

Abstract
Glucocorticoids (GCs) and protein kinase A (PKA)–activating agents (β-adrenergic receptor agonists) are mainstream asthma therapies based on their ability to prevent or reverse excessive airway smooth muscle (ASM) constriction. Their abilities to regulate another important feature of asthma—excessive ASM growth—are poorly understood. Recent studies have suggested that GCs render agents of inflammation such as IL-1β and TNF-α mitogenic to ASM, via suppression of (antimitogenic) induced cyclooxygenase-2–dependent PKA activity. To further explore the mechanistic basis of these observations, we assessed the effects of epidermal growth factor and IL-1β stimulation, and the modulatory effects of GC treatment and PKA inhibition, on the ASM transcriptome by microarray analysis. Results demonstrate that ASM stimulated with IL-1β, in a manner that is often cooperative with stimulation with epidermal growth factor, exhibit a profound capacity to function as immunomodulatory cells. Moreover, results implicate an important role for induced autocrine/paracrine factors (many whose regulation was minimally affected by GCs or PKA inhibition) as regulators of both airway inflammation and ASM growth. Induction of numerous chemokines, in conjunction with regulation of proteases and agents of extracellular matrix remodeling, is suggested as an important mechanism promoting upregulated G protein–coupled receptor signaling capable of stimulating ASM growth. Additional functional assays suggest that intracellular PKA plays a critical role in suppressing the promitogenic effects of induced autocrine factors in ASM. Finally, identification and comparison of GC- and PKA-sensitive genes in ASM provide insight into the complementary effects of β-agonist/GC combination therapies, and suggest specific genes as important targets for guiding the development of new generations of GCs and adjunct asthma therapies.
Keywords: airway smooth muscle, protein kinase A, glucocorticoid, gene expression, G protein–coupled receptors
CLINICAL RELEVANCE
Results from these studies help to understand how gene expression in human airway smooth muscle is regulated by inflammation and by the two principal asthma therapeutics: glucocorticoids and β-agonists. Changes in the airway smooth muscle transcriptome induced by glucocorticoids or protein kinase A inhibition provide insight into how glucocorticoid therapy may promote increased airway smooth muscle mass.
3′-5′-cyclic adenosine monophosphate (cAMP)-dependent protein kinase (PKA) and glucocorticoid (GC)-mediated signaling impact numerous airway smooth muscle (ASM) cell functions via their actions on acute signaling events and through regulation of gene expression. As therapeutic agents, PKA-activating agents and GCs are the principal means of managing the excessive ASM contraction that is the hallmark feature of the obstructive airway disease asthma. Gs-coupled G protein–coupled receptors (GPCRs) capable of PKA activation, including β2-adrenergic receptors (β2AR), negatively regulate ASM contraction by promoting phosphorylation and inhibition of critical molecules that couple excitation and contraction (1). GCs can indirectly affect contraction via suppression of airway inflammation that not only stimulates the production of, but also sensitizes ASM to, contractile stimuli (1).
Aberrant ASM growth is also an important pathologic feature of asthma, although the capacity of PKA- and GC-dependent signaling to regulate ASM growth is less well characterized. Various PKA-activating agents such as β-AR agonists (β-agonists), PGE2, and forskolin have been shown to inhibit mitogen-stimulated growth of ASM cells in culture (2–4). More variable, stimulus-specific effects on ASM growth have been reported for GCs. GCs appear modestly effective in inhibiting ASM growth stimulated by GPCR agonists such as thrombin or leukotriene D4, yet have little effect on growth stimulated by polypeptide growth factors such as epidermal growth factor (EGF) (5–9).
Somewhat surprisingly, we recently discovered that GCs convert IL-1β and TNF-α from inhibitors to strong enhancers of EGF-stimulated ASM growth (3). Mechanistic studies suggested an important link between GC- and PKA-dependent signaling in the regulation of ASM growth by IL-1β and TNF-α. In the absence of GCs, IL-1β (or low concentrations of IL-1β and TNF-α combined) significantly induced cyclooxygenase (COX)-2, PGE2, and intracellular PKA activity, and inhibited growth of EGF-stimulated ASM cells. However, under conditions in which COX-2/PGE2/PKA induction was inhibited by pretreatment with GCs or other inhibitors of COX-2–dependent PGE2 production (10), the cytokines increased EGF-stimulated growth more than 2-fold (3). A similar augmentation of EGF-stimulated ASM growth by IL-1β and TNF-α was effected by direct PKA inhibition, suggesting a critical role of PKA in determining the mitogenic effects of cytokines, and of GC treatment, on ASM.
In this same study, time-dependent analysis of mitogenic signaling pathway activation demonstrated regulation of p42/p44 ERK and PI3K/Akt signaling at late but not early time points. The delayed onset of mitogenic signaling suggested the ability of PKA and GCs to regulate induced gene expression in ASM. Despite the critical role of PKA and GC signaling in determining ASM function, surprisingly little is known regarding PKA- and GC-dependent gene regulation in ASM. In the current study, we provide a comprehensive analysis of the effects of EGF and IL-1β treatment, and of the modulatory effect of GC treatment and direct PKA inhibition, on the ASM transcriptome. Results provide novel insight into the capabilities of ASM to function as an immunomodulatory cell. In addition, data suggest an important role for up-regulated GPCR signaling in mediating the mitogenic effects of cytokines, and via identification of GC- and PKA-sensitive and -insensitive genes provide putative targets for refining current or developing new asthma therapies.
MATERIALS AND METHODS
Generation of Human ASM Cultures
Human ASM cultures were generated from tracheae from unidentified donors as described by Penn and coworkers (11), and were examined in the 5th to 8th passage.
Generation of retroviral-infected human ASM cultures was as described previously (12). Retrovirus for generation of GFP- and PKI-GFP–expressing lines was produced by transfecting GP2-293 cells with pLNCX2-GFP or pLNCX2-PKI-GFP each with pVSV-G vector that encodes the pantropic (VSV-G) envelope protein. Supernatants were harvested 48 hours after transfection and used to infect human ASM cultures. Infected cells typically exhibited greater than 70% GFP expression within 48 hours (direct visualization by fluorescent microscopy) and selection to homogeneity with 200 μg/ml G418 (Sigma Aldrich, St. Louis, MO) was rapid (7 d). Because pharmacologic approaches to PKA inhibition have previously been shown to be either ineffective or subject to significant nonspecific effects (13), experiments herein employ retroviral expression of PKI-GFP previously shown effective in ASM cells (3, 12). For numerous experimental outcomes tested to date (3, 12, 14, 15), we have observed minimal differences in responses between GFP-expressing cells and the naïve culture from which they were derived, suggesting minimal effects of infection per se. Although retroviral infection would undoubtedly alter the ASM transcriptome, its impact on stimulus or group comparisons would be mitigated in part by the retrovirus effect existing for both experimental and control conditions.
RNA Isolation for Microarray Analysis
GFP- and PKI-GFP–expressing cells were plated at 750,000 cells/plate on 10-cm dishes, grown to confluence and arrested in 0.1% bovine serum albumin media for 24 hours before stimulation with vehicle, 10 nM EGF (R&D Systems, Minneapolis, MN), 20 U/ml IL-1β (Roche Applied Science, Indianapolis, IN), or both (E+I) (Figure 1). In all conditions cells were pretreated for 30 minutes with vehicle or 10 nM fluticasone propionate (Flu, kindly provided by GlaxoSmithKline, Raleigh, NC). After 8 hours of stimulation, RNA was isolated from cells by standard TRIzol (Invitrogen, Carlsbad, CA) protocol and purified by phenol/isopropanol precipitation. Ten micrograms of RNA was repurified using the Qiagen (Valencia, CA) columns and quality was measured with Bioanalyzer (Agilent, Santa Clara, CA). The RNA was then reverse transcribed to cDNA, and labeled cRNA was generated. Quality control checks were performed after each step to ensure reactions completed successfully, and that cDNA and cRNA of adequate amount and quality had been generated. Labeled cRNA was then hybridized to U133A microarrays (Affymetrix, Santa Clara, CA), the chips were scanned and .CEL files generated. Changes in ASM transcriptome were analyzed in four different cultures derived from four individual donors and resulted in four replicate arrays each for unstimulated (basal), EGF, and E+I conditions for GFP- and PKI-GFP–expressing cells, and three replicate arrays for IL-1β–stimulated and each Flu-pretreated GFP- and PKI-expressing cells. Given the large number of genes shown to be differentially regulated under the various conditions tested, for practical reasons parallel analyses using real-time PCR were not conducted.
Figure 1.

Experimental design of microarray analysis. Human airway smooth muscle (ASM) cells stably expressing GFP or PKI-GFP chimera (generated as outlined in Materials and Methods) were grown to confluence, arrested for 24 hours in 0.1% bovine serum albumin–containing media, pretreated with vehicle or Fluticasone (10 nM) for 30 minutes, and stimulated with epidermal growth factor (EGF) (10 ng/ml), IL-1β (20 U/ml), or both (E+I) for 8 hours. Cells were lysed in TRIzol and total RNA isolated by phenol/isopropanol precipitation. RNA was processed as outlined in Materials and Methods and gene expression was assessed using the Affymetrix U133A microchip. Gene expression was analyzed in four different cultures derived from four individual donors.
[3H] thymidine incorporation in human ASM cultures was assessed as per the method of Billington and colleagues (14).
BrdU Co-Culture Assay
dsRed-expressing cells were generated in the same manner as GFP- and PKI-GFP–expressing cells (described above) using pLNCX2-dsRed plasmid (Clontech, Mountain View, CA). A mixture containing equal numbers of dsRed- and either GFP- or PKI-GFP–expressing cells were plated in 6-well plates for a total of 400,000 cells/well and grown to confluence. Cells were then serum starved for 24 hours and stimulated with 10 nM EGF, 20 U/ml IL-1β, or both for 72 hours. BrdU (60 μM; Sigma Aldrich, St. Louis, MO) was added to ASM cultures concomitantly with stimuli. After stimulation, cells were harvested by trypsinization, fixed in 10% formalin, and permeabilized with 0.1% Triton-X100/0.01% gelatin for 2 hours. Cells were incubated at 37°C with 300 μM DNase, washed, and then stained with Alexa 647–labeled anti-BrdU antibody (Molecular Probes, Carlsbad, CA). Stained cells were analyzed by flow cytometry on a FACSCalibur (BD Biosciences, San Jose, CA), simultaneously detecting BrdU+, DsRed+, and GFP+ cells.
Effects of conditioned media on ASM Ca2+ mobilization were assessed using media harvested from GFP- and PKI-GFP–expressing cells stimulated for 18 hours with 10 nM EGF and 20 U/ml IL-1β (E+I) in presence or absence of 10 nM Flu. Immediately after harvesting, media were applied to GFP- or PKI-GFP–expressing cells seeded on coverslips and serum starved for 72 hours. Ca2+ imaging was then performed as described previously (16). Briefly, GFP- and PKI-GFP–expressing cells on coverslips were washed and loaded with 5 μM Fura-2 AM (Molecular Probes, Carlsbad, CA) for 30 minutes at 37°C. The cells were washed and maintained in Hank's Balanced Salt Solution (HBSS, pH 7.4) containing 10 mM HEPES, 11 mM glucose, 2.5 mM CaCl2, and 1.2 mM MgCl2. The coverslips were mounted onto an open slide chamber and sealed firmly. Intracellular calcium concentrations were determined using a dual excitation fluorescence photomultiplier system (IonOptix, Milton, MA). The ratio of intensities at 340 nm and 380 nm at each time point was converted into intracellular calcium concentration by extrapolation from a calibration curve derived from the following equation:
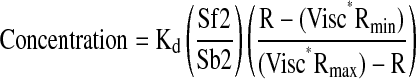 |
in which Kd = dissociation constant (175 nM for fura-2 at room temperature); Visc* = Viscosity-correction factor (0.85 for fura-2); Sf2 and Sb2 the intensities of the 380-nm image in the presence of EGTA and ionophore, respectively; Rmin and Rmax the minimum and maximum 340/380 ratios in the presence of EGTA and ionophore, respectively; and R the ratio of 340 and 380 nm intensities.
The cells were superfused with HBSS and basal calcium concentration measured. Subsequently the cells were stimulated with plain F-12 medium with no phenol red for 10 minutes, followed by the conditioned medium (CM) for 5 minutes, and change in emission signal was recorded. The net calcium response was calculated by subtracting the fluorescence signal in the presence of plain medium from peak fluorescence signal upon stimulation with the CM.
Peripheral Blood Neutrophil Isolation and Chemotaxis Assay
Whole blood was obtained from healthy volunteer subjects by venipuncture under a protocol approved by the Wake Forest University Health Sciences Institutional Review Board. Peripheral blood samples (∼ 60 ml) were collected and neutrophils isolated by a standard protocol using Ficoll-Histopaque density separation. Briefly, blood was mixed with acid citrate dextrose solution followed by Ficoll-Histopaque, and total white blood cells (WBCs) were harvested after aspiration and centrifugation of the top layer. Contaminant red blood cells were lysed by resuspending the cell pellet using ice-cold ddH2O and E-lysis buffer. Total WBCs were subjected to further purification using Ficoll-Hypaque (Sigma) and neutrophils were separated by high speed centrifugation. Cells were resuspended in HBSS with no Ca2+ or Mg+2. The cells were stained using crystal violet or FITC-conjugated anti-CD15 antibody and confirmed to be neutrophils (> 95%).
