retinoid to mediate release of the corepressor paralleled the binding affinity of RARα for that retinoid. For example, RARα possesses little or no measurable affinity for the ret-aldehyde derivatives all-trans-retinal and 9-cis-retinal (49), and consistent with this observation, neither retinal derivative significantly inhibited binding of RARα to the GST-SMRT matrix at hormone concentrations up to 500 nm (Fig. 1). Similarly, all-trans-retinol failed to bind to RARα, and 500 nm all-trans-retinol failed to measurably release the SMRT corepressor from RARα (Fig. 1). In contrast, 13-cis-retinoic acid is bound by RARα, although with a lower affinity than that for all-trans-retinoic acid (49), and 13-cis-retinoic acid released SMRT from RARα at the 500 nm but not at the 50 nm concentration of this hormone (Fig. 1). Although not unexpected, these results support the hypothesis that retinoids mediate release of corepressor exclusively through the ability to bind to the ligand pocket of the receptor.
Intriguingly, there was one notable exception to this general correlation between affinity of the receptor for the ligand and the ability of the ligand to induce corepressor release. The 9-cis-retinoic acid isomer, which has an affinity for RARα virtually equal to that of all-trans-retinoic acid (50), was markedly inefficient in mediating the release of SMRT (Fig. 2A). Under our in vitro conditions, 10-fold more of the 9-cis-retinoic acid preparation was required, relative to all-trans-retinoic acid, to obtain an equivalent inhibition of the SMRT/RARα interaction (Fig. 2B). Both the 9-cis- and all-trans-retinoid preparations were prepared identically, and the concentrations were confirmed spectrophotometrically to ensure that the amounts of each ligand were indeed equal.
Fig. 2. The all-trans- and 9-cis-isomers of retinoic acid have distinct effects on the association between RARα and GST-SMRT.
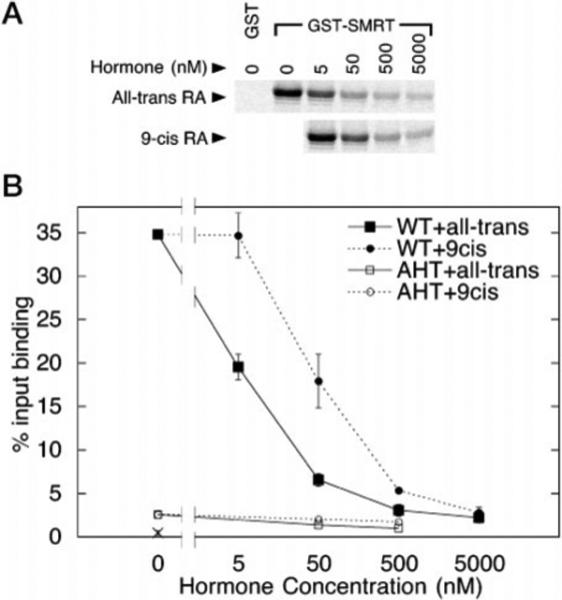
A, an electrophoretogram is depicted illustrating the ability of RARα to bind to GST-SMRT under conditions of increasing retinoid concentration. Radiolabeled, full-length RARα was incubated with GST-SMRT (codons 751−1495) in the presence of different concentrations of all-trans- or 9-cis-retinoic acid, as indicated in the panel. RARα bound to the immobilized GST-SMRT was subsequently washed extensively, eluted with soluble glutathione, and visualized by SDS-PAGE and autoradiography. Binding of radiolabeled RARα to non-recombinant GST (GST), employed as a negative control, was also determined in parallel. B, the binding of RARα to GST-SMRT under conditions of increasing retinoid concentration was quantified. Experiments performed as described for A were quantified by PhosphorImager analysis, and the percentage of the input RARα bound to the GST-SMRT construct at the different hormone concentrations was determined. The average and standard deviation of two or more experiments are presented. Both wild-type RARα (WT) and a mutant form of RARα defective for SMRT association (AHT mutant) were tested. The amount of wild-type RARα bound by a non-recombinant GST construct, employed as a negative control, was also determined (×).
It was conceivable that the in vitro translated RARα protein, as employed in our assay, might be misfolded or in some other manner selectively impaired in the binding of the 9-cis-isomer. Binding of hormone is known to result in formation of a more compact conformation of the nuclear hormone receptors that manifests as an increase in resistance to proteolysis (47, 51, 52). We employed this acquisition of protease resistance as an indicator of receptor ligand occupancy, thereby permitting us to monitor hormone binding by the radiolabeled RARα preparations employed in our GST-pull down protocol (Fig. 3). Notably, as little as 5 nm of either retinoic acid isomer clearly protected the in vitro translated RARα from proteolytic degradation by either elastase or trypsin (Fig. 3). These results suggest that RARα is fully or near-fully occupied by 9-cis-retinoic acid at concentrations of this hormone that fail to mediate release of SMRT corepressor in the GST-pull down assay.
Fig. 3. The all-trans- and 9-cis-isomers of retinoic acid are bound at comparable levels by RARα.

