

Abstract
Glutathionylspermidine is an intermediate formed in the biosynthesis of trypanothione, an essential metabolite in defence against chemical and oxidative stress in the Kinetoplastida. The kinetic mechanism for glutathionylspermidine synthetase (EC 6.3.1.8) from Crithidia fasciculata (CfGspS) obeys a rapid equilibrium random ter-ter model with kinetic constants KGSH = 609 μm, KSpd = 157 μm and KATP = 215 μm. Phosphonate and phosphinate analogues of glutathionylspermidine, previously shown to be potent inhibitors of GspS from Escherichia coli, are equally potent against CfGspS. The tetrahedral phosphonate acts as a simple ground state analogue of glutathione (GSH) (Ki ∼ 156 μm), whereas the phosphinate behaves as a stable mimic of the postulated unstable tetrahedral intermediate. Kinetic studies showed that the phosphinate behaves as a slow-binding bisubstrate inhibitor [competitive with respect to GSH and spermidine (Spd)] with rate constants k3 (on rate) = 6.98 × 104 m−1·s−1 and k4 (off rate) = 1.3 × 10−3 s−1, providing a dissociation constant Ki = 18.6 nm. The phosphinate analogue also inhibited recombinant trypanothione synthetase (EC 6.3.1.9) from C. fasciculata, Leishmania major, Trypanosoma cruzi and Trypanosoma brucei with Kiapp values 20–40-fold greater than that of CfGspS. This phosphinate analogue remains the most potent enzyme inhibitor identified to date, and represents a good starting point for drug discovery for trypanosomiasis and leishmaniasis.
Keywords: drug discovery, enzyme mechanism, glutathionylspermidine synthetase, slow-binding inhibition, trypanothione synthetase
Chagas’ disease, African sleeping sickness and leishmaniasis (cutaneous, mucocutaneous and visceral) are neglected diseases afflicting millions of people worldwide. All of the drugs used to treat these neglected diseases suffer from deficiencies such as poor efficacy, drug resistance, toxicity or high cost of treatment [1]. The parasitic protozoa causing these diseases belong to the order Kinetoplastida, and comparative genomic and biochemical studies have revealed a number of unique metabolic pathways that are being exploited for drug discovery [2]. One of these involves trypanothione [N1,N8-bis(glutathionyl)spermidine] and trypanothione reductase, which replaces not only glutathione/glutathione reductase but also thioredoxin/thioredoxin reductase in mammalian cells [3]. Together with the type I and II tryparedoxin peroxidases [4–6], trypanothione is pivotal in defence against oxidative stress induced by host cell defence mechanisms [7–9] or by redox cycling drugs such as nifurtimox [10,11]. In addition, novel trypanothione-dependent enzymes have been identified, such as trypanothione S-transferase [12] and glyoxalase I and II [13–15], that are probably involved in defence against chemical stress. The pertinence of the effects caused by decreasing trypanothione content and thus increased chemical stress highlight the significance of the biosynthetic enzyme(s) of trypanothione as drug target(s) [16].
Trypanothione is synthesized in these medically important parasites from glutathione (GSH) and spermidine (Spd) by a monomeric C-N ligase [trypanothione synthetase (TryS), EC 6.3.1.9], in a two-step reaction with glutathionylspermidine as an intermediate [17–20]. Both trypanothione reductase and TryS have been shown to be essential for parasite survival [21–25]. However, in the insect parasite, Crithidia fasciculata, TryS forms a heterodimer with the bifunctional glutathionylspermidine synthetase/amidase (GspS, EC 6.3.1.8/GspA, EC 3.5.1.78) [26]. Previous work suggested that each biosynthetic enzyme independently adds successive molecules of GSH to Spd to make trypanothione [26,27]. However, recombinant TryS from C. fasciculata (CfTryS) has been reported subsequently to catalyse both steps of trypanothione synthesis [28]. Although a gene for GspS has not been identified in Trypanosoma brucei, there is a pseudogene in Leishmania major (accession number AJ748279) [19] and putative genes for GspS within the genomes of Leishmania infantum (accession number AM502243) and Trypanosoma cruzi (accession number EAN98995) that remain to be functionally characterized. Genome sequencing information has also highlighted the presence of GSPS in a range of enteric pathogens such as Salmonella and Shigella [29,30]. The mechanism and physiological function of this protein are unknown, but in Escherichia coli it is proposed to be involved in regulation of polyamine levels during growth [31], and a similar function has been postulated for C. fasciculata GspS (CfGspS) [32]. Glutathionylspermidine accumulates only under stationary-phase conditions, and an alternative proposal is that it may be more effective in protecting DNA from oxidant damage than GSH [33]. A previous lack of structural information on this important class of enzymes has been recently resolved with the reported crystal structure of GspS from E. coli (EcGspS), which includes the enzyme in complex with substrate, product and inhibitor [34].
Preliminary enzyme characterization has previously been described for CfGspS [35], as well as kinetic studies on the partially purified native enzyme using an HPLC method [36,37]. Other studies have identified phosphonic and phosphinic acid derivatives of GSH as moderate inhibitors of CfGspS [38]. The most active of these was a phosphonic analogue of GSH (γ-l-Glu-l-Leu-GlyP), which displayed linear noncompetitive inhibition (Ki ∼ 60 μm). This analogue was further improved as an inhibitor of CfGspS by the substitution of the glycine moiety with amino acid analogues, such as diaminopropionic acid (Ki ∼ 7 μm) [39]. Although these inhibitors are excellent lead compounds for drug design against the trypanosomatid parasites, none, as yet, has yielded Ki values in the nanomolar range.
Proteases that catalyse the direct addition of water to proteins or peptides proceed via an unstable tetrahedral intermediate. These enzymes are inhibited by phosphorus-based stable mimics of the intermediate [40]. Such high-affinity analogues are termed transition state analogues or intermediate analogues [41]. Similarly, ATP-dependent ligases involve attack of a nucleophilic substrate on an electrophilic acyl phosphate [42] via a tetrahedral intermediate. These ligases are inhibited by stable analogues of this intermediate [43–45]. Original work on this type of analogue based on glutathionylspermidine was carried out on EcGspS [46,47]. These studies investigated GSH–Spd conjugates (Fig. 1), with the objective of developing enzyme inhibitors that block the biosynthesis of trypanothione [46–51]. The synthetase activity of EcGspS was inhibited by a phosphonate tetrahedral mimic, in a noncompetitive, time-independent manner with Ki ∼ 10 μm [47], and more potently by the phosphinate analogue in a time-dependent manner with Ki* = 8 nm [46,50]. In each case, the phosphorus-based pseudopeptide had no effect on the amidase activity.
Fig. 1.