Chemotaxis assay was performed using Transwell (6.5 mm diameter, 6 μM pore size polycarbonate membrane inserts) plates (Corning, Lowell, MA). The lower chamber was filled with 1 ml plain medium with or without 1 μM IL-8 or 1 μM Formyl-methionyl-leucyl-phenylalanine (FMLP), or the indicated conditioned medium (CM) harvested from human ASM cells. The neutrophil cell suspension was loaded in the top chamber and incubated at 37°C for 3 hours. Cells that migrated to the lower chamber were counted and data normalized using total number of cells migrated in response to plain medium.
Statistical Analysis and Data Presentation
For the transcriptome analysis, we first assessed the quality and variability among chips using the dCHIP software (17). Microarray data were then normalized by the GC-RMA (18) method and differential gene expression was assessed using the affylmGUI (19) package based on the R program. Differential expression of each transcript represented on the Affymetrix U133A microarray, conferred by the stimuli (EGF, IL-1β, E+I) as compared with basal (B), and further modulated by GCs or PKA inhibition, was analyzed and presented in spreadsheet form (Microsoft Office Excel, Redmond, WA). Also reported on these spreadsheets is significance of regulation assessed by B value and P value adjusted using the False Discovery Rate method of Benjamini and Hochberg. The B (Bayes) statistic represents the log odds score that the corresponding gene is differentially expressed and takes into account the variability among replicate arrays. Genes found to be regulated more than 2-fold were further grouped into functional groups using the DAVID (20) and Ingenuity Pathways Analysis (IPA) (Ingenuity Systems, Redwood City, CA) software as described in the study by Hipp and Atala (21), and are reported in tables and figures in the online supplement. Tables 1–5 present the 20 most up-regulated and 20 most down-regulated genes with B values greater than 0 and fold changes greater than 2. In Tables 1–9, multiple accession entries for the same gene were removed to allow for an expanded list (with the entry exhibiting the highest or lowest fold change presented), but are present in the Excel files in the online supplement. Table headers indicate comparison for which differential gene expression is presented (e.g., GFP E-GFP B denotes genes differentially regulated by EGF in GFP-expressing cells as compared with unstimulated condition [basal]). The raw data for each hybridization (.cel files) along with information fulfilling the MIAME standards have been uploaded to the GEO repository (accession #GSE13168) at NCBI.
TABLE 1.
EGF-REGULATED GENE EXPRESSION IN HUMAN ASM*: GENES DIFFERENTIALLY REGULATED BY EGF IN GFP-EXPRESSING CELLS COMPARED WITH THE UNSTIMULATED (BASAL) CONDITION
| Fold Change
|
||||
|---|---|---|---|---|
| Symbol | Name | GFP EGF–GFP Basal | GFP IL-1β–GFP Basal | GFP EGF+IL-1β–GFP Basal |
| DUSP6 | Dual specificity phosphatase 6 | 22.97 | 4.62 | 31.01 |
| F2RL1 | Coagulation factor II (thrombin) receptor-like 1 | 17.60 | −1.17 | 3.17 |
| ISG20 | Interferon stimulated exonuclease gene 20kD | 10.53 | 98.13 | 87.03 |
| SPRY4 | Sprouty homolog 4 (Drosophila) | 10.52 | 2.41 | 14.43 |
| FOSL1 | FOS-like antigen 1 | 9.61 | 6.11 | 8.86 |
| HMGA2 | High mobility group AT-hook 2 | 9.15 | 4.26 | 8.51 |
| SPHK1 | Sphingosine kinase 1 | 7.05 | 4.74 | 10.57 |
| SPRY2 | Sprouty homolog 2 (Drosophila) | 6.66 | 1.88 | 9.50 |
| ANGPTL4 | Angiopoietin-like 4 | 6.53 | 3.59 | 11.48 |
| SLC20A1 | Solute carrier family 20 (phosphate transporter), member 1 | 6.40 | −1.22 | 3.42 |
| TRIB1 | Tribbles homolog 1 (Drosophila) | 6.26 | 2.53 | 6.99 |
| BHLHB2 | Basic helix-loop-helix domain containing, class B, 2 | 6.10 | 5.44 | 5.27 |
| S100A2 | S100 calcium binding protein A2 | 5.80 | 1.08 | 3.80 |
| MICAL2 | Microtubule associated monoxygenase, calponin and LIM domain containing 2 | 5.14 | 2.57 | 5.47 |
| ARHGAP22 | Rho GTPase activating protein 22 | 4.91 | −1.13 | 4.18 |
| MAFF | v-maf musculoaponeurotic fibrosarcoma oncogene homolog F (avian) | 4.90 | 7.30 | 10.81 |
| TUBB2A | Tubulin, beta 2A | 4.05 | 1.92 | 5.25 |
| PHLDA2 | Pleckstrin homology-like domain, family A, member 2 | 3.94 | 4.77 | 7.97 |
| C7orf44 | Chromosome 7 open reading frame 44 | 3.89 | 6.75 | 5.57 |
| HK2 | Hexokinase 2 | 3.83 | 5.84 | 8.05 |
| LOC727942 | Similar to phosphodiesterase 4D interacting protein isoform 2 | −2.98 | −2.70 | −2.62 |
| C20orf19 | Chromosome 20 open reading frame 19 | −3.02 | −3.63 | −4.17 |
| ADRA2A | Adrenergic, alpha-2A-, receptor | −3.08 | −3.40 | −3.41 |
| SMPDL3A | Sphingomyelin phosphodiesterase, acid-like 3A | −3.25 | −2.72 | −5.18 |
| GOLSYN | Golgi-localized protein | −3.32 | −3.34 | −3.33 |
| MAF | v-maf musculoaponeurotic fibrosarcoma oncogene homolog (avian) | −3.34 | −4.14 | −4.52 |
| DAPK1 | Death-associated protein kinase 1 | −3.38 | −3.96 | −5.75 |
| CEP57 | Centrosomal protein 57kDa | −3.68 | −4.33 | −4.55 |
| RHOBTB1 | Rho-related BTB domain containing 1 | −4.02 | −5.02 | −5.04 |
| FRY | Furry homolog (Drosophila) | −4.07 | −5.30 | −5.27 |
| ALDH3A2 | Aldehyde dehydrogenase 3 family, member A2 | −4.15 | −9.07 | −6.26 |
| HLTF | Helicase-like transcription factor | −4.73 | −4.80 | −6.41 |
| MEGF9 | Multiple EGF-like-domains 9 | −5.24 | −5.92 | −10.89 |
| CDKN1C | Cyclin-dependent kinase inhibitor 1C (p57, Kip2) | −7.06 | −8.68 | −12.47 |
| RUNX1T1 | Runt-related transcription factor 1; translocated to, 1 | −7.98 | −10.18 | −13.37 |
| BAMBI | BMP and activin membrane-bound inhibitor homolog (Xenopus laevis) | −8.26 | −5.20 | −10.54 |
| EFNB2 | Ephrin-B2 | −10.04 | −9.03 | −18.03 |
| C10orf10 | Chromosome 10 open reading frame 10 | −10.47 | −4.21 | −57.32 |
| PIK3R1 | Phosphoinositide-3-kinase, regulatory subunit 1 (p85 alpha) | −10.74 | −9.09 | −9.90 |
| NR1D2 | Nuclear receptor subfamily 1, group D, member 2 | −12.16 | −3.26 | −8.77 |
Definition of abbreviations: ASM, airway smooth muscle; EGF, epidermal growth factor.
The 20 most increased and decreased gene transcripts in GFP-expressing cells stimulated for 8 hours with EGF as compared with unstimulated (basal) condition (n = 4). For comparison, regulation by IL-1β and EGF+IL-1β co-stimulation (as compared with unstimulated condition [basal]) is also presented.
TABLE 2.
IL-1β–REGULATED GENE EXPRESSION IN HUMAN ASM*: GENES DIFFERENTIALLY REGULATED BY IL-1β IN GFP-EXPRESSING CELLS COMPARED WITH THE UNSTIMULATED (BASAL) CONDITION
| Fold Change
|
||||
|---|---|---|---|---|
| Symbol | Name | GFP IL-1β–GFP Basal | GFP EGF–GFP Basal | GFP EGF+IL-1β–GFP Basal |
| IL8 | Interleukin 8 | 1,861.0 | 11.8 | 2,493.5 |
| CXCL2 | Chemokine (C-X-C motif) ligand 2 | 1,203.1 | 3.2 | 1,283.0 |
| IL6 | Interleukin 6 (interferon, beta 2) | 709.9 | 9.9 | 915.5 |
| CXCL10 | Chemokine (C-X-C motif) ligand 10 | 608.0 | 1.7 | 93.1 |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 | 601.7 | 3.3 | 799.8 |
| CXCL3 | Chemokine (C-X-C motif) ligand 3 | 545.3 | 3.2 | 1,199.7 |
| MX1 | Myxovirus (influenza virus) resistance 1 | 297.8 | −1.5 | 24.1 |
| CCL20 | Chemokine (C-C motif) ligand 20 | 245.1 | 2.3 | 342.5 |
| TNFAIP3 | Tumor necrosis factor, alpha-induced protein 3 | 225.7 | 3.9 | 224.9 |
| GCH1 | GTP cyclohydrolase 1 (dopa-responsive dystonia) | 214.9 | 1.6 | 123.1 |
| ICAM1 | Intercellular adhesion molecule 1 (CD54) | 202.2 | 4.6 | 215.3 |
| CCL8 | Chemokine (C-C motif) ligand 8 | 199.9 | −1.1 | 23.9 |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 | 189.2 | 2.7 | 198.8 |
| IL1B | Interleukin 1, beta | 164.1 | 2.1 | 543.5 |
| TNFAIP2 | Tumor necrosis factor, alpha-induced protein 2 | 156.5 | 2.0 | 92.8 |
| CCL7 | Chemokine (C-C motif) ligand 7 | 123.3 | 8.9 | 193.6 |
| OAS1 | 2′,5′-oligoadenylate synthetase 1, 40/46kD | 118.0 | −1.0 | 4.9 |
| SOD2 | Superoxide dismutase 2, mitochondrial | 108.7 | 2.8 | 74.5 |
| ISG20 | Interferon stimulated exonuclease gene 20kD | 98.1 | 10.5 | 87.0 |
| NKX3-1 | NK3 homeobox 1 | 91.6 | 1.7 | 63.4 |
| ADH1B | Alcohol dehydrogenase IB (class I), beta polypeptide | −11.9 | −5.1 | −14.4 |
| TSC22D3 | TSC22 domain family, member 3 | −11.7 | −2.8 | −8.9 |
| RUNX1T1 | Runt-related transcription factor 1; translocated to, 1 | −10.2 | −8.0 | −13.4 |
| ALDH3A2 | Aldehyde dehydrogenase 3 family, member A2 | −9.1 | −4.1 | −6.3 |
| EFNB2 | Ephrin-B2 | −9.0 | −10.0 | −18.0 |
| CDKN1C | Cyclin-dependent kinase inhibitor 1C (p57, Kip2) | −8.7 | −7.1 | −12.5 |
| OSR2 | Odd-skipped related 2 (Drosophila) | −8.6 | −6.6 | −32.3 |
| MAF | v-maf musculoaponeurotic fibrosarcoma oncogene homolog | −7.9 | −3.5 | −18.5 |
| C5orf4 | Chromosome 5 open reading frame 4 | −7.1 | −2.7 | −10.6 |
| MAOA | Monoamine oxidase A | −6.5 | −2.9 | −7.5 |
| RCAN2 | Regulator of calcineurin 2 | −6.2 | −3.0 | −8.6 |
| ACOX2 | Acyl-Coenzyme A oxidase 2, branched chain | −6.2 | −2.6 | −5.2 |
| PSIP1 | PC4 and SFRS1 interacting protein 1 | −5.65 | −2.93 | −5.36 |
| PRPS1 | Phosphoribosyl pyrophosphate synthetase 1 | −5.64 | −1.32 | −2.69 |
| ROR1 | Receptor tyrosine kinase-like orphan receptor 1 | −5.63 | −2.36 | −5.15 |
| FOXF2 | Forkhead box F2 | −5.59 | −1.84 | −6.88 |
| FZD2 | Frizzled homolog 2 (Drosophila) | −5.39 | −2.92 | −7.74 |
| TBX2 | T-box 2 | −5.36 | 1.09 | −1.13 |
| BNC2 | Basonuclin 2 | −5.35 | −2.31 | −10.03 |
| CPEB1 | Cytoplasmic polyadenylation element binding protein 1 | −5.30 | −2.08 | −5.99 |
For definition of abbreviations, see Table 1.
The 20 most increased and decreased gene transcripts in GFP-expressing cells stimulated for 8 hours with IL-1β as compared with unstimulated (basal) condition (n = 3). For comparison, regulation by EGF and EGF+IL-1β co-stimulation (as compared with unstimulated condition [basal]) is also presented.
TABLE 3.