A protease resistance assay was employed as a measure of hormone binding by the receptor. Radiolabeled RARα, synthesized by in vitro transcription and translation, was incubated with either hormone-free ethanol carrier or increasing amounts of 9-cis- or all-trans-retinoic acid (RA). Protease (either elastase or trypsin) was then added, and the samples were incubated at room temperature for 10 min. After the reaction was terminated by addition of SDS sample buffer, the samples were resolved by SDS-15% PAGE system, and the RARα-derived peptides resistant to proteolysis were visualized by autoradiography. A, treatment with elastase. B, treatment with trypsin.
To confirm that the differential release of corepressor noted for the 9-cis- and all-trans-isomers of retinoic acid was not an artifact of our particular methodology, we repeated the GST protocol but employing the reciprocal approach of using a GST fusion construct of RARα (synthesized in E. coli) and an in vitro transcribed and translated derivative of SMRT (Fig. 4). Despite the reciprocal methodologies employed, the same pattern was observed as before: all-trans-retinoic acid efficiently released the SMRT-polypeptide from the GST-RARα construct, whereas 9-cis-retinoic acid was comparatively impaired in this property (Fig. 4).
Fig. 4. All-trans- and 9-cis-retinoic acid have distinct effects on the association between SMRT and GST-RARα.
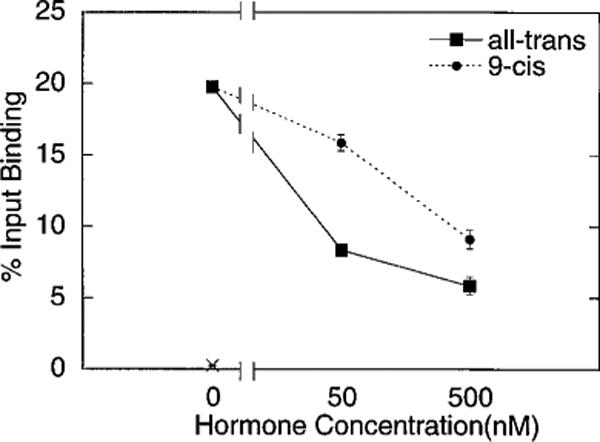
The same general protocol as described for Fig. 2B was repeated but in a reciprocal manner, using radiolabeled SMRT protein synthesized by in vitro transcription and translation and a GST-RARα fusion protein isolated from E. coli. The amount of SMRT protein bound to the GST-RARα construct in the presence of increasing amounts of hormone was quantified. The ability of SMRT to bind to non-recombinant GST, employed as a negative control, is also indicated (×).
Our 9-cis-retinoic acid preparation, obtained commercially, mediated little or no release of corepressor from the receptor at the 5 nm hormone concentration; however, the same 9-cis-retinoic acid preparation did mediate corepressor release at very high hormone concentrations (e.g. greater than 50 nm hormone). This dissociation from corepressor at the highest concentrations of 9-cis-retinoic acid would appear to be inconsistent with the presumption of a single binding site, with a single Kd, for the 9-cis-isomer on the receptor, and inconsistent with our protease resistance data, which indicated substantial or complete ligand occupancy at the 5 nm 9-cis-retinoic acid concentration. Notably, 9-cis-retinoic acid is quite labile chemically, and a variety of enzymatic and non-enzymatic substances can readily mediate isomerization of 9-cis-retinoic acid to the all-trans-isomer, with the equilibrium lying strongly in favor of the latter (e.g. Refs. 53 and 54). Indeed, high pressure liquid chromatography revealed the presence of 5−10% of all-trans-retinoic acid in many of our 9-cis-retinoic acid preparations (data not shown). Although other explanations are possible, we suggest that the release of corepressor observed at the highest 9-cis-retinoic acid concentrations is likely due to contamination by the all-trans-isomer, either due to pre-existing all-trans-retinoic acid in the 9-cis-retinoic acid preparation or due to isomerization of the 9-cis-isomer during the incubation period (see “Discussion”).
The Impaired Ability of 9-cis-Retinoic Acid to Release Corepressor Was Unaltered by Addition of DNA or Mammalian Cell Extracts to the in Vitro Assay and Was Also Observed in a Mammalian Two-hybrid Protocol
Although 9-cis-retinoic acid was impaired in the ability to release corepressor from RARα in vitro, it was possible that this ligand might have a different effect if the RARα was bound to a DNA response element or if additional factors, present in vivo, were included in the GST assay (18). We therefore repeated the in vitro GST-pull down assay in the presence of a large molar excess of an oligonucleotide containing a consensus retinoic acid response element or in the presence of nuclear lysates derived from CV-1 mammalian cells (Fig. 5). Neither the addition of the DNA-binding site nor the presence of mammalian cell extracts altered our initial results; SMRT corepressor was efficiently released by all-trans-, but not 9-cis-, retinoic acid (Fig. 5).
Fig. 5. Addition of DNA or nuclear extracts has no detectable effect on the 9-cis-retinoic acid-refractory association of RARα with SMRT.