Proposed intermediate of glutathionylspermidine and its phosphon(phin)ate analogues.
Here we examined the kinetic mechanism of CfGspS and determined the modality of inhibition and potency of these compounds against CfGspS and the homologous enzyme TryS from various disease-causing parasites.
Results
Initial velocity analysis of the kinetic mechanism of GspS
A matrix of kinetic data was collected in order to determine the kinetic mechanism of GspS. Six families of kinetic data were generated where each ligand (GSH, Spd and ATP) was treated as the varied substrate at different fixed concentrations of another substrate, maintaining a constant saturating concentration of the third substrate [52–55]. The corresponding double reciprocal plots of the data are shown in Fig. 2. A ping-pong mechanism can be ruled out, as the fitted lines of the Lineweaver–Burk plots converge with each combination of substrates. After excluding a ping-pong mechanism, the 16 possible models for rapid equilibrium ter-reactant systems were tested, including random, ordered, and hybrid random–ordered [52]. Statistical tests of each fit revealed that the rapid equilibrium random ter-ter model [see Eqn (1), Experimental procedures] fitted significantly better than any other of the 15 models (P < 10−12). The interaction factors were close to unity in this model, and when the interaction factors were set α = β = γ = 1, the two fits were not significantly different (P > 0.05), but did return ∼ 10-fold lower standard errors for the binding constants. Thus, the simplest model compatible with the data suggests that substrates bind to GspS in any order, without affecting binding of the other substrates, to form a quaternary complex, enzyme–GSH–ATP–Spd. When α = β = γ = 1, the equilibrium dissociation constants for the binding of substrate to the free enzyme are 609 ± 26, 157 ± 5 and 215 ± 8 μm for GSH, Spd and ATP, respectively, and kcat = 22.8 ±0.6 s−1. When GSH and ATP were varied in a constant ratio (10 : 1) versus various concentrations of Spd, they produced a series of Lineweaver–Burk plots that clearly converged (Fig. 3). This indicates that a product release step does not occur between the binding of ATP or GSH and Spd. Thus, the proposed kinetic model for GspS is consistent with a random ter-reactant mechanism, as shown in Fig. 4A.
Fig. 2.

Kinetic analysis of datasets for GspS. Assay details are described in Experimental procedures. The lines represent the global nonlinear fit of the data to the rapid equilibrium random ter-reactant mechanism (Eqn 1) plotted as a Lineweaver–Burk transformation. Reaction rates are reported as catalytic centre activity (s−1).
Fig. 3.

Lineweaver–Burk analysis of the variation of GSH and ATP at a fixed ratio of 1 : 10 versus Spd concentration. The final concentrations of GSH and ATP (mm) for each dataset are displayed on the graph. Reaction rates are reported as catalytic centre activity (s−1).
Fig. 4.

Model of ter-reactant mechanism of GspS catalysis and postulated slow-binding inhibition by the phosphinate mimic. (A) Kinetic mechanism. KGSH, KSpd and KATP are the equilibrium dissociation constants for the binding of substrate to free GspS (E). (B) Postulated structure of the phosphorylated phosphinate mimic.
Inhibition by phosphonate analogue
The compounds used in this study were designed to mimic the unstable tetrahedral intermediate formed during GspS-catalysed synthesis of glutathionylspermidine (Fig. 1). However, as reported for EcGspS [47], no time-dependent inhibition of CfGspS was observed with the phosphonate mimic (Fig. 5), which suggests that this compound is not acting as a mimic of the unstable intermediate, but as a bisubstrate analogue [56] incorporating key functional groups of both GSH and Spd in the inhibitor. This compound behaves as a modest classical linear competitive inhibitor of GspS with respect to GSH (Fig. 5B) with a Ki of 156 ± 13 μm. Note that for classical reversible inhibitors, the rate of product formation is constant provided that there is no significant depletion of substrate or inhibition by product.
Fig. 5.

Linear competitive inhibition of GspS by phosphonate analogue. (A) Progress curves demonstrating the classical competitive inhibition of GspS activity by phosphonate. Assays with GspS were performed in 250 μL of assay buffer with 10 nm GspS, 1 mm Spd, 1 mm GSH, 2 mm ATP and various phosphonate concentrations (0–1000 μm) as indicated. The lines fitted to the data points are linear fits for each of the phosphonate concentrations denoted. The linear regression values for all the data points are ≥ 0.997. (B) Kinetic analysis of GspS inhibition by phosphonate. Assays with GspS were performed in 250 μL of assay buffer with 1 mm Spd, various GSH concentrations (62.5–2000 μm), various phosphonate concentrations (50–400 μm) as indicated, and elevated levels of GspS (200 nm). The lines on the Lineweaver–Burk transformation are the best global nonlinear fit of the data to Eqn (2) describing linear competitive inhibition. Reaction rates are reported as catalytic centre activity (s−1).
Inhibition by phosphinate analogue
In contrast to the simple, linear inhibition shown by the phosphonate, time-dependent inhibition was observed for the phosphinate mimic (Fig. 6A). In reaction mixtures containing a slow-binding inhibitor initiated by the addition of enzyme, the initial velocity v0 is independent of inhibitor concentration, but decreases to a slower steady-state velocity vs that is dependent on inhibitor concentration [41]. These results are consistent with glutathionylspermidine-dependent phosphorylation of the phosphinate (Fig. 4B), as previously demonstrated for the inhibition of EcGspS [34,46,50]. The progress curves for each phosphinate concentration were fitted to Eqn (3) (Experimental procedures) to obtain values for v0, vs and kobs. Values for kobs were then plotted against the inhibitor concentration (Fig. 6B). A linear dependency between [I] and kobs was observed, and was fitted to Eqn (4) (Experimental procedures) to obtain estimates for k3′ and k4. The progress curves used to determine the kobs values were obtained at [S]/Km for GSH of 1.64. The rate constant k3′ (2.64 × 104 m−1·s−1) was subsequently corrected for competition by substrate, yielding k3 = 6.98 × 104 m−1·s−1 (k3 = k3′[1 + [S]/Km]). The y-intercept of Fig. 6B yields an estimate of k4 of 1.3 × 10−3·s−1. Thus, the overall dissociation half-life for the complex is 0.14 h (enzyme–inhibitor complex half-life values were calculated as the ratio of 0.693/k4). For an inhibitor of this type, the dissociation constant (Ki) is then obtained from the ratio of k4/k3, yielding a Ki of 18.6 nm. To confirm the Ki value, v0 and vs obtained at different concentrations of inhibitor were fitted to the equation vs = v0/(1 + [I]/Kiapp) by nonlinear regression, yielding a Kiapp value of 31.1 ± 2.1 nm, and true Ki value was calculated to be 19.0 ± 1.3 nm, using the relationship Ki = Kiapp/(1 + [S]/Km). Thus both methods of determining Ki are in excellent agreement.