EFFECT OF COMBINED EGF+IL-1β STIMULATION ON GENE EXPRESSION IN HUMAN ASM*: GENES DIFFERENTIALLY REGULATED BY EGF+IL-1β IN GFP-EXPRESSING CELLS COMPARED WITH THE UNSTIMULATED (BASAL) CONDITION
| Fold Change
|
||||
|---|---|---|---|---|
| Symbol | Name | GFP EGF+IL-1β–GFP Basal | GFP EGF–GFP Basal | GFP IL-1β–GFP Basal |
| IL8 | Interleukin 8 | 2,493.5 | 11.8 | 1,861.0 |
| CXCL2 | Chemokine (C-X-C motif) ligand 2 | 1,283.0 | 3.2 | 1,203.1 |
| CXCL3 | Chemokine (C-X-C motif) ligand 3 | 1,199.7 | 3.2 | 545.3 |
| IL6 | Interleukin 6 (interferon, beta 2) | 915.5 | 9.9 | 709.9 |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 | 799.8 | 3.3 | 601.7 |
| IL1B | Interleukin 1, beta | 543.5 | 2.1 | 164.1 |
| CCL20 | Chemokine (C-C motif) ligand 20 | 342.5 | 2.3 | 245.1 |
| TNFAIP3 | Tumor necrosis factor, alpha-induced protein 3 | 224.9 | 3.9 | 225.7 |
| ICAM1 | Intercellular adhesion molecule 1 (CD54) | 215.3 | 4.6 | 202.2 |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 | 198.8 | 2.7 | 189.2 |
| CCL7 | Chemokine (C-C motif) ligand 7 | 193.6 | 8.9 | 123.3 |
| GCH1 | GTP cyclohydrolase 1 | 123.1 | 1.6 | 214.9 |
| STC1 | Stanniocalcin 1 | 120.5 | 14.5 | 59.2 |
| CXCL6 | Chemokine (C-X-C motif) ligand 6 | 94.1 | 2.5 | 72.5 |
| CXCL10 | Chemokine (C-X-C motif) ligand 10 | 93.1 | 1.7 | 608.0 |
| TNFAIP2 | Tumor necrosis factor, alpha-induced protein 2 | 92.8 | 2.0 | 156.5 |
| MMP3 | Matrix metallopeptidase 3 | 88.4 | 1.5 | 24.3 |
| ISG20 | Interferon stimulated exonuclease gene 20kDa | 87.0 | 10.5 | 98.1 |
| IL11 | Interleukin 11 | 81.6 | 3.4 | 9.4 |
| BMP2 | Bone morphogenetic protein 2 | 78.9 | 3.1 | 17.6 |
| C10orf10 | Chromosome 10 open reading frame 10 | −57.3 | −10.5 | −4.2 |
| OSR2 | Odd-skipped related 2 (Drosophila) | −32.3 | −6.6 | −8.6 |
| MAF | v-maf musculoaponeurotic fibrosarcoma oncogene homolog | −18.5 | −3.5 | −7.9 |
| EFNB2 | Ephrin-B2 | −18.0 | −10.0 | −9.0 |
| ADH1B | Alcohol dehydrogenase IB (class I), beta polypeptide | −14.4 | −5.1 | −11.9 |
| RUNX1T1 | Runt-related transcription factor 1; translocated to, 1 | −13.4 | −8.0 | −10.2 |
| CDKN1C | Cyclin-dependent kinase inhibitor 1C (p57, Kip2) | −12.5 | −7.1 | −8.7 |
| MEGF9 | Multiple EGF-like-domains 9 | −10.9 | −5.2 | −5.9 |
| C5orf4 | Chromosome 5 open reading frame 4 | −10.6 | −2.7 | −7.1 |
| BAMBI | BMP and activin membrane-bound inhibitor homolog | −10.5 | −8.3 | −5.2 |
| BNC2 | Basonuclin 2 | −10.0 | −2.3 | −5.3 |
| PIK3R1 | Phosphoinositide-3-kinase, regulatory subunit 1 (p85 alpha) | −9.9 | −10.7 | −9.1 |
| TOX | Thymocyte selection-associated high mobility group box | −9.7 | −3.6 | −6.1 |
| TSC22D3 | TSC22 domain family, member 3 | −8.9 | −2.8 | −11.7 |
| NR1D2 | Nuclear receptor subfamily 1, group D, member 2 | −8.77 | −12.16 | −3.26 |
| RCAN2 | Regulator of calcineurin 2 | −8.64 | −2.99 | −6.24 |
| GUCY1B3 | Guanylate cyclase 1, soluble, beta 3 | −8.50 | −4.17 | −4.78 |
| PLEKHA5 | Pleckstrin homology domain containing, family A member 5 | −8.11 | −3.82 | −5.86 |
| CXCL12 | Chemokine (C-X-C motif) ligand 12 (stromal cell-derived factor 1) | −8.10 | −3.17 | −2.02 |
| STC2 | Stanniocalcin 2 | −7.83 | −2.64 | −2.99 |
For definition of abbreviations, see Table 1.
The 20 most increased and decreased gene transcripts in GFP-expressing cells co-stimulated for 8 h with EGF+IL-1β (E+I) as compared with unstimulated (Basal) condition (n = 4). For comparison, regulation by IL-1β and EGF stimulation (as compared with unstimulated condition (B)) is also presented.
TABLE 4.
REGULATION OF EGF-DEPENDENT GENE EXPRESSION IN HUMAN ASM BY FLUTICASONE*: DIFFERENTIAL GENE EXPRESSION IN GFP-EXPRESSING CELLS PRETREATED WITH FLU AND STIMULATED WITH EGF COMPARED WITH GFP-EXPRESSING CELLS STIMULATED WITH EGF ONLY
| Symbol | Name | Fold Change |
|---|---|---|
| ZBTB16 | Zinc finger and BTB domain containing 16 | 43.08 |
| MT1M | Metallothionein 1M | 36.22 |
| C10orf10 | Chromosome 10 open reading frame 10 | 31.17 |
| FAM107A | Family with sequence similarity 107, member A | 31.12 |
| TSC22D3 | TSC22 domain family, member 3 | 30.53 |
| GABBR2 | Gamma-aminobutyric acid (GABA) B receptor, 2 | 22.57 |
| PIK3R1 | Phosphoinositide-3-kinase, regulatory subunit 1 (p85 alpha) | 19.38 |
| SLC19A2 | Solute carrier family 19 (thiamine transporter), member 2 | 18.32 |
| TIPARP | TCDD-inducible poly(ADP-ribose) polymerase | 15.66 |
| KLF9 | Kruppel-like factor 9 | 10.88 |
| TJP2 | Tight junction protein 2 (zona occludens 2) | 10.62 |
| PHF17 | PHD finger protein 17 | 10.46 |
| IMPA2 | Inositol(myo)-1(or 4)-monophosphatase 2 | 10.03 |
| ITGA10 | Integrin, alpha 10 | 9.94 |
| MT1X | Metallothionein 1X | 9.44 |
| CRISPLD2 | Cysteine-rich secretory protein LCCL domain containing 2 | 8.51 |
| MMD | Monocyte to macrophage differentiation-associated | 8.48 |
| GADD45B | Growth arrest and DNA-damage-inducible, beta | 8.47 |
| HSD11B1 | Hydroxysteroid (11-beta) dehydrogenase 1 | 8.27 |
| FOXO1 | Forkhead box O1 | 8.16 |
| F2RL1 | Coagulation factor II (thrombin) receptor-like 1 | −12.88 |
| LIF | Leukemia inhibitory factor (cholinergic differentiation factor) | −12.36 |
| IER3 | Immediate early response 3 | −11.75 |
| ZHX2 | Zinc fingers and homeoboxes 2 | −11.65 |
| FST | Follistatin | −10.55 |
| BDKRB2 | Bradykinin receptor B2 | −10.31 |
| BHLHB2 | Basic helix-loop-helix domain containing, class B, 2 | −9.98 |
| DUSP6 | Dual specificity phosphatase 6 | −8.51 |
| GDF15 | Growth differentiation factor 15 | −7.06 |
| GPER | G protein-coupled estrogen receptor 1 | −6.77 |
| CXCL12 | Chemokine (C-X-C motif) ligand 12 (stromal cell-derived factor 1) | −6.67 |
| SYNJ2 | Synaptojanin 2 | −5.72 |
| GATA6 | GATA binding protein 6 | −5.60 |
| ARL4C | ADP-ribosylation factor-like 4C | −5.34 |
| CNIH3 | Cornichon homolog 3 (Drosophila) | −5.00 |
| SOX9 | SRY (sex determining region Y)-box 9 (campomelic dysplasia, autosomal sex-reversal) | −4.96 |
| STK17A | Serine/threonine kinase 17a | −4.17 |
| ZMIZ1 | Zinc finger, MIZ-type containing 1 | −4.08 |
| SHB | Src homology 2 domain containing adaptor protein B | −4.06 |
| FZD7 | Frizzled homolog 7 (Drosophila) | −3.82 |
For definition of abbreviations, see Table 1.
The 20 most increased and decreased gene transcripts in GFP-expressing cells pre-treated with Flu for 30 minutes and stimulated with EGF for 8 hours as compared with GFP-expressing cells stimulated with EGF only (n = 3).
TABLE 5.
REGULATION OF IL-1β–DEPENDENT GENE EXPRESSION IN HUMAN ASM BY FLUTICASONE*: DIFFERENTIAL GENE EXPRESSION IN GFP-EXPRESSING CELLS PRETREATED WITH FLU AND STIMULATED WITH IL-1β COMPARED WITH GFP-EXPRESSING CELLS STIMULATED WITH IL-1β ONLY
| Symbol | Name | Fold Change |
|---|---|---|
| TSC22D3 | TSC22 domain family, member 3 | 119.88 |
| FAM107A | family with sequence similarity 107, member A | 76.11 |
| C10orf10 | chromosome 10 open reading frame 10 | 63.03 |
| ZBTB16 | zinc finger and BTB domain containing 16 | 39.71 |
| MT1M | metallothionein 1M | 28.37 |
| C13orf15 | chromosome 13 open reading frame 15 | 25.74 |
| ZNF22 | zinc finger protein 22 (KOX 15) | 23.13 |
| GLUL | glutamate-ammonia ligase (glutamine synthetase) | 17.94 |
| PIK3R1 | phosphoinositide-3-kinase, regulatory subunit 1 (p85 alpha) | 17.46 |
| MAOA | monoamine oxidase A | 12.78 |
| PHF17 | PHD finger protein 17 | 10.97 |
| CTGF | connective tissue growth factor | 10.67 |
| TIPARP | TCDD-inducible poly(ADP-ribose) polymerase | 10.26 |
| NET1 | neuroepithelial cell transforming gene 1 | 9.70 |
| NEDD4 | neural precursor cell expressed, developmentally downregulated 4 | 8.79 |
| FKBP5 | FK506 binding protein 5 | 8.49 |
| GADD45B | growth arrest and DNA-damage-inducible, beta | 7.95 |
| FOXO1 | forkhead box O1 | 7.67 |
| IRS2 | insulin receptor substrate 2 | 7.65 |
| LRRC16 | leucine rich repeat containing 16 | 7.44 |
| CCL8 | chemokine (C-C motif) ligand 8 | −123.87 |
| IL1B | interleukin 1, beta | −109.09 |
| PTGS2 | prostaglandin-endoperoxide synthase 2 | −106.33 |
| MX2 | myxovirus (influenza virus) resistance 2 (mouse) | −85.45 |
| IFIT1 | interferon-induced protein with tetratricopeptide repeats 1 | −81.37 |
| MX1 | myxovirus (influenza virus) resistance 1, interferon-inducible protein p78 | −76.52 |
| RTP4 | receptor (chemosensory) transporter protein 4 | −49.07 |
| CCL7 | chemokine (C-C motif) ligand 7 | −48.37 |
| IFIT3 | interferon-induced protein with tetratricopeptide repeats 3 | −46.54 |
| OAS1 | 2′,5′-oligoadenylate synthetase 1, 40/46 kD | −45.78 |
| IFI44 | interferon-induced protein 44 | −40.49 |
| ISG20 | interferon stimulated exonuclease gene 20 kD | −34.94 |
| OAS2 | 2′-5′-oligoadenylate synthetase 2, 69/71 kD | −33.60 |
| HERC6 | hect domain and RLD 6 | −33.57 |
| RSAD2 | radical S-adenosyl methionine domain containing 2 | −33.56 |
| TNFSF10 | tumor necrosis factor (ligand) superfamily, member 10 | −32.83 |
| IFIT2 | interferon-induced protein with tetratricopeptide repeats 2 | −27.36 |
| LIF | leukemia inhibitory factor (cholinergic differentiation factor) | −27.06 |
| IFI44L | interferon-induced protein 44-like | −26.14 |
| GPRC5A | G protein-coupled receptor, family C, group 5, member A | −24.79 |
Definition of abbreviation: ASM, airway smooth muscle.
The 20 most increased and decreased gene transcripts in GFP-expressing cells pretreated with Flu for 30 minutes and stimulated with IL-1β for 8 hours as compared with GFP-expressing cells stimulated with IL-1β only (n = 3).
TABLE 6.