Radiolabeled, full-length RARα was incubated with GST-SMRT (codons 751−1495) in the absence of any additions, in the presence of 50 nm 9-cis-retinoic acid, in the presence of 50 nm 9-cis-retinoic acid plus nuclear extracts derived from 6 × 105 CV-1 cells, or in the presence of 9-cis-retinoic acid plus 200 ng of a deoxyoligonucleotide containing a strong binding site for RARα (βRARE). The amount of RARα bound to the GST-SMRT protein was quantified; the average of duplicate experiments and the range are presented.
We next employed a mammalian cell two-hybrid protocol to determine if the isomer-specific differences in corepressor release observed in vitro were also observed in vivo. For this assay, a GAL4-DNA binding domain (GAL4-DBD) fusion of SMRT was cointroduced into CV-1 cells together with a GAL4-activation domain (GAL4-AD) fusion of RARα. An interaction between the GAL4-DBD-SMRT and GAL4AD-RARα proteins results in reconstitution of a functional GAL4 transcription factor, which we measured by assaying the expression of a GAL4 (17-mer) luciferase reporter gene (Fig. 6). Neither of the GAL4-fusion constructs alone are able to stimulate luciferase reporter expression under these conditions (Fig. 6 and data not shown); similarly, introduction of mutations into either SMRT or into RARα that are known to disrupt the SMRT/RAR interaction in vitro also impair the two-hybrid interaction (55). The strong two-hybrid interaction observed for SMRT and RARα in the transfected CV-1 cells was severely inhibited by the addition of all-trans-retinoic acid to the culture medium, with 5 nm all-trans-retinoic acid reducing the luciferase activity by more than 50%. By comparison, 9-cis-retinoic acid was significantly impaired in this inhibition, with 25 nm 9-cis-retinoic acid required to produce an inhibition equivalent to that produced by 5 nm of the all-trans-isomer. Therefore our results in vivo appear to parallel those in vitro and demonstrate a differential release of SMRT corepressor in response to 9-cis-, versus all-trans-, retinoic acid.
Fig. 6. The distinctive effects of all-trans- and 9-cis-retinoic acid on the RARα/SMRT interaction are also observed in a mammalian two-hybrid assay.
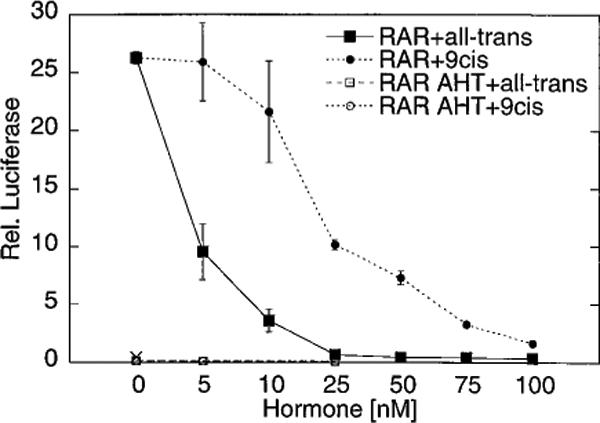
A pSG5-GAL4DBD-SMRT fusion vector was introduced into CV-1 cells together with a pSG5-GAL4AD-RARα fusion vector and a GAL4 (17-mer) luciferase reporter. The cells were subsequently incubated in the presence of different concentrations of either all-trans- or 9-cis-retinoic acid, as indicated. The cells were harvested, and the relative luciferase activity was determined and normalized to that of a pCMV-lacZ plasmid introduced as an internal control. Both a wild-type RARα construct (RAR) and a mutant RARα construct defective for SMRT association (RAR-AHT) were tested. The activity of an empty pSG5-GAL4AD construct, used in place of the pSG5-GAL4AD-RARα fusion as a negative control, was also determined (×). The data represent the average and range of duplicate experiments.
The Differential Effects of 9-cis- and All-trans-retinoic Acid Are Also Observed Using the Oncogenic PML-RARα Protein
Human acute promyelocytic leukemias are associated with chromosomal translocations that create chimeric polypeptides in which the N terminus of RARα is been replaced by novel protein sequences, such as that of PML (promyelocytic leukemia protein) (56). The ability of these chimeric RARα derivatives to tether corepressor has been implicated in their oncogenic activities; consistent with this hypothesis, treatment with all-trans-retinoic acid both releases SMRT from PML-RARα and induces differentiation in leukemic cells arising from PML-RARα translocations, leading to clinical regression (37-39, 56). We therefore examined the response of PML-RARα to 9-cis-retinoic acid. Notably, 9-cis-retinoic acid failed to release SMRT from PML-RARα under conditions in which the all-trans-isomer mediated significant release of SMRT (Fig. 7). We conclude that the differential corepressor release observed for all-trans- and 9-cis-retinoic acid extends to the oncogenic PML-RARα oncoprotein.
Fig. 7. The all-trans- and 9-cis-isomers of retinoic acid also have distinct effects on the association between SMRT and the PML-RARα oncoprotein.