Fig. 6.

Slow-binding inhibition of GspS by phosphinate analogue. (A) Assays with GspS were performed as described in Experimental procedures with 15 nm GspS, and various phosphinate concentrations (0–1 μm) as indicated, with 1 mm each GSH and Spd. (B) Determination of the association rate k3′ from the plot of kobs as a function of phosphinate concentration. The line represents a linear fit of kobs and [I] values (phosphinate concentrations). The kobs values were calculated from Eqn (4), and the line predicts a slope (k3′) of 0.026 μm−1 s−1.
An alternative approach was used to obtain an independent estimate of k4. In this method, the enzyme was preincubated with excess inhibitor ([I] ≥ 10 [E]), and the reaction was then initiated with substrate. Under these conditions, a slow release of inhibitor is observed until a steady state is reached. Provided that there is no significant enzyme inactivation, substrate depletion or product inhibition, this steady state should be identical to the steady state established when initiating with enzyme [57]. High concentrations of enzyme and inhibitor were preincubated for 1 h to allow the system to reach equilibrium. Subsequent dilution into a large volume of assay mix containing saturating substrate concentrations causes dissociation of the enzyme–inhibitor complex with regain of activity. Under these conditions, provided that the initial rate v0 and the effective inhibitor concentration are approximately equal to zero, the rate of recovery of full enzyme activity will provide k4. When maintaining [I] > [E] ([I] = 250 nm, [E] = 20 nm), it proved impossible to measure enzyme activity upon 100-fold dilution into the assay mixture. Instead, high concentrations of inhibitor (200 μm), enzyme (20 μm) and ATP (400 μm) were preincubated on ice for 60 min and then applied to a desalting column to remove all free inhibitor. The following reactions were then analysed: (a) enzyme-only control (Fig. 7, open circles); (b) the complete inhibition reaction, enzyme + inhibitor + ATP (Fig. 7, open squares); and (c) inhibitor-only control added to an equal volume of the enzyme-only control sample (Fig. 7, closed circles). The inhibitor-only control progress curve is linear and matches that of the enzyme-only control, demonstrating that essentially no inhibitor has passed through the resin. The regain of activity experiment (Fig. 7, open squares) clearly shows that an enzyme-bound inhibitor complex passes through the column and undergoes very slow dissociation upon dilution into the assay mixture. Under these conditions, both v0 and the free inhibitor concentration should be negligible in the final assay, so that the rate of recovery of activity provides the value for k4. After fitting of the data to Eqn (3) (Experimental procedures), a k4 value of 1.36 ± 0.06 × 10−3·s−1 was obtained, in excellent agreement with the value obtained previously by varying the concentration of phosphinate and initiating with enzyme.
Fig. 7.

Rate constant (k4) for dissociation of the GspS–phosphinate complex. Three samples were preincubated for 60 min in 100 mm Hepes (pH 7.3) on ice. The first contained GspS (20 μm) only, the second GspS (20 μm) with excess phosphinate (200 μm) and Mg2+-ATP (400 μm), and the third inhibitor/Mg2+-ATP only (i.e. no enzyme). All samples were desalted, and the flow-through was added to the coupled assay reaction mix in the following combination: flow-through of sample 1 (enzyme-only control, ○); flow-through of sample 2 (GspS preincubated with excess phosphinate, □); and flow-through of sample 1 plus sample 3 (i.e. a control showing that unbound inhibitor is completely removed using the column method, •), The rate constant associated with the regeneration of enzymatic activity (k4) was determined as described in the text.
Modality of inhibition
The mode of inhibition of the slow-binding phosphinate was determined by examining the effect of varying each substrate on the value of kobs at a fixed inhibitor concentration [58]. For a competitive inhibitor, kobs decreases in a hyperbolic fashion with increasing concentrations of substrate. This is observed with GSH or Spd as varied substrate (Fig. 8, closed and opened circles). For a noncompetitive inhibitor, kobs is independent of substrate concentration (i.e. kobs = k), whereas for an uncompetitive inhibitor, kobs increases in a hyperbolic fashion with increasing concentrations of substrate. As kobs increases with increasing concentrations of ATP (Fig. 8, closed squares), this suggests uncompetitive inhibition. These data were then fitted to the appropriate equation for either competitive inhibition [Eqn (5), Experimental procedures] or uncompetitive inhibition [Eqn (6), Experimental procedures]. The respective Km values for GSH, Spd and ATP are 400 ± 80 μm, 120 ± 40 μm and 130 ± 26 μm, in reasonable agreement with the respective Km values determined directly in the substrate matrix experiment above. Thus, the phosphinate inhibitor behaves as a slow-binding competitive bisubstrate inhibitor with respect to GSH and Spd, but not ATP. The latter observation is consistent with the hypothesis that an electrophilic acyl phosphate is formed by reaction of ATP and GSH. The acyl phosphate then reacts with Spd to form an unstable tetrahedral intermediate, which is mimicked by the stable tetrahedral phosphinate inhibitor. The nucleotide is not a component of the unstable tetrahedral intermediate, and therefore the phosphinate would not be expected to compete with ATP in binding to the enzyme.
Fig. 8.

Modality of inhibition by phosphinate analogue. The effect of varying GSH (•), Spd (○) and ATP (▪) on kobs was determined at a fixed concentration of phosphinate. The hyperbolic fits were obtained using either Eqn (5) for competitive inhibition (for GSH and Spd) or Eqn (6) for uncompetitive inhibition (for ATP).
To determine whether the phosphinate is turned over by CfGspS in the presence of ATP, the activity of the enzyme (100 nm) was determined in the absence of GSH or Spd plus or minus 1 μm phosphinate over 30 min. After correction for the background rate due to auto-oxidation of NADPH and hydrolysis of ATP in the coupling system, the net rates of endogenous ATPase activity (∼ 0.01% of kcat) in the presence and absence of inhibitor are 1.4 (± 0.9) × 10−3 and 3.0 (± 1.5) × 10−3·s−1, respectively (mean of three determinations). This shows that the inhibitor is not turned over by the enzyme. However, this method is insufficiently sensitive to detect a single phosphorylation event.