REGULATION OF E+I-DEPENDENT GENE EXPRESSION IN HUMAN ASM BY FLUTICASONE*: DIFFERENTIAL GENE EXPRESSION IN GFP-EXPRESSING CELLS PRETREATED WITH FLU AND STIMULATED WITH EGF+IL-1β AS COMPARED TO GFP-EXPRESSING CELLS STIMULATED WITH EGF+IL-1β ONLY
| Symbol | Name | Fold Change |
|---|---|---|
| C10orf10 | chromosome 10 open reading frame 10 | 196.79 |
| TSC22D3 | TSC22 domain family, member 3 | 55.58 |
| FAM107A | family with sequence similarity 107, member A | 27.61 |
| ZBTB16 | zinc finger and BTB domain containing 16 | 27.16 |
| MT1M | metallothionein 1M | 25.36 |
| ZNF22 | zinc finger protein 22 (KOX 15) | 13.36 |
| CTGF | connective tissue growth factor | 11.07 |
| FOXO1 | forkhead box O1 | 10.67 |
| WISP1 | WNT1 inducible signaling pathway protein 1 | 10.32 |
| SLC19A2 | solute carrier family 19 (thiamine transporter), member 2 | 10.07 |
| IRS2 | insulin receptor substrate 2 | 9.94 |
| IMPA2 | inositol(myo)-1(or 4)-monophosphatase 2 | 9.33 |
| TIPARP | TCDD-inducible poly(ADP-ribose) polymerase | 9.24 |
| PER1 | period homolog 1 (Drosophila) | 8.26 |
| TMEM164 | transmembrane protein 164 | 7.91 |
| SAA2 | serum amyloid A2 | 7.69 |
| PRUNE2 | prune homolog 2 (Drosophila) | 7.60 |
| MAOA | monoamine oxidase A | 7.50 |
| GLUL | glutamate-ammonia ligase (glutamine synthetase) | 7.31 |
| PLXNA2 | plexin A2 | 7.15 |
| BCL2A1 | BCL2-related protein A1 | −64.26 |
| IL11 | interleukin 11 | −45.69 |
| PTGS2 | prostaglandin-endoperoxide synthase 2 | −32.43 |
| BMP2 | bone morphogenetic protein 2 | −30.59 |
| IL33 | interleukin 33 | −25.07 |
| HBEGF | heparin-binding EGF-like growth factor | −22.81 |
| GPRC5A | G protein-coupled receptor, family C, group 5, member A | −21.44 |
| CCL8 | chemokine (C-C motif) ligand 8 | −18.45 |
| IFIT1 | interferon-induced protein with tetratricopeptide repeats 1 | −18.19 |
| IL13RA2 | interleukin 13 receptor, alpha 2 | −17.38 |
| CH25H | cholesterol 25-hydroxylase | −15.16 |
| NR4A2 | nuclear receptor subfamily 4, group A, member 2 | −14.54 |
| LIF | leukemia inhibitory factor (cholinergic differentiation factor) | −12.94 |
| IL1B | interleukin 1, beta | −12.87 |
| FST | follistatin | −9.46 |
| LPXN | leupaxin | −9.10 |
| TRAF1 | TNF receptor-associated factor 1 | −8.68 |
| DUSP10 | dual specificity phosphatase 10 | −8.37 |
| ID2 | inhibitor of DNA binding 2, dominant negative helix-loop-helix protein | −8.12 |
| PLK2 | polo-like kinase 2 (Drosophila) | −7.63 |
For definition of abbreviations, see Table 1.
The 20 most increased and decreased gene transcripts in GFP-expressing cells pretreated with Flu for 30 minutes and co-stimulated with EGF+IL-1β for 8 hours as compared with GFP-expressing cells co-stimulated with EGF+IL-1β only (n = 3).
TABLE 7.
REGULATION OF EGF+IL-1β–DEPENDENT GENE EXPRESSION BY PKA INHIBITION*: DIFFERENTIAL GENE EXPRESSION IN PKI-GFP–EXPRESSING CELLS STIMULATED WITH EGF+IL-1β FOR 8 HOURS AS COMPARED TO GFP-EXPRESSING CELLS STIMULATED WITH EGF+IL-1β
| Symbol | Name | Fold Change |
|---|---|---|
| RGS4 | Regulator of G-protein signaling 4 | 13.93 |
| DACT1 | Dapper, antagonist of beta-catenin, homolog 1 | 13.59 |
| SLC14A1 | Solute carrier family 14 (urea transporter), member 1 | 10.08 |
| KCNJ2 | Potassium inwardly-rectifying channel, subfamily J, member 2 | 9.64 |
| CNIH3 | Cornichon homolog 3 (Drosophila) | 9.29 |
| FGF5 | Fibroblast growth factor 5 | 7.14 |
| NET1 | Neuroepithelial cell transforming gene 1 | 6.60 |
| MEOX1 | Mesenchyme homeobox 1 | 6.11 |
| SOCS2 | Suppressor of cytokine signaling 2 | 4.68 |
| RGS5 | Regulator of G-protein signaling 5 | 4.34 |
| TMEM16A | Transmembrane protein 16A | 4.16 |
| C3orf52 | Chromosome 3 open reading frame 52 | 4.15 |
| MYL9 | Myosin, light chain 9, regulatory | 2.83 |
| TPM1 | Tropomyosin 1 (alpha) | 2.71 |
| NRG1 | Neuregulin 1 | 2.49 |
| DAB2 | Disabled homolog 2, mitogen-responsive phosphoprotein | 2.48 |
| PML | Promyelocytic leukemia | 2.29 |
| ZFP36L2 | Zinc finger protein 36, C3H type-like 2 | 2.29 |
| SAMD4A | Sterile alpha motif domain containing 4A | 2.14 |
| PALLD | Palladin, cytoskeletal associated protein | 2.10 |
| AVPI1 | Arginine vasopressin-induced 1 | −8.75 |
| ID2 | Inhibitor of DNA binding 2, dominant negative helix-loop-helix protein | −4.59 |
| MYBL1 | v-myb myeloblastosis viral oncogene homolog (avian)-like 1 | −3.96 |
| FAM110B | Family with sequence similarity 110, member B | −3.65 |
| C14orf132 | Chromosome 14 open reading frame 132 | −2.20 |
| SLCO4A1 | Solute carrier organic anion transporter family, member 4A1 | −2.17 |
| TSC22D1 | TSC22 domain family, member 1 | −2.10 |
| CTSL1 | Cathepsin L1 | −1.98 |
| WWC3 | WWC family member 3 | −1.97 |
Definition of abbreviations: EGF, epidermal growth factor; PKA, protein kinase A.
The 20 most increased and decreased gene transcripts in PKI-GFP–expressing cells stimulated with EGF+IL-1β for 8 hours as compared with GFP-expressing cells co-stimulated with EGF+IL-1β (n = 3). Only nine down-regulated genes met the criteria of significance and 2-fold change.
TABLE 8.
EGF+IL-1B–REGULATED TRANSCRIPTS AFFECTED SIMILARLY BY FLUTICASONE AND PKA INHIBITION*
| Fold Change
|
|||
|---|---|---|---|
| Symbol | Name | GFP FluEGF+IL-1β–GFP EGF+IL-1β | PKI EGF+IL-1β–GFP EGF+IL-1β |
| C10orf10 | Chromosome 10 open reading frame 10 | 196.79 | 2.55 |
| C13orf15 | Chromosome 13 open reading frame 15 | 14.92 | 2.25 |
| CTGF | Connective tissue growth factor | 11.07 | 3.03 |
| TIPARP | TCDD-inducible poly(ADP-ribose) polymerase | 9.24 | 3.31 |
| GRAMD3 | GRAM domain containing 3 | 7.63 | 2.34 |
| MBNL1 | Muscleblind-like (Drosophila) | 6.29 | 2.49 |
| DNAJB4 | DnaJ (Hsp40) homolog, subfamily B, member 4 | 5.45 | 2.53 |
| ZFP36L2 | Zinc finger protein 36, C3H type-like 2 | 4.59 | 3.43 |
| CYR61 | Cysteine-rich, angiogenic inducer, 61 | 4.56 | 6.99 |
| KLF7 | Kruppel-like factor 7 (ubiquitous) | 4.41 | 2.61 |
| ALCAM | Activated leukocyte cell adhesion molecule | 4.36 | 2.03 |
| TACC1 | Transforming, acidic coiled-coil containing protein 1 | 3.72 | 2.21 |
| POLR3C | Polymerase (RNA) III (DNA directed) polypeptide C (62 kD) | 3.51 | 2.01 |
| CDC42EP3 | CDC42 effector protein (Rho GTPase binding) 3 | 3.47 | 3.15 |
| F3 | Coagulation factor III (thromboplastin, tissue factor) | 3.41 | 3.63 |
| USP33 | Ubiquitin specific peptidase 33 | 3.40 | 2.02 |
| ZFAND5 | Zinc finger, AN1-type domain 5 | 3.39 | 2.13 |
| TJP2 | Tight junction protein 2 (zona occludens 2) | 3.36 | 3.17 |
| FAM69A | Family with sequence similarity 69, member A | 3.09 | 3.41 |
| HMGA2 | High mobility group AT-hook 2 | 2.87 | 2.83 |
| C5orf30 | Chromosome 5 open reading frame 30 | 2.83 | 3.09 |
| FOXO3 | Forkhead box O3 | 2.82 | 2.08 |
| PHACTR2 | Phosphatase and actin regulator 2 | 2.80 | 2.17 |
| STX7 | Syntaxin 7 | 2.79 | 2.26 |
| NR2F2 | Nuclear receptor subfamily 2, group F, member 2 | 2.69 | 2.12 |
| ADD3 | Adducin 3 (gamma) | 2.69 | 2.02 |
| SWAP70 | SWAP-70 protein | 2.66 | 2.12 |
| TRAK2 | Trafficking protein, kinesin binding 2 | 2.40 | 2.39 |
| SHANK2 | SH3 and multiple ankyrin repeat domains 2 | 2.39 | 2.40 |
| RAB22A | RAB22A, member RAS oncogene family | 2.38 | 2.05 |
| DPP8 | Dipeptidyl-peptidase 8 | 2.38 | 2.29 |
| CLASP1 | Cytoplasmic linker associated protein 1 | 2.37 | 2.29 |
| NET1 | Neuroepithelial cell transforming gene 1 | 2.32 | 6.60 |
| PPM2C | Protein phosphatase 2C, magnesium-dependent, catalytic subunit | 2.32 | 3.03 |
| LBH | Limb bud and heart development homolog (mouse) | 2.28 | 2.59 |
| RSL1D1 | Ribosomal L1 domain containing 1 | 2.24 | 2.12 |
| AKAP13 | A kinase (PRKA) anchor protein 13 | 2.20 | 2.41 |
| SPRY4 | Sprouty homolog 4 (Drosophila) | 2.19 | 3.05 |
| SEMA5A | Sema domain, seven thrombospondin repeats (type 1 and type 1-like), transmembrane domain (TM) and short cytoplasmic domain, (semaphorin) 5A | 2.18 | 2.02 |
| EIF4G3 | Eukaryotic translation initiation factor 4 gamma, 3 | 2.18 | 2.39 |
| ZFP106 | Zinc finger protein 106 homolog (mouse) | 2.16 | 2.99 |
| SMAD4 | SMAD family member 4 | 2.13 | 2.11 |
| FILIP1L | Filamin A interacting protein 1-like | 2.12 | 2.22 |
| KLF6 | Kruppel-like factor 6 | 2.08 | 2.51 |
| EIF3A | Eukaryotic translation initiation factor 3, subunit A | 2.07 | 2.16 |
| AHNAK | AHNAK nucleoprotein | 2.05 | 2.55 |
| TTC3 | Tetratricopeptide repeat domain 3 | 2.04 | 2.24 |
| MLL | Myeloid/lymphoid or mixed-lineage leukemia | 2.02 | 2.27 |
| SLCO4A1 | Solute carrier organic anion transporter family, member 4A1 | −2.03 | −2.17 |
| PGF | Placental growth factor | −2.05 | −2.17 |
| CES1 | Carboxylesterase 1 | −2.19 | −2.52 |
| NR4A1 | Nuclear receptor subfamily 4, group A, member 1 | −2.37 | −2.07 |
| TLE4 | Transducin-like enhancer of split 4 | −2.38 | −2.07 |
| HIST2H2BE | Histone cluster 2, H2be | −2.38 | −2.70 |
| SMAD6 | SMAD family member 6 | −2.39 | −2.38 |
| OLFML1 | Olfactomedin-like 1 | −2.40 | −2.40 |
| RPLP2 | Ribosomal protein, large, P2 | −2.41 | −2.04 |
| PTPRE | Protein tyrosine phosphatase, receptor type, E | −2.47 | −2.81 |
| FOXC1 | Forkhead box C1 | −2.58 | −3.31 |
| TNFSF10 | Tumor necrosis factor (ligand) superfamily, member 10 | −2.59 | −2.03 |
| SLC39A8 | Solute carrier family 39 (zinc transporter), member 8 | −2.78 | −2.36 |
| GDF15 | Growth differentiation factor 15 | −2.90 | −2.09 |
| PGBD5 | PiggyBac transposable element derived 5 | −3.25 | −2.52 |
| OASL | 2′-5′-oligoadenylate synthetase-like | −3.26 | −3.06 |
| SLC2A3 | Solute carrier family 2 (facilitated glucose transporter), member 3 | −3.39 | −2.10 |
| AVPI1 | Arginine vasopressin-induced 1 | −3.55 | −8.75 |
| ZEB1 | Zinc finger E-box binding homeobox 1 | −3.61 | −2.85 |
| MYBL1 | v-myb myeloblastosis viral oncogene homolog (avian)-like 1 | −3.63 | −3.96 |
| DAAM1 | Dishevelled associated activator of morphogenesis 1 | −3.91 | −2.22 |
| PDE4B | Phosphodiesterase 4B, cAMP-specific | −4.02 | −2.23 |
| PDE4D | Phosphodiesterase 4D, cAMP-specific | −4.80 | −2.78 |
| LYPD3 | LY6/PLAUR domain containing 3 | −4.84 | −3.45 |
| FLRT3 | Fibronectin leucine rich transmembrane protein 3 | −5.03 | −2.64 |
| TSC22D1 | TSC22 domain family, member 1 | −5.13 | −2.10 |
| NGFB | Nerve growth factor, beta polypeptide | −5.34 | −2.03 |
| TNFRSF1B | Tumor necrosis factor receptor superfamily, member 1B | −7.32 | −2.04 |
| ID2 | Inhibitor of DNA binding 2, dominant negative helix-loop-helix protein | −8.12 | −3.07 |
| FST | Follistatin | −9.46 | −2.75 |
| CCL7 | Chemokine (C-C motif) ligand 7 | −11.87 | −3.10 |
| NR4A2 | Nuclear receptor subfamily 4, group A, member 2 | −14.54 | −4.00 |
| CCL8 | Chemokine (C-C motif) ligand 8 | −18.45 | −2.76 |
| GPRC5A | G protein–coupled receptor, family C, group 5, member A | −21.44 | −2.10 |
| CXCR7 | Chemokine (C-X-C motif) receptor 7 | −22.33 | −6.89 |
| IL33 | Interleukin 33 | −25.07 | −3.77 |
For definition of abbreviations, see Table 7.