Radiolabeled PML-RARα was incubated with GST-SMRT (codons 751−1495) in the presence of different concentrations of all-trans- or 9-cis-retinoic acid, as indicated in the panel. PML-RARα bound to the immobilized GST-SMRT was subsequently washed extensively, eluted with soluble glutathione, and quantified by SDS-PAGE and PhosphorImager analysis. Binding of radiolabeled PML-RARα to non-recombinant GST, employed as a negative control, was also determined in parallel (×).
The Differential Ability of 9-cis- and All-trans-retinoic Acid to Release Corepressor May Manifest as Differences in the Transcriptional Activity of RARα
Given the different capacities of 9-cis- and all-trans-retinoic acid to release corepressor, it was plausible that these two hormone ligands might exhibit distinct transcriptional activation profiles in cells. We therefore examined the ability of these different ligands to convert RARα from a repressor to an activator. We initially employed a GAL4-DBD fusion of RARα, restricted to the hormone binding domain, together with a GAL (17-mer) reporter plasmid, so as to minimize interference by endogenous RARs and RXRs present in the CV-1 cells (Fig. 8A). As expected, introduction of the GAL4DBD-RARα fusion in the absence of hormone repressed the expression of the Gal 17-mer reporter construct (Fig. 8A). Addition of all-trans-retinoic acid converted the GAL4DBD-RARα from a repressor into an activator, with approximately 100 nm all-trans-retinoic acid required for induction of maximal reporter gene activity. In contrast, significantly more 9-cis-retinoic acid, 1000 nm, was required for maximal activation under identical circumstances. This differential response to the two hormone isomers is consistent with our in vitro data and suggests that an impaired release of corepressor at low 9-cis-retinoic acid concentrations may attenuate the ability of the GAL4-RARα construct to toggle from a repressor to an activator.
Fig. 8. All-trans- and 9-cis-retinoic acid differ in the ability to induce RARα-mediated transcription.

A, the 9-cis-isomer was impaired in inducing transcriptional activation by a GAL4DBD-RARα fusion protein on a GAL4 (17-mer) reporter. A pSG5-GAL4AD-RARα fusion vector was introduced into CV-1 cells together with the GAL4 (17-mer) luciferase reporter. The cells were subsequently incubated in the presence of increasing amounts of either all-trans- (filled columns) or 9-cis-retinoic (hatched columns) acid, as indicated. The cells were harvested, and the relative luciferase (Rel. Luciferase) activity was determined and normalized to that of a pCMV-lacZ plasmid introduced as an internal control. The activity of an empty pGAL4DBD construct, used in place of the pSG5-GAL4DBD-RARα fusion as a negative control, was also determined (open columns). The data represent the average and range of duplicate experiments. B, the 9-cis-isomer was more effective than all-trans-retinoic acid in inducing transcription activation by the native RARα on a β-RARE-luciferase reporter. A pSG5 construct expressing native RARα was introduced into CV-1 cells together with a thymidine kinase promoter-luciferase reporter containing three copies of the β-RARE response element. The cells were treated with hormone and harvested as in A, and the relative luciferase activity was determined and normalized to that of a pCMV-lacZ plasmid. The activity of an empty pSG-5 construct was also determined (open columns). The data represent the average and range of duplicate experiments.
We repeated these experiments but using a native RARα construct and a reporter containing a retinoic acid response element (Fig. 8B). In contrast to our results with the GAL4DBD-RARα derivative, native RARα actually activated transcription more efficiently in response to the 9-cis-retinoic acid than to the all-trans-isomer (Fig. 8B). However, it should be noted that in the absence of hormone the native RARα also failed to function as a transcriptional repressor in the CV-1 cell background (Fig. 8B; compare the levels of reporter expression in the absence and presence of receptor at the zero hormone concentration). This may be due to the differing natures of the two different reporter constructs or to the presence of an additional activation domain (denoted AF-1) in the N terminus of the native RARα that is not present in the GAL4DBD-RARα fusion (57). Whatever the precise mechanistic basis of this phenomenon, it appears that the response of RARα to 9-cis-retinoic acid can be measurably different from that of all-trans-retinoic acid, depending on the target gene and the nature of the receptor construct.
All-trans- and 9-cis-Retinoic Acid Confer Indistinguishable Effects on the Conformation of the C-terminal Helix 12 of RARα
A change in the conformation of the C-terminal helix 12 of RARs and thyroid hormone receptors has been proposed to play an important role in the release of corepressor (24, 41-47). To determine if 9-cis- and all-trans-retinoic acid differ in the ability to invoke this conformational change in helix 12, we applied an exopeptidase technique. If, as proposed from crystallographic analysis, the C terminus of the unliganded RARα is in an extended conformation, it might be expected that the unliganded receptor would be relatively susceptible to degradation by an exopeptidase such as carboxypeptidase Y. Conversely, if the C terminus of RARα becomes more sequestered from solvent on addition of hormone, the liganded receptor would be expected to be more resistant to carboxypeptidase Y degradation (47). These predictions were fulfilled in experiments utilizing all-trans-retinoic acid; RARα was significantly more susceptible to carboxypeptidase Y in the absence versus in the presence of the all-trans-isomer, suggesting that the C terminus of the receptor is more sequestered in the presence of the all-trans-isomer (Fig. 9). Intriguingly, a virtually indistinguishable protection from carboxypeptidase Y degradation could be observed at analogous concentrations of 9-cis-retinoic acid (Fig. 9). We conclude that both all-trans- and 9-cis-retinoic acid confer detectable sequestration of the receptor C terminus, and that the differences in corepressor release observed for these two ligands must therefore reside in alterations to the conformation of the receptor not visualized by our exopeptidase assay. These differences may represent alterations in the ligand-induced folding of other domains of the RARα or may represent alterations in the conformation of helix 12 that cannot be resolved by the carboxypeptidase Y methodology.