Inhibition of TryS with phosphinate
Having established that CfGspS is potently inhibited by the phosphinate inhibitor, it remained to be determined whether the homologous enzyme, TryS, could also be inhibited in a similar manner. Owing to the various pH optima, Km values for substrates and GSH substrate inhibition profiles of the various TryS enzymes to be compared (C. fasciculata, L. major, T. cruzi and T. brucei), a uniform assay was used for IC50 determination, i.e. 2 mm Spd, 0.2 mm GSH, 2 mm ATP, 100 mm (K+) Hepes (pH 7.2). This allows for direct comparison of the data collected for all the enzymes under conditions that approximate to the physiological metabolite levels found in these organisms [59]. In this study, IC50 values, slope factors and Kiapp values were determined and found to be 20–40-fold less that that of CfGspS (Table 1). In all cases, the slope factor was approximately 1, indicating simple binding at a single site for all the enzymes tested.
Table 1.
Inhibition constants of phosphinate against GspS and various TryS enzymes. All assays were performed under conditions approximating to the intracellular physiological state (i.e. pH 7.2, 2 mm Spd, 0.2 mm GSH and 2 mm Mg2+-ATP), and were initiated with 100 nm each enzyme in the presence of various phosphinate concentrations. IC50 values and slope factors are from the inhibition profiles determined from Eqn (7), and Kiapp values were determined from the tight-binding inhibition equation (Eqn 8). The errors represent the standard error of the fit to the appropriate equation.
| Enzyme |
|||||
|---|---|---|---|---|---|
| Inhibition constants | CfGspS | CfTryS | L. major TryS | T. cruzi TryS | T. brucei TryS |
| IC50 (nm) | 72 ± 6 | 1380 ± 380 | 650 ± 25 | 530 ± 20 | 1300 ± 50 |
| Slope factor | 1.2 ± 0.1 | 1.1 ± 0.3 | 1.1 ± 0.04 | 1.1 ± 0.04 | 1.1 ± 0.05 |
| Kiapp (nm) | 29 ± 5 | 1330 ± 350 | 580 ± 30 | 490 ± 20 | 1200 ± 500 |
Discussion
An understanding of the kinetic and chemical mechanism of GspS and TryS involved in the biosynthesis of glutathionylspermidine and trypanothione is crucial for the design of inhibitors against these potential drug targets. TryS is particularly challenging in this respect, as these enzymes display pronounced high substrate inhibition by GSH and form glutathionylspermidine as an intermediate [17–19]. CfGspS does not display substrate inhibition by GSH [35,60], and therefore provides a convenient simple model for this class of ATP-dependent C-N ligases.
The kinetic dataset for CfGspS fits best to a rapid equilibrium random ter-ter reaction mechanism, and definitively excludes a mechanism where either: (a) ADP is released after phosphorylation of GSH prior to binding of Spd; or (b) ADP is released following formation of a phosphorylated enzyme intermediate (ping-pong) prior to binding of GSH or Spd. In this respect, the mechanism for CfGspS is similar to that for γ-glutamylcysteine synthetase from T. brucei [53]. However, unlike the case with γ-glutamylcysteine synthetase, we did not detect any marked influence of prior binding of one substrate on the equilibrium dissociation constants of the other substrates [that is, the interaction factors α, β and γ were all close to unity, and statistical analysis did not favour their inclusion in Eqn (1)] (Experimental procedures) [52].
Our results are also broadly in agreement with a previous study which concluded that partially purified CfGspS follows a rapid equilibrium random order mechanism with interaction factors close to unity [37]. However, we were unable to reconcile the peptide sequence data reported by Flohéet al. with our own, as it corresponded to our sequence for CfTryS. This discrepancy was later corrected in an erratum by Flohé’s group [36], but raised a second discrepancy concerning CfTryS. In our hands, heterologous expression of CfTryS did not yield active proteins, whereas Flohé’s group reported that authentic CfTryS was able to catalyse the synthesis of trypanothione from GSH, Spd and ATP [28], similar to our findings for TryS from T. brucei, L. major and T. cruzi [17–19]. To resolve this remaining discrepancy, we have repeated our initial study. The newly cloned enzyme was found to differ at position 89, with a serine replacing an asparagine in the original construct (AF006615). The homogeneously pure soluble protein was found to be active with either GSH or glutathionylspermidine, and the product with either substrate was confirmed to be trypanothione by HPLC analysis (data not shown). The reason for our previous failure [27] to detect this activity by heterologous expression in yeast is not apparent, but may have been due to a cloning or PCR error involving this S89N mutation. Nonetheless, we now agree entirely with the report by Comini et al. [28] that CfTryS is capable of catalysing both steps in the biosynthesis of trypanothione from GSH and Spd.
A kinetic mechanism has not been determined for the E. coli enzyme, but a reaction mechanism has been proposed in which the glycine carboxylate of GSH is initially phosphorylated by the γ-phosphate of ATP to form an acyl phosphate, and this is followed by nucleophilic attack of the N1-primary amine of Spd on the acyl phosphate, leading to the formation of an unstable tetrahedral intermediate [46,48,49]. Structural studies on EcGspS in complex with substrates and inhibitors provide strong support for this model [34]. Of particular note was the observation that the slow-binding phosphinate inhibitor [46,50] had been phosphorylated by ATP to form the tetrahedral phosphinophosphate in the active site, as previously postulated [51]. In addition, a disordered domain in the apoenzyme was observed to adopt an ordered conformation over the active site when bound with substrates or inhibitor. Our kinetic studies indicate that all three substrates have to bind to the enzyme prior to catalysis. This suggests that formation of the quaternary complex induces closure of the lid domain over the active site to form a catalytically competent complex, thereby preventing access of water to hydrolyse the acyl phosphate intermediate.
Our kinetic analysis shows that the phosphonate analogue displays classical, linear competitive inhibition with respect to GSH, with a modest Ki of 156 μm against CfGspS, as compared to the mixed-type pattern (Ki and Ki′ of 6 and 14 μm, respectively) reported for EcGspsS [47]. In contrast, the phosphinate displays slow tight-binding inhibition with a Ki of 19 nm, similar to the Ki* of 8 nm for the E. coli enzyme [46]. Our studies also demonstrate that this inhibitor behaves as a mimic of the unstable tetrahedral intermediate that is proposed to form during the GspS-catalysed reaction as originally postulated [51]. At first sight, the uncompetitive behaviour of the phosphinate inhibitor rather than noncompetitive behaviour is not consistent with a rapid equilibrium random mechanism. However, such an inhibition pattern would be expected if the inhibitor underwent binding followed by a single phosphorylation event, as suggested by the kinetic behaviour observed in this study and others [46,50] and confirmed in the crystal structure of this inhibitor bound in the active site of EcGspS [34]. The glutathionylspermidine phosphinate analogue is also a potent inhibitor of TryS enzymes from L. major, T. cruzi and T. brucei; when assayed under identical conditions approximating to intracellular concentrations, TryS enzymes are approximately 20-fold less sensitive than CfGspS. Although the phosphinate showed no growth-inhibitory activity at 100 μm over 72 h of exposure against L. major promastigotes, T. cruzi epimastigotes and T. brucei procyclics, various chemical modifications could enhance cellular penetration, e.g. acyloxy ester prodrugs [61].