All EGF+IL-1β–regulated genes affected similarly by Flu (GFP FluEI-GFP EI) and PKA inhibition (PKI EI-GFP EI). GFP FluEI-GFP EI denotes comparison of gene expression in GFP-expressing cells pretreated with Flu and stimulated with EGF+IL-1β as compared with. GFP-expressing cells stimulated with EGF+IL-1β only. PKI EI-GFP EI denotes comparison of gene expression in PKI-expressing cells stimulated with EGF+IL-1β as compared with. GFP-expressing cells stimulated with EGF+IL-1β.
TABLE 9.
DISPARATE EFFECTS OF FLUTICASONE AND PKA INHIBITION ON EGF+IL-1β–DEPENDENT GENES*
| Symbol | Name | PKI EGF+IL-1β–GFP EGF+IL-1β up | GFP FluEGF+IL-1β–GFP EGF+IL-1β down |
|---|---|---|---|
| RGS4 | Regulator of G-protein signaling 4 | 12.38 | −6.12 |
| SLC14A1 | Solute carrier family 14 (urea transporter), member 1 | 10.08 | −2.05 |
| RGS5 | Regulator of G-protein signaling 5 | 3.89 | −2.58 |
| MMP10 | Matrix metallopeptidase 10 (stromelysin 2) | 3.60 | −3.78 |
| SLC5A3 | Solute carrier family 5 (inositol transporters), member 3 | 3.55 | −2.43 |
| DIO2 | Deiodinase, iodothyronine, type II | 3.18 | −3.59 |
| NRG1 | Neuregulin 1 | 3.01 | −2.79 |
| LRRC32 | Leucine rich repeat containing 32 | 2.93 | −2.24 |
| IFIT3 | Interferon-induced protein with tetratricopeptide repeats 3 | 2.91 | −2.98 |
| NAV3 | Neuron navigator 3 | 2.78 | −2.85 |
| AMIGO2 | Adhesion molecule with Ig-like domain 2 | 2.65 | −5.72 |
| EHD1 | EH-domain containing 1 | 2.64 | −2.72 |
| MMP12 | Matrix metallopeptidase 12 (macrophage elastase) | 2.48 | −6.70 |
| HBEGF | Heparin-binding EGF-like growth factor | 2.45 | −14.96 |
| FGF2 | Fibroblast growth factor 2 (basic) | 2.44 | −4.52 |
| SMAD3 | SMAD family member 3 | 2.43 | −3.60 |
| GBP1 | Guanylate binding protein 1, interferon-inducible, | 2.39 | −3.18 |
| HAS2 | Hyaluronan synthase 2 | 2.36 | −4.23 |
| PLK2 | Polo-like kinase 2 (Drosophila) | 2.27 | −7.63 |
| MMP1 | Matrix metallopeptidase 1 (interstitial collagenase) | 2.27 | −28.50 |
| CSF1 | Colony-stimulating factor 1 (macrophage) | 2.23 | −2.93 |
| LEPREL1 | Leprecan-like 1 | 2.09 | −3.54 |
| CSF2 | Colony-stimulating factor 2 | 2.08 | −2.89 |
| COPA | Coatomer protein complex, subunit alpha | 2.08 | −4.77 |
| TRAF1 | TNF receptor-associated factor 1 | 2.05 | −8.68 |
| BPGM | 2,3-bisphosphoglycerate mutase | 2.04 | −2.20 |
| PRRX1 | Paired related homeobox 1 | 2.03 | −3.50 |
| KCTD12 | Potassium channel tetramerisation domain containing 12 | −3.12 | 2.01 |
| AKR1C2 | Aldo-keto reductase family 1, member C2 | −3.01 | 3.02 |
| GAS1 | Growth arrest–specific 1 | −2.94 | 3.85 |
| FADS1 | Fatty acid desaturase 1 | −2.84 | 2.22 |
| TGFBR3 | Transforming growth factor, beta receptor III | −2.62 | 2.50 |
| ADAMTS5 | ADAM metallopeptidase with thrombospondin type 1 motif, 5 | −2.29 | 2.38 |
| ADAMTS1 | ADAM metallopeptidase with thrombospondin type 1 motif, 1 | −2.22 | 2.02 |
| SLC19A2 | Solute carrier family 19 (thiamine transporter), member 2 | −2.08 | 10.07 |
| CEBPD | CCAAT/enhancer binding protein (C/EBP), delta | −2.07 | 2.32 |
For definition of abbreviations, see Table 7.
Comparison of effects of PKI expression versus Flu pretreatment on gene transcript level changes effected by EGF+IL-1β (relative to that in GFP-expressing cells stimulated with EGF+IL-1β).
For other experiments, statistically significant differences among groups were assessed by either ANOVA with Fisher's PLSD post hoc analysis (Statview 4.5; Abacus Concepts, Berkeley, CA) or by t test for paired samples, with P values < 0.05 sufficient to reject the null hypothesis.
RESULTS
To examine changes in human ASM gene expression that occur under disparate mitogenic conditions effected by growth factors, cytokines, glucocorticoids, and PKA inhibition, microarray analyses were conducted in parallel with [3H]-thymidine incorporation assays using ASM cultures derived from four separate donors. As previously reported (3), IL-1β was capable of inhibiting EGF-stimulated growth in GFP-expressing cells (Figure 2). However, in PKI- or in GFP-expressing cells pretreated with Flu, IL-1β greatly enhanced EGF-stimulated proliferation.
Figure 2.

Glucocorticoid (GC) treatment and protein kinase A (PKA) inhibition regulate mitogenic effects of IL-1β. Effect of PKA inhibition via heterologous expression of PKA inhibitor (PKI) or pretreatment with GC on IL-1β-, EGF-, and EGF+IL-1β–stimulated [3H]thymidine incorporation in human ASM. Data represent mean ± SE values, n = 4.
To examine the changes in gene expression associated with these conditions, cultures were stimulated for 8 hours with vehicle, EGF, IL-1β, or E+I with or without Flu pretreatment and total RNA harvested for transcriptome analysis using the Affymetrix U133A chip. An 8-hour time point was chosen in an attempt to capture gene expression changes induced by both early (acute signaling events) and delayed (induced autocrine-dependent) signaling events.
Chip Quality and Clustering Analysis
Microarray data were first analyzed with dChip (17) to ensure adequate chip quality, assess variability among replicate samples (biological variability), and assess the effect of donor line origin and processing time (batch effect). The analyses reported adequate chip quality and no batch effects were observed (data not shown). Clustering analysis of chip-to-chip variability among replicates within group suggests small variance (see Figure E1 in the online supplement) and differences present likely reflect biological variability associated with use of cells from different donors. Additional information regarding the chip-to-chip variability is available within the normalized signal matrix generated by GCRMA and from the individual .cel files enclosed within the GEO submission. Subsequent clustering analyses based on genes similarly expressed among all chips (variability of 0.5–100 and present call 50 or 20%, respectively) indicated grouping of samples into two main categories with two subcategories each (Figure 3): (1) samples treated with vehicle, EGF, or Flu with both GFP and PKI cells stimulated with EGF clustering in subgroup 1a, while subcategory 1b contained basal and Flu treated samples; and (2) samples treated with IL-1β or E+I with or without Flu, in which GFP and PKI cells treated with IL-1β or E+I clustered into 2a and all samples stimulated in presence of Flu grouped in 2b. This pattern of clustering suggests that while Flu has minimal effect on genes expressed under the unstimulated (basal) condition, Flu strongly modulates gene expression regulated by IL-1β, EGF, and E+I. EGF-stimulated samples clustered separately from IL-1β– and E+I- stimulated conditions; hence EGF appears to regulate a distinct set of genes corresponding to the proliferative phenotype. Clustering of IL-1β and E+I samples together suggests a common gene expression profile, with IL-1β having a dominant effect modulated by EGF on the human ASM transcriptome. This pattern of clustering was taken into consideration during subsequent analyses of differential gene expression, and the analyses focused on differences observed within and between EGF and IL-1β/E+I groups with respect to cell growth/proliferation and inflammation. Of note, the transcriptome analysis also indicated regulation by Flu and PKA of genes involved in metabolic processes in ASM cells. However, due to the breadth of the findings, these analyses are not discussed in the current manuscript.
Figure 3.

Clustering analysis of microarray data with dCHIP. Clustering analysis based on similarly regulated genes (variability 0.5–100 and present call 50%) revealed clustering into two groups with the following subgroups: (1a) Flu or Flu- and EGF-stimulated cells; (1b) unstimulated and EGF-stimulated cells; (2a) IL-1β– or E+I-stimulated cells; (2b) IL-1β– or E+I-stimulated cells in presence of Flu. Stars by sample name indicate number of replicates per condition + 1.
Regulation of Gene Expression by EGF, IL-1β, and E+I
EGF stimulation of human ASM cells resulted in differential regulation of genes involved in processes including cell death, cell cycle, cell growth and proliferation, cell development, and gene expression (analysis by Bio Functions IPA; Figure E2), consistent with the known mitogenic effect of EGF. Additional analysis using DAVID indicated activation of MAPK, JAK-STAT, and Wnt signaling pathways (Table E1). The top 20 induced- and 20 most down-regulated genes with B values > 0 are listed in Table 1 (and complete differential expression data presented in Table E2). Consistent with the known mitogenic effect of EGF, among the most significantly induced genes were those encoding proteins involved in cell proliferation (FOS-like antigen 1, early growth response 1), whereas numerous genes encoding negative regulators of cell cycle progression (including S100 calcium binding protein A2, ephrin B2, and cyclin-dependent kinase inhibitor 1C [p57/Kip2]) were down-regulated. Also induced were genes known to be involved in the negative feedback to mitogenic signaling, including dual specificity phosphate (DUSP)-1 and -6 and sprouty homolog-2 and -4.
Stimulation with IL-1β induced expression of numerous inflammatory genes (Tables 2 and E3), many of which were similarly induced by E+I treatment, including COX-2– and PKA-dependent genes previously shown to be modulated by COX-2 induction (see below). The overrepresented biological functions among the set of genes regulated by IL-1β included cell death, cell growth and proliferation, and cell cycle (Bio Functions IPA analysis; Figure E3), as well as inflammatory response and chemokine and cytokine signaling (DAVID analysis; Table E4). Our results are in agreement with previous studies describing those genes regulated in ASM cells stimulated 4 or 24 hours with IL-1β (22, 23).
Co-stimulation with IL-1β and EGF regulated expression of both mitogenic and inflammatory genes (Tables 3 and E5). As with IL-1β, E+I–up-regulated genes included those encoding numerous chemokines (of both the CCL and CXCL families), interleukins (including IL-8, IL-6, and IL-11), and proteases (see below), as well as other genes associated with the inflammatory process. EGF had a variable effect on the IL-1β–dependent genes, while IL-1β had a minimal effect on EGF-regulated genes involved in the progression of cell proliferation, as most were similarly regulated by E+I. However, E+I typically induced higher expression (in terms of fold change, relative to EGF-stimulated cells) of transcripts encoding negative regulators of mitogenic signaling (e.g., DUSP1 and -6, sprouty 4). Both Bio Functions IPA and DAVID analyses (Figure E4 and Table E6) indicated differential regulation of genes involved in cell death, cell cycle, cell morphology, cell growth and proliferation, and (negative) regulation of progression through cell cycle in addition to cytokine and chemokine signaling and inflammatory response. Most importantly, E+I stimulation strongly induced abundance of transcripts encoding cytoplasmic phospholipase A2, COX-2, and PGE synthase (PGES), consistent with analysis of protein induction described previously (10) These changes lead to increased production of PGE2 and activation of PKA in human ASM cells, which we have previously demonstrated to be critical to the growth inhibitory effect of IL-1β and TNF-α on human ASM (3). Increases in phosphodiesterases (PDE) 4B and 4D further reflected activation of PKA, as these genes are established to be PKA-dependent and comprise an important negative feedback regulation of Gαs-coupled receptor signaling (24). Interestingly, both IL-1β alone and E+I also regulated genes involved in the EGF signaling pathway, including those encoding the EGF receptor and downstream intermediates in the mitogen-activated protein kinase (MAPK) and PI3K pathways. Given our functional data (Figure 2 and Ref. 3), these transcriptome data suggest that IL-1β is capable of enhancing the signaling of EGF by positively regulating elements of the EGF receptor pathway. However, the consequences of this regulation are overridden by the induction of COX-2/PGES and the associated increase in PKA activity.