Fig. 9. Both all-trans- and 9-cis-retinoic acid induce sequestration of the C terminus of RARα.

A carboxypeptidase Y resistance assay was employed as a measure of the accessibility of the RARα C terminus to solvent under different hormone conditions (47). Radiolabeled RARα, synthesized by in vitro transcription and translation, was incubated with either hormone-free ethanol carrier or increasing amounts of 9-cis- or all-trans-retinoic acid. Carboxypeptidase Y was then added (4 μg per reaction), and the samples were incubated for 10 min at room temperature. After the reaction was terminated by addition of SDS-sample buffer, the samples were resolved by SDS-PAGE, and the amount of full-length (undegraded) RARα remaining was quantified by PhosphorImager analysis. The results of duplicate experiments, and the range, are presented.
DISCUSSION
Different Ligands That Bind to RARα Can Invoke Distinct Functional Responses
Binding of hormone by nuclear hormone receptors is believed to result in a series of conformational changes in the ligand binding domain of these polypeptides. These conformational changes consist of a global compaction of the polypeptide in a tight induced-fit about the hormone ligand, a flip in the conformation of an “omega loop,” and a reorientation in the position of the C-terminal helix 12 (41-46). Concomitant with these rearrangements, and presumably mediated by them, the receptor loses its affinity for the SMRT/N-CoR corepressor complex and acquires instead the ability to bind certain coactivator polypeptides. In essence, hormone provides a hydrophobic core, around which the receptor polypeptide chain assumes its final conformation. This close approximation between hormone and liganded receptor suggests that structurally distinct ligands might induce distinct receptor conformations, which in turn might display different propensities to recruit or release transcriptional cofactors. To test this hypothesis, we examined the ability of different retinoid ligands to release the SMRT corepressor complex from RARα; we chose this particular experimental model due to the important role of corepressor in RAR function, the accessibility of a wide assortment of well characterized retinoid ligands, and the availability of a high resolution crystal-derived structure for the liganded RARα polypeptide (5, 42).
Consistent with our hypothesis, we observed that although both 9-cis- and all-trans-retinoic acid bind to RARα with near equal affinities, only the all-trans-isomer induces the release of SMRT corepressor efficiently. We could demonstrate this differential corepressor release both in a GST assay in vitro and in a mammalian two-hybrid protocol in vivo. RARα displays this hormone-specific differential SMRT release at concentrations of all-trans- and 9-cis-retinoic acid that, by protease assay, result in an indistinguishable ligand occupancy of the receptor by the two isomers. We interpret these results to suggest that the differential release of SMRT corepressor is an allosteric phenomenon and that binding of all-trans-retinoic acid by RARα confers a set of conformational changes in the receptor that result in corepressor release; binding of 9-cis-retinoic acid, in contrast, fails to invoke one or more components of this set of conformational changes and thus fails to release corepressor in our assay.
We have considered a variety of alternative explanations for our observations that might arise from experimental artifact. As previously noted, we confirmed that the RARα preparation employed in our in vitro assays actually binds 9-cis- and all-trans-retinoic acid with comparable avidities. We also demonstrated that the impaired release of corepressor observed in vitro was mimicked in a two-hybrid assay in vivo. We were concerned that the all-trans- and 9-cis-retinoic acid might be differentially adsorbed by components of the in vitro translation mix or by the walls of the assay tubes, thereby lowering the effective concentration of the 9-cis-hormone available for binding by the receptor. Although our protease protection assay (demonstrating equal occupancy at nominally equal hormone concentrations) would appear to argue against both of these scenarios as potential artifacts, we also repeated our assays using siliconized tubes with no alteration in the results.2 Similarly, we also modified the experimental protocol so as to first bind the radiolabeled RARα to the GST-SMRT matrix; we then washed the matrix extensively and subsequently challenged the now-purified GST-SMRTzRARα complex with retinoid; once again, the all-trans-retinoic acid efficiently released receptor from the corepressor, whereas the 9-cis-isomer was significantly impaired in this function. We conclude that the difference we observed in the ability of all-trans- and 9-cis-retinoic acid to release corepressor from RARα is most likely an authentic reflection of an actual biological difference in the properties of these two hormone isomers.