An alignment of EcGspS with CfGspS and other TryS proteins reveals some other interesting features (Fig. 9). First, despite the trypanosomatid proteins having < 30% identity and < 45% similarity, all three residues involved in binding Mg2+ (green triangles) and three of four involved in binding ATP (red triangles) are absolutely conserved. Second, four of five residues interacting with GSH (blue triangles) in the productive binding mode are also conserved. Third, two of three residues implicated in binding of the Spd moiety of the phosphinate inhibitor (yellow triangles) are also conserved. Fourth, Pai et al. also noted a nonproductive binding mode (black triangles), where GSH forms a mixed disulfide with Cys338 and an isopeptide bond between the glycine moiety of GSH and Lys607 of the protein. However, this is clearly not required for catalysis in the trypanosomatid enzymes, as neither residue is conserved in any of these enzymes. Finally, the E. coli enzyme is a homodimer, whereas the trypanosomatid TryS enzymes are monomeric, or heterodimeric in the case of CfTryS and CfGspS. In this case, the residues that interact between monomers in EcGspS (black circles) are hardly conserved at all. One other interesting difference between EcGspS and CfGspS is that the latter enzyme has an additional 100 amino acids. The alignment in Fig. 9 highlights a number of insertions that are dispersed throughout the sequence of CfGspS. These include an insertion of 17 amino acids in the amidase domain and two in the synthetase domain (one of 14 amino acids and the other of 39 amino acids). It may be that these additional insertions in CfGspS are required for its heterodimeric interactions with CfTryS.
Fig. 9.

Conservation of key functional residues identified for EcGspS in CfGspS and TryS. The GenBank/EMBL/DDBJ accession numbers used to generate the alignment using t-coffee are: EcGspS (U23148), CfGspS (U66520), CfTryS (AF006615), L. major TryS (AJ311570), T. cruzi TryS (AF311782) and T. brucei TryS (AJ347018). Absolutely conserved residues are marked in bold; coloured residues indicate side chain interactions in EcGspS with substrates or inhibitors [33]. Green triangles, residues involved in binding Mg2+; red triangles, three of four residues involved in binding ATP; blue triangles, four of five residues interacting with GSH; yellow triangles, two of three residues implicated in binding of the Spd moiety of the phosphinate inhibitor; black triangles, nonproductive binding mode, where GSH forms a mixed disulfide with Cys338 and an isopeptide bond between the glycine moiety of GSH and Lys607 of the protein; black circles, residues that interact between monomers in EcGspS. Only the relevant C-terminal region of the synthetase domain is shown.
From the above analysis, it is not immediately obvious why the phosphinate inhibitor is ∼ 20-fold less potent against the TryS enzymes than against CfGspS and EcGspS. Possibly, the substitution of Asp610, which is involved in recognition of the N8-amine of Spd, for a proline in TryS (methionine in CfGspS) is a critical factor. Alternatively, the fact that TryS has to accommodate either N1-glutathionylspermidine or N8-glutathionylspermidine as well as Spd in the polyamine-binding site may be a significant factor. The current ligand-free structure of L. major TryS [62] is not helpful in resolving these issues, and substrates or inhibitors in complex with TryS are needed. In the meantime, the phosphinate inhibitors represent a valuable starting point for further development of drug-like inhibitors against this target.
Experimental procedures
Materials
All chemicals were of the highest grade available from Sigma-Aldrich (Gillingham, UK), Roche Diagnostics Ltd (Burgess Hill, UK) or Calbiochem (Merck Biosciences, Nottingham, UK). The phosphonate and phosphinate analogues of glutathionylspermidine were synthesized as previously described [49,51]. The structure and purity of both compounds were confirmed by NMR, high-resolution MS and elemental analysis.
Expression and purification of GspS
Recombinant GspS was prepared using a 60 L fermenter, and purified to greater than 98% homogeneity as described previously [35], except that a HiLoad Q Sepharose 16/10 column (GE Healthcare, Amersham, UK) was used in the final step. Active fractions were pooled, buffer was exchanged into 100 mm (K+) Hepes containing 0.01% sodium azide, 1 mm dithiothreitol and 1 mm EDTA, and the sample concentration was determined using the calculated extinction coefficient of 99 370 at 280 nm. Aliquots of GspS were then flash frozen and stored in aliquots at −80 °C.
Expression and purification of TryS enzymes
TryS enzymes from T. brucei, L. major and T. cruzi were prepared as described previously [17–19]. In addition, we were able to obtain functionally active CfTryS by generating a new construct in a modified pET15b vector in which the thrombin cleavage site had been replaced by a TEV protease cleavage site. The ORF was PCR amplified from C. fasciculata genomic DNA using the sense primer 5′-CATATGGCG TCC GCT GAG CGT GTG CCG G-3′, which includes an NdeI site (underlined) and a start codon (in bold), and the antisense primer 5′-GGA TCC TTACTC ATC CTC GGC GAG CTT G-3′, which includes a stop codon (in bold) and a BamHI site (underlined); the PCR product was subsequently cloned, via pCR-Blunt II-TOPO (Invitrogen, Paisley, UK), into the NdeI/BamHI site of pET15bTEV. Sequencing of three independent clones revealed that the sequence was almost identical to the sequence previously deposited for CfTryS (AF006615), except that serine replaced asparagine at position 89 of the ORF. This construct, CfTryS_pET15bTEV, was transformed into BL21(DE3)pLysS-competent cells (Novagen, Merck Biosciences); typically, cultures were then grown in Terrific Broth at 37 °C to D600 nm ≥ 1.2, cooled to 22 °C, induced with a final concentration of 0.5 mm isopropyl-β-d-thiogalactoside, and grown for an additional 16 h. Purification of recombinant protein was achieved using two chromatographic steps [5 mL His-Trap (GE Healthcare), TEV protease cleavage (2 h, 30 °C), followed by a HiLoad Q Sepharose 16/10 HP column (GE Healthcare)].