In addition, IL-1β and E+I stimulation could potentially regulate ASM proliferation through their effects on expression of numerous proteases, their receptors, and inhibitors. In the E+I condition, we observed significant induction of many of these genes, including those encoding MMP-1 and -3, plasminogen activator/urokinase receptor, and tissue factor pathway inhibitor 2. Moreover, several other transcripts encoding proteases or protease inhibitors exhibited a greater than 2-fold increase or decrease, yet due to variability among cultures failed to achieve a positive B value (Tables E3 and E5). These findings suggest a potential profound effect on extracellular matrix (ECM) remodeling. ECM provides a structural support for the cells and has been shown to bind and trap a variety of growth factors and other bioactive molecules. Increased protease activity can promote cell proliferation through effects on cell adhesion and integrin signaling (25) as well as via release of pro-mitogenic ligands (reviewed in Ref. 26). In addition, proteases can directly, or by activation of other proteases, stimulate cell surface receptors (e.g., GPCRs including PARs) leading to enhanced cell proliferation, as discussed below. We also observed that IL-1β– and E+I-dependent regulation of proteases was accompanied by enhanced expression of transcripts encoding collagens Vα3 and XIIIα1, and suppression of laminin α2 mRNA. Both collagen and laminin ECM proteins have been shown to regulate growth factor–stimulated ASM cell proliferation (27, 28). Thus, ECM remodeling and subsequent changes in ECM composition could be another mechanism through which inflammatory mediators regulate ASM growth.
Collectively, these data support and extend the emerging concept of ASM cell as an immunomodulatory cell in the airway (29). ASM cells had previously been regarded as cells whose singular purpose was to contract to regulate airway patency. However, recent studies (reviewed in Refs. 30–32) indicate that ASM cells can actively respond to the environmental signals by remodeling the ECM, and secrete numerous inflammatory mediators (e.g., cytokines, interleukins, prostaglandins) and growth factors. These synthetic abilities position ASM cells as modulators of the inflammatory process and key participants in coordinating the inflammatory cell influx into the asthmatic lung.
Fluticasone-Regulated Genes in ASM Cells
In cells stimulated with EGF, IL-1β, or E+I, Flu pretreatment greatly modulated gene expression with the largest effect on genes demonstrated to be involved in inflammation (see below) (Tables 4–6 and E7–E10). EGF-dependent gene expression modulated by Flu was most significantly associated with oxidative stress response, GC receptor signaling, chemokine signaling (Figure E5), and cell interaction with ECM (Table E11). Flu regulated many EGF-dependent genes involved in cell proliferation, such as those encoding cyclin D3, Fos, and PI3K p110α. However, Flu regulation of EGF-dependent genes important to cell proliferation and cell cycle regulation showed no obvious pattern, consistent with the small net mitogenic effect on ASM cells (Figure 2 and Ref. 3).
In cells stimulated with IL-1β or E+I, Flu suppressed expression of inflammatory genes such as those encoding interleukins (IL-1β, -33, -11), chemokines (CCL-7, CCL-8, CXCL-11), and cytokine receptors (IL-13Rα, IL-15Rα), as well as other inflammatory regulators including COX-2, PGE and prostacyclin synthases, and TNF-α– and interferon-signaling regulatory molecules. These observations are in agreement with suppressive effects of GCs on inflammatory gene expression induced by IL-1β, TNF-α, or IFN-γ in both bronchial epithelial and ASM cells (22, 33). In many instances, however, although expression of these genes was reduced by Flu, it remained significantly elevated compared with that observed for the unstimulated condition. Among the genes only minimally (and not significantly) suppressed by Flu were those encoding colony-stimulating factors 1 and 3, IL-8, CXCL-1 and -5, and CCL2, which primarily target neutrophils, basophils, and eosinophils (34). That GCs fail to effectively limit ASM production of neutrophil chemoattractive agents is consistent with the failure of GCs in managing chronic obstructive pulmonary disease.
To further examine the ability of Flu to suppress expression of ASM chemotactic agents, conditioned media were harvested from ASM cells stimulated 18 hours with EGF+IL-1β in the presence/absence of Flu. The chemotactic effect of these media on purified human neutrophils was tested using a standard Boyden chamber assay (Figure 4). Results demonstrate that (E+I) conditioned media promoted a variable but strong chemotaxis of neutrophils. Consistent with the minimal effect of Flu on the levels of induced transcripts encoding chemotactic agents, Flu pretreatment had but a small, nonsignificant effect on neutrophil chemotaxis induced by conditioned media.
Figure 4.

Effects of fluticasone treatment of ASM on chemotactic effect of conditioned media from ASM. Conditioned media were harvested from GFP or PKI-GFP–expressing ASM cells treated for 18 hours with vehicle (basal) or EGF (10 ng/ml) plus IL-1β (20 U/ml) (E+I) ± 10 nM Flu. These media were tested for the ability to promote chemotaxis of human neutrophils purified (∼ 95% purity) from peripheral blood as described in Materials and Methods. Formyl-methionyl-leucyl-phenylalanine (FMLP; 1 μM) and IL-8 (1 μM) were used as positive controls. Data represent mean ± SE values, n = 4.
Comparison of the Flu effect on IL-1β– and E+I-regulated genes revealed that even though these stimuli regulate a similar set of genes, the suppressive effect of Flu was typically greater in cells stimulated with IL-1β alone (Table E12). This differential effect of Flu was observed for not only IL-1β– and E+I-dependent inflammatory (cytokines, interleukins, PG synthesis, interferon signaling) genes but also for other genes. Whether these results reflect an antagonistic effect of EGF on GC and GC receptor function (perhaps mediated by p42/p44 MAPK activity [35]), or a competitive dynamic at the level of transcriptional regulation of genes, is unclear.
Consistent with the promitogenic effect of Flu pretreatment on E+I-stimulated cells, Flu modulated expression of certain mitogenic and cell cycle regulatory genes (e.g., those encoding k-Ras, PI3K Cα, cyclins A1 and D3) and modulated the positive effects of IL-1β on genes encoding EGF signaling pathway proteins (described above) (Table E10). However, regulation of these transcripts was variable among cultures and not statistically significant (B < 0). With respect to E+I-regulated genes significantly regulated by Flu, the analysis indicated enrichment in transcription factors and their regulators. Numerous transcripts encoding zinc finger–containing proteins, such as ZBTB16, ZNF22, or PFH17, were increased by Flu pretreatment. In addition, Flu modulated the transcripts of ion-binding protein metallothionein 1M (MT1M), forkhead box O1 (FoxO1), and inhibitor of DNA binding 2 (ID2), proteins that regulate transcription factor activity. Interestingly, some of the gene products most highly induced by Flu pretreatment included putative positive regulators of cell proliferation: C10orf10, Fam107A, and Wisp1.
Flu pretreatment also significantly induced expression of transcripts encoding MMP-19, vinculin, integrins (α5 and α10), and collagen IV α1 in E+I-stimulated cells. As discussed above, ECM binding directly to integrins has the capacity to regulate cell proliferation; thus, modulation by Flu of the ECM composition, or expression of integrins and other proteins constituting focal adhesions, may be another mechanism by which Flu can regulate cell growth. Interestingly, Flu also increased levels of SMARCD2 mRNA encoding a protein involved in chromatin structure modification in an actin-dependent fashion.
Perhaps most importantly, however, Flu strongly and significantly attenuated the expression of COX-2, PGES, and cPLA2, resulting in suppression of PGE2 production and subsequent activation of PKA. Collectively, these data suggest that Flu promotes E+I-stimulated ASM proliferation due to potent suppression of PKA activation, via increased activity of transcription factors necessary for cell cycle progression, and via alterations in cell–matrix interaction. The extent to which regulation of mitogenic and cell cycle genes by Flu is an indirect effect caused by regulation of genes promoting increased intracellular PKA activity is explored in the analysis of effects of PKA inhibition below.
PKA-Regulated Genes in ASM Cells
We have previously asserted that effects of Flu on proliferation of human ASM cells are mediated through suppression of the COX-2/PGE2/PKA pathway and that PKA is a strong antimitogenic effector in these cells. Because PKA regulates gene expression via regulation of canonical mitogenic signaling pathways and activation of CREB and associated regulators, we evaluated PKA-dependent gene expression in cells stimulated with EGF, IL-1β, and E+I employing a heterologous stable expression of PKA-inhibiting peptide, PKI (Tables 7 and E14–17). Inhibition of PKA in cells stimulated with EGF was associated with regulation of cell movement, cell growth and proliferation, and cell-to-cell signaling (IPA analysis, Figure E6). However, despite apparent regulation of mitogenic gene expression that would suggest augmented growth, there was only small effect on cell proliferation in thymidine incorporation assays (Figure 2 and Ref. 3).
PKA inhibition had minimal effect on IL-1β–dependent expression of inflammatory genes, affecting only a small number of genes encoding chemokines, interleukins, and proteases (Table E16). Because PKI-expressing cells, in contrast to control cells, exhibited significant thymidine incorporation upon stimulation with IL-1β (Figure 2), we assessed PKI effects on IL-1β–regulated genes. Interestingly, PKA inhibition modulated expression of few cell proliferation/cell cycle genes, and such regulation was variable among cultures. Moreover, the (direction of) regulation of these transcripts was not consistent with a clear promitogenic profile (i.e., some promitogenic genes were down, whereas some antimitogenic genes were up).
Although stimulation with EGF and IL-1β causes the most profound activation of PKA (due to pronounced COX-2 and PGE2 induction) (10, 36), the effect of PKA inhibition on the ASM transcriptome remained limited in these cells (Tables 7 and E17). A handful of genes (including ID2, PLM, and EIF1B) known to be important to cell proliferation/cell cycling were significantly regulated, whereas numerous other genes exhibited a greater than 2-fold increase or decrease but failed to achieve a significant B value due to variability among cultures. Such variability is consistent with our previous observations that IL-1β–mediated PKA activation, and the functional consequences, indeed exhibit variability among human ASM cultures (3). Thus, the importance of targeting PKA-regulated gene expression in asthma therapy may be restricted to specific subpopulations, and will require a more comprehensive analysis of the (variability in) effects of PKA on the transcriptome than that afforded herein. Regarding the role of PKA in regulating growth in ASM stimulated with EGF+IL-1β, various interpretations of our data might be considered. First, those proteins encoded by transcripts identified as differentially regulated by PKI may prove critical in mediating ASM growth (and other ASM synthetic functions). Second, additional important effects of transcriptome regulation by PKA may occur at later time points and be missed by our analysis. This possibility must be considered in light of the fact that in other cell types, cAMP- and PKA-mediated inhibition of cell proliferation appears mediated primarily by cell cycle arrest associated with increased expression of p21/Cip1 and p27/Kip1 and decreased expression of D-type cyclins (reviewed in Refs. 37 and 38) and changes in the expression of these genes may be more evident at later time points. Finally, it is probable that PKA regulates important mitogenic signaling events that either do not impact the ASM transcriptome or elicit subtle, secondary transcriptional events. Such regulation may involve phosphorylation and regulation of cell cycle proteins (e.g., retinoblastoma protein [pRb] and its homologs p107 and p130) that enable cell cycle progression. Interestingly, in PKA-knockout mice cyclin D1 protein, but not mRNA levels, are increased (39). In addition, PKA has been shown capable of suppressing proteins important in enabling the induction of new protein synthesis critical to cell division (40). Ribosomal S6 protein, elongation factor-2 (EF-2), poly A–binding protein, and releasing factor 1 have all been identified as PKA substrates (41). PKA phosphorylation of Elongation factor-2 kinase (eEF-2K) has been shown to negatively regulate mRNA translation via the phosphorylation and inactivation of elongation factor-2 (eEF-2) (42, 43). In addition, PKA is known to inhibit numerous cytosolic mitogenic signaling pathways (activated by either growth factors or GPCRs) through acute phosphorylation of both receptors and their downstream effectors. It is interesting to note that both RGS5 and RGS6 are induced by PKA inhibition in EGF+IL-1β–stimulated cells. These proteins are feedback regulators of Gq/Gi-coupled receptor signaling, and are upregulated when Gq or Gi signaling is increased. In EGF+IL-1β–stimulated ASM cells, Gq or Gi signaling stimulated by the induction of numerous chemokines and other GPCR agonists (see below) can be further increased by relieving the constraint imposed by PKA-mediated phosphorylation of GPCRs, G proteins, and PLC (1).