Although impaired in corepressor release at moderate (<50 nm) hormone concentrations, 9-cis-retinoic acid did release corepressor from RARα at high (>50 nm) hormone concentrations. This may be simply due to isomerization of a small percentage of the 9-cis-retinoic acid to the all-trans-form prior to or during the experimental protocol; notably the chemical equilibrium lies greatly in favor of this transition and many common chemical reagents can help catalyze this isomerization (53, 54). Alternatively, although less likely, we cannot exclude the possibility that there are actually two populations of RARα in our preparation as follows: one that binds 9-cis-retinoic acid with a high affinity, but fails to release corepressor, and one that binds 9-cis-retinoic acid with a lower affinity but does, in response, release corepressor. These two RARα populations might represent receptor dimers versus monomers, for example, or might represent two conformationally distinct forms of RARα monomer (e.g. Refs. 58-60). Whatever the molecular basis for this phenomenon, our studies indicate that the effects of all-trans- and 9-cis-retinoic acid on RARα are clearly distinguishable at physiological concentrations of hormone.
Although somewhat unanticipated, our results are not without precedent. Crystallographic analysis of the estrogen receptor indicates that the C-terminal helix 12 domain of this receptor assumes a dramatically different conformation when the receptor is bound to a ligand agonist versus an antagonist (44). As a consequence, the antagonist-bound estrogen receptor is unable to efficiently bind certain coactivators and is impaired in transcriptional activation. Notably, the 9-cis-retinoic acid isomer displays a significantly different topography, manifested as an orthogonal bend in the center of the retinoid side chain, from that of the all-trans-isomer. Although the specific conformation of RARα bound to 9-cis-retinoic acid has not been experimentally determined, molecular modeling and genetic analysis suggests that the 9-cis-isomer can be accommodated in the binding pocket of RARα but that the 9-cis-isomer would differ in its contacts with receptor helices 3, 11, and 12 relative to those mediated by the all-trans-isomer (42, 61). Given that these helices play important roles in the allosteric changes observed for RARα on binding of all-trans-retinoic acid (41-46), it is plausible that binding of the 9-cis-isomer may result in a distinct, if subtle, reorientation of one or more of these essential helices, resulting in impaired release of the SMRT corepressor.
The Differing Abilities of 9-cis- and All-trans-Retinoic Acid to Induce Corepressor Release May Reflect Different Roles for These Two Hormones in the Biology of RARα
Although 9-cis-retinoic acid hormone was first identified as a naturally occurring ligand for the retinoid X receptors (RXRs), 9-cis-retinoic acid is bound by RARs at a substantially higher affinity than by RXRs, and the affinity of RARs for the 9-cis-isomer is comparable to the affinity of RARs for its archetypic ligand, all-trans-retinoic acid (50). Therefore, although 9-cis-retinoic acid has traditionally been thought of as an inducer of RXR-mediated responses, this same retinoid isomer also represents an extremely potent mediator of RAR-mediated responses. What might be the evolutionary rationale of having two distinct, naturally occurring hormone isomers able to bind to a single given receptor? Our results suggest that the 9-cis-isomer might play a previously unanticipated role: that of an allosteric effector able to bind to RARα but impaired in the ability to release corepressor. Thus, the transcriptional response of RARα to 9-cis-retinoic acid might be anticipated to be different from that to all-trans-isomer, at least in certain contexts, and this might allow physiologically distinct roles for the two isomers in RAR biology. Our work suggests that 9-cis-retinoic acid exhibits an impaired ability to toggle GAL4DBD-RARα constructs from repressors to activators of transcription. However, this impaired response to 9-cis-retinoic acid is not invariably observed under all conditions. In fact, native RARα on a retinoic response element exhibited the opposite effect, with the 9-cis-retinoic acid isomer conferring transcriptional activation at lower hormone levels than required for all-trans-retinoic acid. There are several plausible explanations for this apparent discrepancy. For example, the retinoic response elements employed in the latter experiments are likely to bind RXR/RAR heterodimers, and the RXR component may mediate the enhanced response to the 9-cis-retinoic acid. Alternatively, an N-terminal, hormone-independent transcriptional activation domain, denoted AF-1, is present in the native RARα but absent in our GAL4DBD-RARα construct and may be responsible for overcoming the effect of bound corepressor by activating transcription even in the presence of a bound corepressor. It is also possible that other differences in the nature of the reporter constructs, or the precise conditions employed, generate the distinct retinoid responses we report here. Whatever the mechanistic basis, it appears that distinct retinoids can invoke distinct transcriptional responses once bound to RARα and thus are potentially able to mediate distinct physiological consequences. We suggest that the multiplicity of retinoids isolated from nature reflects this multiplicity of allosteric effects that these retinoids can invoke in their cognate receptors.
Acknowledgments
We thank P. Chambon for generously providing the RARα molecular clone adapted for use in this research and C. Meares for advice and providing access to a high pressure liquid chromatograph.
Footnotes
This work was supported by Public Health Services/National Institutes of Health Grants R37 CA-53394 and R01 DK-53528. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: RARs, retinoic acid receptors; GST, glutathione S-transferase; PAGE, polyacrylamide gel electrophoresis; DBD, DNA binding domain; AD, activation domain; RXR, retinoid X receptors; PML, promyelocytic leukemia; RARE, retinoic acid response element.