Assay conditions for the kinetic mechanism of GspS
All kinetic assays were performed at 25 °C using an assay system that couples ADP production to NADH oxidation at 340 nm [35]. Each assay contained 100 mm (K+) Hepes (pH 7.3), 0.2 mm NADH, 1 mm phosphoenolpyruvate, 5 mm dithiothreitol or Tris(2-carboxyethyl)phosphine hydrochloride, 0.5 mm EDTA, 10 mm MgSO4, 2 U·mL−1l-lactate dehydrogenase, and 2 U·mL−1 pyruvate kinase (both coupling enzymes were from rabbit muscle, and purchased from Roche), with varying amounts of ATP, GSH and Spd in a total volume of 1 mL. Rates are expressed in moles of substrate utilized per second per mole of enzyme. To determine the kinetic mechanism, data were collected for GspS at a range of substrate concentrations. A complete matrix of rates as a function of substrate concentration (ATP, 31.25–500 μm; GSH, 62.5–1000 μm; and Spd 62.5–1000 μm) was gathered, so that for any given concentration of any one substrate the rates were measured over the entire range of the remaining two substrates. When fixed concentrations of each of these substrates were used, the final concentrations for ATP, GSH and Spd were 0.5, 1 and 1 mm respectively, unless otherwise stated. The assay was initiated by adding GspS (300 nm) and, after a lag of 10 s, the linear decrease in absorbance was monitored for up to 1 min. Data were then globally fitted by nonlinear regression to all possible models for rapid equilibrium ter-reactant systems [52]. The goodness of fit for each model was compared statistically using the F-test and kinetic constants obtained by fitting to Eqn (1):
 |
(1) |
This equation describes a rapid equilibrium random ter-ter system, where KGSH, KSpd and KATP are the equilibrium dissociation constants for the binding of substrate with free enzyme, and α, β and γ are the interaction factors between Spd and ATP, GSH and ATP, and GSH and Spd, respectively.
Inhibitors and enzyme inhibition assays
Inhibitor studies employed the coupled assay described above. Possible inhibition of the coupling enzyme system was excluded by substituting glucose and hexokinase for GspS or TryS in the assays, in which case no enzyme inhibition was observed. Reactions were typically carried out as described for the kinetic mechanism, with GSH and Spd (both at 1 mm) and ATP (2 mm) concentrations kept constant. Substrates and inhibitors were preincubated for 10 min before initiation of the reaction with GspS (10–20 nm). Data for the phosphonate analogue were fitted to the Michaelis–Menten equation for competitive inhibition (Eqn 2) when GSH was varied, and analysed using the program grafit:
 |
(2) |
For time-dependent inhibition by the phosphinate analogue, the progress curves at different inhibitor concentrations can be described by Eqn (3):
| (3) |
where [P] is the product concentration at time t, v0 and vs are the initial and final steady-state rates, and kobs is the apparent first-order rate constant for the establishment of the final steady-state equilibrium. The resulting values for kobs were plotted as a function of inhibitor concentration, I, and fitted to Eqn (4) to obtain estimates of k3′ and k4:
| (4) |
The rate constant k4, for the dissociation of the enzyme–inhibitor complex, was also measured directly from the time-dependent recovery of enzyme activity. GspS (20 μm) was preincubated, with or without phosphinate (200 μm) and Mg2+-ATP (400 μm), in 30 μL of assay buffer at 4 °C for 1 h, in order to reach equilibrium. A sample containing only inhibitor and Mg2+-ATP sample was also included as an internal control to verify efficient retention of the phosphinate by the column. Following preincubation, samples were applied to 0.5 mL of Zeba desalt spin columns (Pierce) and centrifuged to remove unbound inhibitor (1500 g, 2 min, 22 °C). Subsequently, 2 μL of each sample was diluted (1 : 500) into the complete enzyme assay mixture and the absorbance change was monitored. The recovery of enzymatic activity was measured at 340 nm using the coupled assay described above.
To determine the modality of inhibition by the phosphinate, assays were carried out in a reaction mixture of 1 mL containing 1 μm inhibitor in addition to the other components of the coupled assay. When GSH was varied, ATP and Spd were kept constant at 2 and 10 mm respectively; when Spd was varied, ATP and GSH were kept constant at 2 and 10 mm respectively; and when ATP was varied, GSH and Spd were kept constant at 1 mm. The reaction mix was left for 5 min at 25 °C, and the reaction was then initiated with CfGspS (20 nm) and monitored for 15 min. These data were then fitted to the appropriate equation [58] for either competitive inhibition (Eqn 5)
| (5) |
or uncompetitive inhibition (Eqn 6)
| (6) |
where k is the value for kobs in the absence of substrate, and Km is the binding constant for the varied substrate S.
IC50 data were also gathered for representative recombinant TryS enzymes (T. cruzi, T. brucei and L. major) [17–19], using more physiological-like conditions, i.e. 2 mm Spd, 0.2 mm GSH, 2 mm ATP, and 100 mm (K+) Hepes (pH 7.2) (the remainder of the components of the coupled assay were as previously above) and various phosphinate concentrations (0–10 μm). Reactions were initiated using 100 nm each enzyme, and the change in absorbance was monitored for 30 min. The resulting steady-state rates were then fitted to the following two-parameter equation (Eqn 7), where the lower data limit is 0, i.e. the data are background corrected, and the upper data limit is 100, i.e. the data are range corrected.
 |
(7) |
In this equation, s is a slope factor. The equation assumes that y falls with increasing [I]. The Kiapp values of the inhibitor against each enzyme were determined using the following tight-binding inhibition equation [41] (Eqn 8), where the enzyme concentration [E] was fixed at 100 nm:
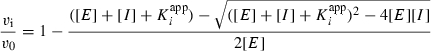 |
(8) |
Acknowledgments
We would like to thank Dr Jon Nunes for preliminary enzyme–inhibitor studies. This work is supported by the Wellcome Trust (grant numbers WT 079838 and WT 083481).