Common/Differential Gene Regulation by Fluticasone and PKA Inhibition
Comparison of differential regulation of gene expression in human ASM cells by Flu and PKA inhibition demonstrated a set of similarly regulated genes that occurred under most stimulatory conditions (Tables 8, E18, E19). For such genes, whose regulation by Flu promotes a promitogenic or proinflammatory ASM phenotype, counteractive effects of inhaled β-agonists (promoting competitive PKA activity) may be critical for achieving effective asthma management, and may underlie the therapeutic advantage of GC and β-agonist combination therapy (44). In EGF+IL-1β–stimulated cells, transcripts similarly regulated by Flu or PKA inhibition included those encoding proteins involved in the inflammatory process (including CCL and CXCL chemokines, interleukins), cAMP-dependent signaling (phosphodiesterases), and growth regulation (see below). Some of the genes similarly regulated by Flu and PKA inhibition, such as PDE4B and ID2, exhibited a comparable magnitude of regulation (fold change). However, several genes, including IL-33, CCL8, and IL-1α and β, were more profoundly affected by Flu. Previous studies have identified some of these genes, including those including follistatin (45, 46) and connective tissue growth factor (47, 48), as both GC and PKA dependent. Our findings suggest the existence of numerous genes whose regulation of transcript abundance by GC is mediated in part through suppression of inflammation-induced PKA activity.
In addition, DAVID analysis identified genes that were regulated by Flu but not by PKA inhibition, or vice versa, in cells stimulated with IL-1β, EGF, or E+I (Table E20). The genes regulated by Flu but not PKI included those encoding proteins involved in inflammatory response, chemotaxis, apoptosis, and regulation of proliferation and progression through cell cycle, pathways regulating focal adhesion, actin cytoskeleton, and interactions of ECM or cytokines with cell receptors (Table E21). Genes regulated by PKA inhibition, but not by Flu pretreatment, also included those involved in cell proliferation, apoptosis, and pathways regulating ECM interactions with receptors, cell communication, and focal adhesion (Table E22). Also, selectively regulated by PKA inhibition were genes involved in regulation of cell size and signal transduction. The PKI-induced Flu-insensitive genes included those encoding chemokines (CX3CL1, CXC11, CSF1), cell cycle regulators (cyclin M1), mitogens (FGF5), and mitogenic pathway intermediates (Cyr61, RasGAP, and multiple MAPKKs), and ADAM and MMP proteases (ADAM-10, -19, and MMP-3, -10). Each of these proteins could contribute to asthma pathology via effects on inflammatory cells, ASM cell proliferation, or ECM remodeling. As PKA-dependent genes, these genes represent potentially important therapeutic targets of (PKA-activating) β-agonists. β-agonist–, PKA-mediated regulation of these genes may be most critical in the context of concomitant GC treatment, during which GC suppression of inflammation-induced COX-2/PGE2/PKA activity would be promitogenic. Interestingly, for IL-1β–regulated genes, all Flu-induced genes were also regulated by PKI, suggesting that the major effect of Flu on these genes may be due to inhibition of PKA activation.
We also observed that some genes under the IL-1β–, EGF-, and E+I- stimulatory conditions were regulated in the opposite direction by Flu and PKI (Tables 9, E23, and E24). These genes include those encoding proteases (MMP-1, -10, -12, and ADAMTS-1, -5), growth factors (heparin binding EGF-like growth factor, FGF2 and -5, CSF1 and -2), solute transporters (urea, inositol, thiamine, potassium), and GPCR-signaling regulators (RGS4 and -5). This set of genes is likely similarly regulated by GCs and β-agonists in a redundant or a reinforcing manner.
Because ASM cells stimulated with E+I in the presence of Flu or PKA inhibition exhibit enhanced growth, we compared the effects of Flu and PKI on expression of mitogenic regulators (Table E13). Interestingly, from this list the only promitogenic genes regulated by both Flu and PKA, and in a similar manner, were C13ORF15, CYR61, and ID2. Thus, our analysis suggests that GC/PKA-effected transcriptome changes that mediate or are associated with the promitogenic effects of GC treatment or PKA inhibition appear to be limited. Alternatively, mechanisms proposed above involving effects of suppressed PKA activity on protein translation, cell cycle control, and mitogenic signaling pathways may contribute significantly.
Role of Autocrine Factors and GPCR Signaling in Promoting Enhanced Proliferation of ASM Cells
Our laboratory has previously shown that Gq protein–coupled receptor signaling enhances (15, 49), while Gs-coupled receptor signaling attenuates (3), growth factor-stimulated ASM proliferation. We thus compared the expression of genes encoding proteins relevant to GPCR signaling in control, Flu-pretreated, or PKI-expressing cells stimulated with E+I (Table E25), conditions associated with attenuated or enhanced ASM proliferation (Figure 2). E+I stimulation resulted in decreased expression of transcripts encoding PI3K p85α, induction of PDE4B and D, DUSP1 and 6, and proteases (listed above), as well as differential regulation of genes encoding various GPCRs. Flu pretreatment resulted in suppression of transcripts of DUSP6 and PDE4B, and increased expression of Gαq and MMP19. In contrast, PKA inhibition increased expression of transcripts of RGS4 and 5. The disparate effects of Flu and PKA on RGS4 and -5 are interesting. Although our data suggest that GCs appear capable of directly regulating RGS4 and -5 independent of Gαq/Gαi signaling, the induction of RGS4 and -5 in PKI-expressing cells likely reflects the up-regulation of classical negative feedback regulation due to an increase in Gαq- and Gαi-coupled receptor signaling (50). Conversely, in PKI-expressing cells the observed decrease in PDE4B/D likely reflects the loss of negative feedback regulation for cAMP-dependent signaling, due to the reduction in cAMP coupling to its downstream effector PKA.
As noted above, E+I stimulation enhanced expression of genes encoding inflammatory agents such as cytokines, chemokines, and proteases (as compared with EGF or IL-1β stimulations alone) that can activate GPCRs. Although Flu suppressed expression of some of these genes, in many instances induction remained significant. Recently, expression of functional chemokine receptors CXCR1, CXCR2 (51), CCR7 (52), CCR1 (53), and CCR3 (54) has been demonstrated in ASM cells. Stimulation of these GPCRs resulted in ASM contraction and migration, and regulation of Ca2+ mobilization, suggesting receptor coupling to Gαq or Gαi. In addition, as discussed above, cytokine-induced expression of proteases can lead to activation of Gαq/Gαi-coupled PARs. Under conditions of suppressed PKA activity (by Flu treatment or PKI expression), Gαq/Gαi-coupled receptor signaling is further augmented. Such signaling, in conjunction with that stimulated by polypeptide growth factors, has been shown to contribute to the observed enhanced human ASM cell proliferation (14, 15).
To examine the ability of induced autocrine factors to stimulate Gαq/Gαi signaling in ASM cells, we tested the ability of conditioned media from GFP- or PKI-expressing cells to stimulate Ca2+ mobilization in ASM cells (Figures 5A and 5B). Media harvested from cells treated 18 hours with E+I promoted Ca2+ mobilization in both (quiesced) GFP- and PKI-expressing cells, with a significantly greater peak Ca2+ flux being observed in the PKI-expressing cells. Peak Ca2+ values were similar when using media harvested from GFP- or PKI-expressing cells. The ability of conditioned media to stimulate greater Ca2+ mobilization in PKI-expressing cells is consistent with the ability of PKA activity to antagonize Gq/Gi-dependent signaling at multiple junctures upstream of store-dependent Ca2+ release (1).
Figure 5.

Effects of autocrine/juxtacrine factors induced in ASM cells. (A and B) Conditioned media were harvested from GFP- and PKI-expressing cells stimulated for 18 hours with EGF (10 ng/ml) plus IL-1β (20 U/ml) and used to stimulate [Ca2+]i mobilization in GFP- and PKI-expressing cells as described in Materials and Methods. (A) Representative tracing and (B) cumulative results (mean ± SE values, n = 3) are presented. (C) dsRed-expressing cells were co-cultured with GFP- or PKI-GFP–expressing cells, stimulated with EGF (10 ng/ml), IL-1β (20 U/ml), or both in presence of BrdU (60 μM). BrdU incorporation was evaluated in dsRed+ and GFP+ cell populations. Data represent mean ± SE values, n = 4.
To further explore the capacity of autocrine factors to regulate ASM proliferation, we conducted co-culture experiments using GFP-, dsRed-, and PKI-GFP chimera–expressing cells and BrdU incorporation as a measure of cell proliferation stimulated with vehicle, IL-1β, EGF, or E+I. We reasoned that should autocrine factors produced by PKI-expressing cells be sufficient to augment EGF-stimulated cell growth, they would be capable of increasing BrdU incorporation of co-cultured dsRed-expressing (i.e., control) cells in a paracrine/juxtacrine manner. Results depicted in Figure 5C demonstrate that co-cultured dsRed- and GFP-expressing cells stimulated with E+I both exhibit lesser BrdU incorporation than when stimulated with EGF alone. In experiments in which dsRed- and PKI-GFP–expressing cells were co-cultured, control (dsRed) cells still exhibited lesser BrdU incorporation when stimulated with E+I compared with stimulation with EGF alone, whereas IL-1β predictably increased EGF-stimulated BrdU incorporation in PKI-GFP–expressing cells. These results suggest that the release of factors from PKI-GFP–expressing cells is not sufficient to augment EGF-stimulated growth and that inhibition of intracellular PKA activity in an absolute requirement. Collectively, data from Figure 5 suggest minimal differences in potential promitogenic autocrine factors released from GFP- versus PKI-expressing cells, and that the cellular response to such factors is dependent on the level of cellular PKA activity.
In summary, our studies provide a comprehensive analysis of the differential regulation of human ASM gene expression by GCs and PKA, in the context of mitogenic (EGF) and inflammatory (IL-1β and E+I) stimuli. In ASM cells, EGF primarily regulates expression of mitogenic effectors, whereas IL-1β strongly induces expression of proinflammatory genes including cytokines, chemokines, interleukins, PG synthesis enzymes, and other inflammatory regulators. Combination of the two conditions (E+I) results in enhanced expression of many inflammatory mediators, with variable effects on genes regulating ASM growth. The most important regulatory effect of IL-1β and E+I stimulation on ASM growth appears to be the induction of COX-2/PGES. The consequent PGE2 synthesis resulting in increased cellular PKA activity exerts profound control over the mitogenic capacity of both growth factors and GPCR agonists. The induction of numerous proteases and GPCR agonists by IL-1β suggests a potential mechanism underlying the cooperative nature of growth factors and cytokines in regulating ASM growth.
Clearly, however, limitations exist in our ability to interpret the functional significance of the induction of ASM mRNAs encoding cytokines, chemokines, and other autocrine/paracrine factors. Obvious limitations include the fact that changes in mRNA do not necessarily reflect changes in protein expression. Moreover, transcriptome changes observed after 8 hours of stimulation may fail to identify potential important regulatory effects at shorter and longer durations of IL-1β stimulation or GC treatment. Jarai and colleagues (23) compared changes in the ASM transcriptome that occur with 4 hours and 24 hours of stimulation with IL-1β. Although many genes were significantly up-regulated at both time points, differences in the magnitude of increase, and evidence of differential gene regulation, were frequently observed. Finally, the ASM transcriptome changes and their functional consequences depend on the complex environment of the airway in which ASM reside, which is impossible to replicate in in vitro models.
Through identification and comparison of genes differentially expressed under condition of GC treatment and PKA inhibition, our findings provide insight into mechanisms of action, with respect to ASM growth regulation, of the two most common anti-asthma drugs: inhaled steroids and β-agonists. The profile of Flu-sensitive and -insensitive genes provides a number of potential drug targets to guide the development of new generations of GCs or add-on therapies capable of addressing pathogenic mechanisms unaffected, or promoted, by GCs. Finally, our results further strengthen the concept of ASM as an important immunomodulatory cell in the airway, capable of generating numerous paracrine agents affecting multiple resident and infiltrating airway cell types involved in the orchestration of airway inflammation.
Supplementary Material
Acknowledgments
The authors thank Reen Wu for advice in data analysis and presentation.
This study was funded by National Institutes of Health grant HL58506 (to R.B.P.) and an independent investigator grant from GlaxoSmithKline (to R.B.P.).