S.-H. Hong and M. L. Privalsky, unpublished data.
REFERENCES
- 1.Lazar MA. Endocr. Rev. 1993;14:184–193. doi: 10.1210/edrv-14-2-184. [DOI] [PubMed] [Google Scholar]
- 2.Ribeiro RC, Apriletti JW, West BL, Wagner RL, Fletterick RJ, Schaufele F, Baxter JD. Ann. N. Y. Acad. Sci. 1993;758:366–389. doi: 10.1111/j.1749-6632.1995.tb24843.x. [DOI] [PubMed] [Google Scholar]
- 3.Tsai MJ, O'Malley BW. Annu. Rev. Biochem. 1994;63:451–483. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 4.Beato M, Herrlich P, Schutz G. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 5.Kastner P, Mark M, Chambon P. Cell. 1995;83:859–869. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- 6.Mangelsdorf DJ, Evans RM. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 7.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Damm K, Thompson CC, Evans RM. Nature. 1989;339:593–597. doi: 10.1038/339593a0. [DOI] [PubMed] [Google Scholar]
- 9.Graupner G, Wills KN, Tzukerman M, Zhang X, Pfahl M. Nature. 1989;340:653–656. doi: 10.1038/340653a0. [DOI] [PubMed] [Google Scholar]
- 10.Sap J, Munoz A, Schmitt H, Stunnenberg H, Vennstrom B. Nature. 1989;340:242–244. doi: 10.1038/340242a0. [DOI] [PubMed] [Google Scholar]
- 11.Baniahmad A, Kohne AC, Renkawitz R. EMBO J. 1992;11:1015–1023. doi: 10.1002/j.1460-2075.1992.tb05140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen H-W, Privalsky ML. Mol. Cell. Biol. 1993;13:5970–5980. doi: 10.1128/mcb.13.10.5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casanova J, Helmer E, Selmi-Ruby S, Qi JS, Au-Flieger M, Desai-Yajnik V, Koudinova N, Yarm F, Raaka BM, Samuels HH. Mol. Cell. Biol. 1994;14:5756–5765. doi: 10.1128/mcb.14.9.5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baniahmad A, Leng X, Burris TP, Tsai SY, Tsai MJ, O'Malley BW. Mol. Cell. Biol. 1995;15:76–86. doi: 10.1128/mcb.15.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung L. Mol. Endocrinol. 1996;10:1167–1177. doi: 10.1210/mend.10.10.9121485. [DOI] [PubMed] [Google Scholar]
- 16.Chen JD, Evans RM. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 17.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamel Y, Soderstrom M, Glass CK, Rosenfeld MG. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 18.Kurokawa R, Soderstrom M, Horlein A, Halachmi S, Brown M, Rosenfeld MG, Glass CK. Nature. 1995;377:451–454. doi: 10.1038/377451a0. [DOI] [PubMed] [Google Scholar]
- 19.Downes M, Burke LJ, Bailey PJ, Muscat GE. Nucleic Acids Res. 1996;2:4379–4386. doi: 10.1093/nar/24.22.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sande S, Privalsky ML. Mol. Endocrinol. 1996;10:813–825. doi: 10.1210/mend.10.7.8813722. [DOI] [PubMed] [Google Scholar]
- 21.Seol W, Mahon MJ, Lee YK, Moore DD. Mol. Endocrinol. 1996;10:1646–1655. doi: 10.1210/mend.10.12.8961273. [DOI] [PubMed] [Google Scholar]
- 22.Zamir I, Harding HP, Atkins GB, Horlein A, Glass CK, Rosenfeld M, Lazar MA. Mol. Cell. Biol. 1996;16:5458–5465. doi: 10.1128/mcb.16.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson TA, Richer JK, Bain DL, Takimoto GS, Tung L, Horwitz KB. Mol. Endocrinol. 1997;11:693–705. doi: 10.1210/mend.11.6.0004. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Leo C, Schroen DJ, Chen JD. Mol. Endocrinol. 1997;11:2025–2037. doi: 10.1210/mend.11.13.0028. [DOI] [PubMed] [Google Scholar]
- 25.Shibata H, Nawaz Z, Tsai SY, O'Malley BW. Mol. Endocrinol. 1997;11:714–724. doi: 10.1210/mend.11.6.0002. [DOI] [PubMed] [Google Scholar]
- 26.Smith CL, Nawaz Z, O'Malley BW. Mol. Endocrinol. 1997;11:657–666. doi: 10.1210/mend.11.6.0009. [DOI] [PubMed] [Google Scholar]
- 27.Yoh SM, Chatterjee VKK, Privalsky ML. Mol. Endocrinol. 1997;11:470–480. doi: 10.1210/mend.11.4.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Zamir I, Lazar MA. Mol. Cell. Biol. 1997;17:6887–6897. doi: 10.1128/mcb.17.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pazin MJ, Kadonaga JT. Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- 30.Wolffe AP. Nature. 1997;387:16–17. doi: 10.1038/387016a0. [DOI] [PubMed] [Google Scholar]
- 31.Fondell JD, Roy AL, Roeder RG. Genes Dev. 1993;7:1400–1410. doi: 10.1101/gad.7.7b.1400. [DOI] [PubMed] [Google Scholar]