Glossary
Abbreviations
- CfGspS
Crithidia fasciculata glutathionylspermidine synthetase
- CfTryS
Crithidia fasciculata trypanothione synthetase
- EcGspS
Escherichia coli glutathionylspermidine synthetase
- GSH
glutathione
- GspA
glutathionylspermidine amidase
- GspS
glutathionylspermidine synthetase
- Spd
spermidine
- TryS
trypanothione synthetase
References
- 1.Nwaka S, Hudson A. Innovative lead discovery strategies for tropical diseases. Nat Rev Drug Discov. 2006;5:941–955. doi: 10.1038/nrd2144. [DOI] [PubMed] [Google Scholar]
- 2.Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 3.Krauth-Siegel RL, Comini MA. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim Biophys Acta. 2008;1780:1236–1248. doi: 10.1016/j.bbagen.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Flohé L, Steinert P, Hecht HJ, Hofmann B. Tryparedoxin and tryparedoxin peroxidase. Methods Enzymol. 2002;347:244–258. doi: 10.1016/s0076-6879(02)47024-3. [DOI] [PubMed] [Google Scholar]
- 5.König J, Fairlamb AH. A comparative study of type I and type II tryparedoxin peroxidases in Leishmania major. FEBS J. 2007;274:5643–5658. doi: 10.1111/j.1742-4658.2007.06087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlecker T, Comini MA, Melchers J, Ruppert T, Krauth-Siegel RL. Catalytic mechanism of the glutathione peroxidase-type tryparedoxin peroxidase of Trypanosoma brucei. Biochem J. 2007;405:445–454. doi: 10.1042/BJ20070259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fairlamb AH, Henderson GB. Metabolism of trypanothione and glutathionylspermidine in trypanosomatids. In: Chang K-P, Snary D, editors. Host–Parasite Cellular and Molecular Interactions in Protozoal Infections. Berlin: Springer-Verlag/NATO ASI Series, Vol. H11; 1987. pp. 29–40. [Google Scholar]
- 8.Fairlamb AH, Cerami A. Metabolism and functions of trypanothione in the Kinetoplastida. Annu Rev Microbiol. 1992;46:695–729. doi: 10.1146/annurev.mi.46.100192.003403. [DOI] [PubMed] [Google Scholar]
- 9.Krauth-Siegel RL, Schöneck R. Trypanothione reductase and lipoamide dehydrogenase as targets for a structure-based drug design. FASEB J. 1995;9:1138–1146. doi: 10.1096/fasebj.9.12.7672506. [DOI] [PubMed] [Google Scholar]
- 10.Henderson GB, Ulrich P, Fairlamb AH, Rosenberg I, Pereira M, Sela M, Cerami A. ‘Subversive’ substrates for the enzyme trypanothione disulfide reductase: alternative approach to chemotherapy of Chagas’ disease. Proc Natl Acad Sci USA. 1988;85:5374–5378. doi: 10.1073/pnas.85.15.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jockers-Scherubl MC, Schirmer RH, Krauth-Siegel RL. Trypanothione reductase from Trypanosoma cruzi: catalytic properties of the enzyme and inhibition studies with trypanocidal compounds. Eur J Biochem. 1989;180:267–272. doi: 10.1111/j.1432-1033.1989.tb14643.x. [DOI] [PubMed] [Google Scholar]
- 12.Vickers TJ, Fairlamb AH. Trypanothione S-transferase activity in a trypanosomatid ribosomal elongation factor 1B. J Biol Chem. 2004;279:27246–27256. doi: 10.1074/jbc.M311039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irsch T, Krauth-Siegel RL. Glyoxalase II of African trypanosomes is trypanothione-dependent. J Biol Chem. 2004;279:22209–22217. doi: 10.1074/jbc.M401240200. [DOI] [PubMed] [Google Scholar]
- 14.Ariza A, Vickers TJ, Greig N, Armour KA, Dixon MJ, Eggleston IM, Fairlamb AH, Bond CS. Specificity of the trypanothione-dependent Leishmania major glyoxalase I: structure and biochemical comparison with the human enzyme. Mol Microbiol. 2006;59:1239–1248. doi: 10.1111/j.1365-2958.2006.05022.x. [DOI] [PubMed] [Google Scholar]
- 15.Greig N, Wyllie S, Vickers TJ, Fairlamb AH. Trypanothione-dependent glyoxalase I in Trypanosoma cruzi. Biochem J. 2006;400:217–223. doi: 10.1042/BJ20060882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sousa SM, Ferreira AE, Tomas AM, Cordeiro C, Ponces FA. Quantitative assessment of the glyoxalase pathway in Leishmania infantum as a therapeutic target by modelling and computer simulation. FEBS J. 2005;272:2388–2398. doi: 10.1111/j.1742-4658.2005.04632.x. [DOI] [PubMed] [Google Scholar]
- 17.Oza SL, Tetaud E, Ariyanayagam MR, Warnon SS, Fairlamb AH. A single enzyme catalyses formation of trypanothione from glutathione and spermidine in Trypanosoma cruzi. J Biol Chem. 2002;277:35853–35861. doi: 10.1074/jbc.M204403200. [DOI] [PubMed] [Google Scholar]
- 18.Oza SL, Ariyanayagam MR, Aitcheson N, Fairlamb AH. Properties of trypanothione synthetase from Trypanosoma brucei. Mol Biochem Parasitol. 2003;131:25–33. doi: 10.1016/s0166-6851(03)00176-2. [DOI] [PubMed] [Google Scholar]
- 19.Oza SL, Shaw MP, Wyllie S, Fairlamb AH. Trypanothione biosynthesis in Leishmania major. Mol Biochem Parasitol. 2005;139:107–116. doi: 10.1016/j.molbiopara.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Comini M, Menge U, Flohé L. Biosynthesis of trypanothione in Trypanosoma brucei brucei. Biol Chem. 2003;384:653–656. doi: 10.1515/BC.2003.072. [DOI] [PubMed] [Google Scholar]
- 21.Dumas C, Ouellette M, Tovar J, Cunningham ML, Fairlamb AH, Tamar S, Olivier M, Papadopoulou B. Disruption of the trypanothione reductase gene of Leishmania decreases its ability to survive oxidative stress in macrophages. EMBO J. 1997;16:2590–2598. doi: 10.1093/emboj/16.10.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tovar J, Cunningham ML, Smith AC, Croft SL, Fairlamb AH. Down-regulation of Leishmania donovani trypanothione reductase by heterologous expression of a trans-dominant mutant homologue: effect on parasite intracellular survival. Proc Natl Acad Sci USA. 1998;95:5311–5316. doi: 10.1073/pnas.95.9.5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krieger S, Schwarz W, Ariyanayagam MR, Fairlamb AH, Krauth-Siegel RL, Clayton C. Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Mol Microbiol. 2000;35:542–552. doi: 10.1046/j.1365-2958.2000.01721.x. [DOI] [PubMed] [Google Scholar]