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2008-0266OC on December 4, 2008
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Deshpande DA, Penn RB, Targeting G. Protein-coupled receptor signaling in asthma. Cell Signal 2006;18:2105–2120. [DOI] [PubMed] [Google Scholar]
- 2.Bonacci JV, Stewart AG. Regulation of human airway mesenchymal cell proliferation by glucocorticoids and beta2-adrenoceptor agonists. Pulm Pharmacol Ther 2006;19:32–38. [DOI] [PubMed] [Google Scholar]
- 3.Misior AM, Yan H, Pascual RM, Deshpande DA, Panettieri RA Jr, Penn RB. Mitogenic effects of cytokines on smooth muscle are critically dependent on protein kinase A and are unmasked by steroids and cyclooxygenase inhibitors. Mol Pharmacol 2008;73:566–574. [DOI] [PubMed] [Google Scholar]
- 4.Stewart AG, Harris T, Fernandes DJ, Schachte LC, Koutsoubos V, Guida E, Ravenhall CE, Vadiveloo P, Wilson JW. Beta2-adrenergic receptor agonists and CAMP arrest human cultured airway smooth muscle cells in the G(1) phase of the cell cycle: role of proteasome degradation of cyclin D1. Mol Pharmacol 1999;56:1079–1086. [DOI] [PubMed] [Google Scholar]
- 5.Dixon ER, Weinberg JA, Lew DB. Effect of dexamethasone on bovine airway smooth muscle cell proliferation. J Asthma 1999;36:519–525. [DOI] [PubMed] [Google Scholar]
- 6.Schramm CM, Omlor GJ, Quinn LM, Noveral JP. Methylprednisolone and isoproterenol inhibit airway smooth muscle proliferation by separate and additive mechanisms. Life Sci 1996;59:L9–L14. [DOI] [PubMed] [Google Scholar]
- 7.Stewart AG, Fernandes D, Tomlinson PR. The effect of glucocorticoids on proliferation of human cultured airway smooth muscle. Br J Pharmacol 1995;116:3219–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vlahos R, Stewart AG. Interleukin-1alpha and tumour necrosis factor-alpha modulate airway smooth muscle DNA synthesis by induction of cyclo-oxygenase-2: inhibition by dexamethasone and fluticasone propionate. Br J Pharmacol 1999;126:1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vlahos R, Lee KS, Guida E, Fernandes DJ, Wilson JW, Stewart AG. Differential inhibition of thrombin- and EGF-stimulated human cultured airway smooth muscle proliferation by glucocorticoids. Pulm Pharmacol Ther 2003;16:171–180. [DOI] [PubMed] [Google Scholar]
- 10.Pascual RM, Carr E, Seeds MC, Guo M, Panettieri RA, Peters SP, Penn RB. Regulatory features of interleukin-1beta-mediated prostaglandin E2 synthesis in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 2006;290:501–508. [DOI] [PubMed] [Google Scholar]
- 11.Penn RB, Pascual RM, Kim Y-M, Mundell SJ, Krymskaya VP, Panettieri RA Jr, Benovic JL. Arrestin specificity for G protein-coupled receptors in human airway smooth muscle. J Biol Chem 2001;276:32648–32656. [DOI] [PubMed] [Google Scholar]
- 12.Guo M, Pascual RM, Wang S, Fontana MF, Valancius CA, Panettieri RA Jr, Tilley SL, Penn RB. Cytokines regulate beta-2-adrenergic receptor responsiveness in airway smooth muscle via multiple PKA- and EP2 receptor-dependent mechanisms. Biochemistry 2005;44:13771–13782. [DOI] [PubMed] [Google Scholar]
- 13.Penn RB, Parent JL, Pronin AN, Panettieri RA Jr, Benovic JL. Pharmacological inhibition of protein kinases in intact cells: antagonism of beta adrenergic receptor ligand binding by H-89 reveals limitations of usefulness. J Pharmacol Exp Ther 1999;288:428–437. [PubMed] [Google Scholar]
- 14.Billington CK, Kong KC, Bhattacharyya R, Wedegaertner PB, Panettieri RA, Chan TO, Penn RB. Cooperative regulation of P70S6 kinase by receptor tyrosine kinases and G protein-coupled receptors augments airway smooth muscle growth. Biochemistry 2005;44:14595–14605. [DOI] [PubMed] [Google Scholar]
- 15.Kong KC, Billington CK, Gandhi U, Panettieri RA Jr, Penn RB. Cooperative mitogenic signaling by G protein-coupled receptors and growth factors is dependent on G(q/11). FASEB J 2006;20:1558–1560. [DOI] [PubMed] [Google Scholar]
- 16.Deshpande DA, Pascual RM, Wang SW, Eckman DM, Riemer EC, Funk CD, Penn RB. PKC-dependent regulation of the receptor locus dominates functional consequences of cysteinyl leukotriene type 1 receptor activation. FASEB J 2007;21:2335–2342. [DOI] [PubMed] [Google Scholar]
- 17.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA 2001;98:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Z, Irizarry RA. Stochastic models inspired by hybridization theory for short oligonucleotide arrays. J Comput Biol 2005;12:882–893. [DOI] [PubMed] [Google Scholar]
- 19.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004;3:3–20. [DOI] [PubMed] [Google Scholar]
- 20.Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 2003;4:3. [PubMed] [Google Scholar]
- 21.Hipp J, Atala A. GeneChips in stem cell research. Methods Enzymol 2006;420:162–224. [DOI] [PubMed] [Google Scholar]
- 22.Hakonarson H, Halapi E, Whelan R, Gulcher J, Stefansson K, Grunstein MM. Association between IL-1beta/TNF-alpha-induced glucocorticoid-sensitive changes in multiple gene expression and altered responsiveness in airway smooth muscle. Am J Respir Cell Mol Biol 2001;25:761–771. [DOI] [PubMed] [Google Scholar]
- 23.Jarai G, Sukkar M, Garrett S, Duroudier N, Westwick J, Adcock I, Chung KF. Effects of interleukin-1beta, interleukin-13 and transforming growth factor-beta on gene expression in human airway smooth muscle using gene microarrays. Eur J Pharmacol 2004;497:255–265. [DOI] [PubMed] [Google Scholar]
- 24.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 2007;76:481–511. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen TT, Ward JP, Hirst SJ. Beta1-integrins mediate enhancement of airway smooth muscle proliferation by collagen and fibronectin. Am J Respir Crit Care Med 2005;171:217–223. [DOI] [PubMed] [Google Scholar]
- 26.Gueders MM, Foidart JM, Noel A, Cataldo DD. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: potential implications in asthma and other lung diseases. Eur J Pharmacol 2006;533:133–144. [DOI] [PubMed] [Google Scholar]
- 27.Bonacci JV, Harris T, Wilson JW, Stewart AG. Collagen-induced resistance to glucocorticoid anti-mitogenic actions: a potential explanation of smooth muscle hyperplasia in the asthmatic remodelled airway. Br J Pharmacol 2003;138:1203–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirst SJ, Twort CH, Lee TH. Differential effects of extracellular matrix proteins on human airway smooth muscle cell proliferation and phenotype. Am J Respir Cell Mol Biol 2000;23:335–344. [DOI] [PubMed] [Google Scholar]
- 29.Tliba O, Amrani Y, Panettieri RA Jr. Is airway smooth muscle the “missing link” modulating airway inflammation in asthma? Chest 2008;133:236–242. [DOI] [PubMed] [Google Scholar]
- 30.Fernandes DJ, Bonacci JV, Stewart AG. Extracellular matrix, integrins, and mesenchymal cell function in the airways. Curr Drug Targets 2006;7:567–577. [DOI] [PubMed] [Google Scholar]
- 31.Hirst SJ, Martin JG, Bonacci JV, Chan V, Fixman ED, Hamid QA, Herszberg B, Lavoie JP, McVicker CG, Moir LM, et al. Proliferative aspects of airway smooth muscle. J Allergy Clin Immunol 2004;114:2–17. [DOI] [PubMed] [Google Scholar]
- 32.Parameswaran K, Willems-Widyastuti A, Alagappan VK, Radford K, Kranenburg AR, Sharma HS. Role of extracellular matrix and its regulators in human airway smooth muscle biology. Cell Biochem Biophys 2006;44:139–146. [DOI] [PubMed] [Google Scholar]
- 33.Pawliczak R, Logun C, Madara P, Barb J, Suffredini AF, Munson PJ, Danner RL, Shelhamer JH. Influence of IFN-gamma on gene expression in normal human bronchial epithelial cells: modulation of IFN-gamma effects by dexamethasone. Physiol Genomics 2005;23:28–45. [DOI] [PubMed] [Google Scholar]
- 34.Busse WW, Lemanske RF Jr. Asthma. N Engl J Med 2001;344:350–362. [DOI] [PubMed] [Google Scholar]
- 35.Tsitoura DC, Rothman PB. Enhancement of MEK/ERK signaling promotes glucocorticoid resistance in CD4+ T cells. J Clin Invest 2004;113:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pascual RM, Billington CK, Hall IP, Panettieri RA Jr, Fish JE, Peters SP, Penn RB. Mechanisms of cytokine effects on G protein-coupled receptor-mediated signaling in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 2001;281:1425–1435. [DOI] [PubMed] [Google Scholar]
- 37.Stork PJ, Schmitt JM. Crosstalk between CAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 2002;12:258–266. [DOI] [PubMed] [Google Scholar]
- 38.Zambon AC, Zhang L, Minovitsky S, Kanter JR, Prabhakar S, Salomonis N, Vranizan K, Dubchak I, Conklin BR, Insel PA. Gene expression patterns define key transcriptional events in cell-cycle regulation by CAMP and protein kinase A. Proc Natl Acad Sci USA 2005;102:8561–8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nadella KS, Kirschner LS. Disruption of protein kinase a regulation causes immortalization and dysregulation of D-type cyclins. Cancer Res 2005;65:10307–10315. [DOI] [PubMed] [Google Scholar]
- 40.Haccard O, Jessus C. Redundant pathways for Cdc2 activation in Xenopus oocyte: either cyclin B or Mos synthesis. EMBO Rep 2006;7:321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sugimoto I, Li Z, Sakamoto Y, Ito S, Hashimoto E. Mass-spectrometric identification of proteins detected in forskolin-stimulated Xenopus laevis oocytes using antibody against phospho-(Ser/Thr) CAMP-dependent protein kinase substrate. Biomed Res 2007;28:231–238. [DOI] [PubMed] [Google Scholar]
- 42.Diggle TA, Subkhankulova T, Lilley KS, Shikotra N, Willis AE, Redpath NT. Phosphorylation of elongation factor-2 kinase on serine 499 by CAMP-dependent protein kinase induces Ca2+/calmodulin-independent activity. Biochem J 2001;353:621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutzkow KB, Lahne HU, Naderi S, Torgersen KM, Skalhegg B, Koketsu M, Uehara Y, Blomhoff HK. Cyclic AMP inhibits translation of cyclin D3 in T lymphocytes at the level of elongation by inducing EEF2-phosphorylation. Cell Signal 2003;15:871–881. [DOI] [PubMed] [Google Scholar]
- 44.Miller-Larsson A, Selroos O. Advances in asthma and COPD treatment: combination therapy with inhaled corticosteroids and long-acting beta 2-agonists. Curr Pharm Des 2006;12:3261–3279. [DOI] [PubMed] [Google Scholar]
- 45.Leclerc N, Luppen CA, Ho VV, Nagpal S, Hacia JG, Smith E, Frenkel B. Gene expression profiling of glucocorticoid-inhibited osteoblasts. J Mol Endocrinol 2004;33:175–193. [DOI] [PubMed] [Google Scholar]
- 46.Michel U, Farnworth P. Follistatin steady state messenger ribonucleic acid levels in confluent cultures of a rat renal mesangial cell line are regulated by multiple factors. Endocrinology 1992;130:3684–3693. [DOI] [PubMed] [Google Scholar]
- 47.Dammeier J, Beer HD, Brauchle M, Werner S. Dexamethasone is a novel potent inducer of connective tissue growth factor expression. implications for glucocorticoid therapy. J Biol Chem 1998;273:18185–18190. [DOI] [PubMed] [Google Scholar]
- 48.Stratton R, Rajkumar V, Ponticos M, Nichols B, Shiwen X, Black CM, Abraham DJ, Leask A. Prostacyclin derivatives prevent the fibrotic response to TGF-beta by inhibiting the Ras/MEK/ERK pathway. FASEB J 2002;16:1949–1951. [DOI] [PubMed] [Google Scholar]
- 49.Krymskaya VP, Orsini MJ, Eszterhas A, Benovic JL, Panettieri RA, Penn RB. Potentiation of human airway smooth muscle proliferation by receptor tyrosine kinase and G protein-coupled receptor activation. Am J Respir Cell Mol Biol 2000;23:546–554. [DOI] [PubMed] [Google Scholar]
- 50.Penn RB, Benovic JL. Regulation of heterotrimeric G protein signaling in airway smooth muscle. Proc Am Thorac Soc 2008;5:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Govindaraju V, Michoud MC, Al-Chalabi M, Ferraro P, Powell WS, Martin JG. Interleukin-8: novel roles in human airway smooth muscle cell contraction and migration. Am J Physiol Cell Physiol 2006;291:C957–C965. [DOI] [PubMed] [Google Scholar]
- 52.Kaur D, Saunders R, Berger P, Siddiqui S, Woodman L, Wardlaw A, Bradding P, Brightling CE. Airway smooth muscle and mast cell-derived CC chemokine ligand 19 mediate airway smooth muscle migration in asthma. Am J Respir Crit Care Med 2006;174:1179–1188. [DOI] [PubMed] [Google Scholar]
- 53.Joubert P, Lajoie-Kadoch S, Welman M, Dragon S, Letuvee S, Tolloczko B, Halayko AJ, Gounni AS, Maghni K, Hamid Q. Expression and regulation of CCR1 by airway smooth muscle cells in asthma. J Immunol 2008;180:1268–1275. [DOI] [PubMed] [Google Scholar]
- 54.Joubert P, Lajoie-Kadoch S, Labonte I, Gounni AS, Maghni K, Wellemans V, Chakir J, Laviolette M, Hamid Q, Lamkhioued B. CCR3 expression and function in asthmatic airway smooth muscle cells. J Immunol 2005;175:2702–2708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.