- 32.Fondell JB, Brunel F, Hisatake K, Roeder RG. Mol. Cell. Biol. 1996;16:281–287. doi: 10.1128/mcb.16.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muscat GEO, Burke LJ, Downes M. Nucleic Acids Res. 1998;26:2899–2907. doi: 10.1093/nar/26.12.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong C-W, Privalsky ML. Mol. Cell. Biol. 1998;18:5500–5510. doi: 10.1128/mcb.18.9.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang W-M, Inouye C, Zeng Y, Bearss D, Seto E. Proc. Natl. Acad. Sci. U. S. A. 1996;93:12845–12850. doi: 10.1073/pnas.93.23.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhordain P, Albagli O, Lin RJ, Ansieau S, Quief S, Leutz A, Kerckaert JP, Evans RM, Leprince D. Proc. Natl. Acad. Sci. U. S. A. 1997;94:10762–10767. doi: 10.1073/pnas.94.20.10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong S-H, David G, Wong CW, Dejean A, Privalsky ML. Proc. Natl. Acad. Sci. U. S. A. 1997;94:9028–9033. doi: 10.1073/pnas.94.17.9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I, Seiser C, Grignani F, Lazar MA, Minucci S, Pelicci PG. Nature. 1998;391:815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- 39.Lin RJ, Nagy L, Inoue S, Shao W, Miller. WH, Jr., Evans RM. Nature. 1998;391:811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- 40.Luo RX, Postigo AA, Dean DC. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- 41.Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Nature. 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- 42.Renaud J-P, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- 43.Wagner RL, Apriletti JW, McGrath ME, West BL, Baxter JD, Fletterick RJ. Nature. 1995;378:690–697. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- 44.Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom P, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 45.Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Proc. Natl. Acad. Sci. U. S. A. 1998;95:5998–6003. doi: 10.1073/pnas.95.11.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams SP, Sigler PB. Nature. 1998;393:392–396. doi: 10.1038/30775. [DOI] [PubMed] [Google Scholar]
- 47.Lin BC, Hong S-H, Krig S, Yoh SM, Privalsky ML. Mol. Cell. Biol. 1997;17:6131–6138. doi: 10.1128/mcb.17.10.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong S-H, Wong C-W, Privalsky ML. Mol. Endocrinol. 1998;12:1161–1171. doi: 10.1210/mend.12.8.0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crettaz M, Baron A, Siegenthaler G, Hunziker W. Biochem. J. 1990;272:391–397. doi: 10.1042/bj2720391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Allenby G, Bocquel M-T, Saunders M, Kazmer S, Speck J, Rosenberger M, Lovey A, Kastner P, Grippo JF, Chambon P, Levin AA. Proc. Natl. Acad. Sci. U. S. A. 1993;90:30–34. doi: 10.1073/pnas.90.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keidel S, LeMotte P, Apfel C. Mol. Cell. Biol. 1994;14:287–298. doi: 10.1128/mcb.14.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leng X, Tsai SY, O'Malley BW, Tsai MJ. J. Steroid Biochem. Mol. Biol. 1993;46:643–661. doi: 10.1016/0960-0760(93)90306-h. [DOI] [PubMed] [Google Scholar]
- 53.Shih TW, Lin TH, Shealy YF, Hill DL. Drug Metab. Dispos. 1997;25:27–32. [PubMed] [Google Scholar]
- 54.Chen H, Juchau MR. Drug Metab. Dispos. 1998;26:222–228. [PubMed] [Google Scholar]
- 55.Wong CW, Privalsky ML. Mol. Cell. Biol. 1998;18:5724–5733. doi: 10.1128/mcb.18.10.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pandolfi PP. Haematologica. 1996;81:472–482. [PubMed] [Google Scholar]
- 57.Leid M, Kastner P, Chambon P. Trends Biochem. Sci. 1992;17:427–433. doi: 10.1016/0968-0004(92)90014-z. [DOI] [PubMed] [Google Scholar]
- 58.Leng X, Tsai SY, O'Malley BW, Tsai MJ. J. Steroid Biochem. Mol. Biol. 1993;46:643–661. doi: 10.1016/0960-0760(93)90306-h. [DOI] [PubMed] [Google Scholar]
- 59.Claret F-X, Antakly T, Karin M, Saatcioglu F. Mol. Cell. Biol. 1996;16:219–227. doi: 10.1128/mcb.16.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schulman IG, Li C, Schwabe JWR, Evans RM. Genes Dev. 1997;11:299–308. doi: 10.1101/gad.11.3.299. [DOI] [PubMed] [Google Scholar]
- 61.Tate BF, Allenby G, Janocha R, Kazmer S, Speck J, Sturzenbecker LJ, Abarzua P, Levin AA, Grippo JF. Mol. Cell. Biol. 1994;14:2323–2330. doi: 10.1128/mcb.14.4.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]