- 24.Comini MA, Guerrero SA, Haile S, Menge U, Lunsdorf H, Flohé L. Validation of Trypanosoma brucei trypanothione synthetase as drug target. Free Radic Biol Med. 2004;36:1289–1302. doi: 10.1016/j.freeradbiomed.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Ariyanayagam MR, Oza SL, Guther ML, Fairlamb AH. Phenotypic analysis of trypanothione synthetase knockdown in the African trypanosome. Biochem J. 2005;391:425–432. doi: 10.1042/BJ20050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith K, Nadeau K, Walsh C, Fairlamb AH. Purification of glutathionylspermidine and trypanothione synthetases from Crithidia fasciculata. Protein Sci. 1992;1:874–883. doi: 10.1002/pro.5560010705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tetaud E, Manai F, Barrett MP, Nadeau K, Walsh CT, Fairlamb AH. Cloning and characterization of the two enzymes responsible for trypanothione biosynthesis in Crithidia fasciculata. J Biol Chem. 1998;273:19383–19390. doi: 10.1074/jbc.273.31.19383. [DOI] [PubMed] [Google Scholar]
- 28.Comini M, Menge U, Wissing J, Flohé L. Trypanothione synthesis in Crithidia revisited. J Biol Chem. 2005;280:6850–6860. doi: 10.1074/jbc.M404486200. [DOI] [PubMed] [Google Scholar]
- 29.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du FY, et al. Complete genome sequence of Salmonella enterica serovar typhimurium LT2. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 30.Jin Q, Yuan ZH, Xu JG, Wang Y, Shen Y, Lu WC, Wang JH, Liu H, Yang J, Yang F, et al. Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 2002;30:4432–4441. doi: 10.1093/nar/gkf566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tabor H, Tabor CW. Isolation, characterization and turnover of glutathionylspermidine from Escherichia coli. J Biol Chem. 1975;250:2648–2654. [PubMed] [Google Scholar]
- 32.Shim H, Fairlamb AH. Levels of polyamines, glutathione and glutathione–spermidine conjugates during growth of the insect trypanosomatid Crithidia fasciculata. J Gen Microbiol. 1988;134:807–817. doi: 10.1099/00221287-134-3-807. [DOI] [PubMed] [Google Scholar]
- 33.Smith K, Borges A, Ariyanayagam MR, Fairlamb AH. Glutathionylspermidine metabolism in Escherichia coli. Biochem J. 1995;312:465–469. doi: 10.1042/bj3120465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pai CH, Chiang BY, Ko TP, Chou CC, Chong CM, Yen FJ, Chen SJ, Coward JK, Wang AHJ, Lin CH. Dual binding sites for translocation catalysis by Escherichia coli glutathionylspermidine synthetase. EMBO J. 2006;25:5970–5982. doi: 10.1038/sj.emboj.7601440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oza SL, Ariyanayagam MR, Fairlamb AH. Characterization of recombinant glutathionylspermidine synthetase/amidase from Crithidia fasciculata. Biochem J. 2002;364:679–686. doi: 10.1042/BJ20011370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koenig K, Menge U, Kiess M, Wray V, Flohé L. Convenient isolation and kinetic mechanism of glutathionylspermidine synthetase from Crithidia fasciculata. J Biol Chem. 2005;280:7407. doi: 10.1074/jbc.272.18.11908. Vol. 272 (1997) 11908–11915. [DOI] [PubMed] [Google Scholar]
- 37.Koenig K, Menge U, Kiess M, Wray V, Flohé L. Convenient isolation and kinetic mechanism of glutathionylspermidine synthetase from Crithidia fasciculata. J Biol Chem. 1997;272:11908–11915. doi: 10.1074/jbc.272.18.11908. [DOI] [PubMed] [Google Scholar]
- 38.Verbruggen C, De Craecker S, Rajan P, Jiao XY, Borloo M, Smith K, Fairlamb AH, Haemers A. Phosphonic acid and phosphinic acid tripeptides as inhibitors of glutathionylspermidine synthetase. Bioorg Med Chem Lett. 1996;6:253–258. [Google Scholar]
- 39.Amssoms K, Oza SL, Augustyns K, Yamani A, Lambeir A, Bal G, Van der Veken P, Fairlamb AH, Haemers A. Glutathione-like tripeptides as inhibitors of glutathionylspermidine synthetase. Part 2: Substitution of the glycine part. Bioorg Med Chem Lett. 2002;12:2703–2705. doi: 10.1016/s0960-894x(02)00538-3. [DOI] [PubMed] [Google Scholar]
- 40.Radzicka A, Wolfenden R. Transition-state and multisubstrate analog inhibitors. Methods Enzymol. 1995;249:284–312. doi: 10.1016/0076-6879(95)49039-6. [DOI] [PubMed] [Google Scholar]
- 41.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 42.Banerjee RV, Shane B, McGuire JJ, Coward JK. Dihydrofolate synthetase and folylpolyglutamate synthetase – direct evidence for intervention of acyl phosphate intermediates. Biochemistry. 1988;27:9062–9070. doi: 10.1021/bi00425a027. [DOI] [PubMed] [Google Scholar]
- 43.Valiaeva N, Bartley D, Konno T, Coward JK. Phosphinic acid pseudopeptides analogous to glutamyl-gamma-glutamate: synthesis and coupling to pteroyl azides leads to potent inhibitors of folylpoly-gamma-glutamate synthetase. J Org Chem. 2001;66:5146–5154. doi: 10.1021/jo010283t. [DOI] [PubMed] [Google Scholar]
- 44.McGuire JJ, Haile WH, Valiaeva N, Bartley D, Guo JX, Coward JK. Potent inhibition of human folylpolyglutamate synthetase by a phosphinic acid mimic of the tetrahedral reaction intermediate. Biochem Pharmacol. 2003;65:315–318. doi: 10.1016/s0006-2952(02)01602-7. [DOI] [PubMed] [Google Scholar]
- 45.Bartley DM, Coward JK. A stereoselective synthesis of phosphinic acid phosphapeptides corresponding to glutamyl-gamma-glutamate and incorporation into potent inhibitors of folylpoly-gamma-glutamyl synthetase. J Org Chem. 2005;70:6757–6774. doi: 10.1021/jo0507439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin C-H, Chen S, Kwon DS, Coward JK, Walsh CT. Aldehyde and phosphinate analogs of glutathione and glutathionylspermidine: potent, selective binding inhibitors of the E. coli bifunctional glutathionylspermidine synthetase/amidase. Chem Biol. 1997;4:859–866. doi: 10.1016/s1074-5521(97)90118-6. [DOI] [PubMed] [Google Scholar]
- 47.Kwon DS, Lin CH, Chen SJ, Coward JK, Walsh CT, Bollinger JM., Jr Dissection of glutathionylspermidine synthetase/amidase from Escherichia coli into autonomously folding and functional synthetase and amidase domains. J Biol Chem. 1997;272:2429–2436. doi: 10.1074/jbc.272.4.2429. [DOI] [PubMed] [Google Scholar]
- 48.Bollinger JM, Jr, Kwon DS, Huisman GW, Kolter R, Walsh CT. Glutathionylspermidine metabolism in Escherichia coli. Purification, cloning, overproduction, and characterization of a bifunctional glutathionylspermidine synthetase/amidase. J Biol Chem. 1995;270:14031–14041. doi: 10.1074/jbc.270.23.14031. [DOI] [PubMed] [Google Scholar]
- 49.Chen SJ, Lin CH, Kwon DS, Walsh CT, Coward JK. Design, synthesis, and biochemical evaluation of phosphonate and phosphonamidate analogs of glutathionylspermidine as inhibitors of glutathionylspermidine synthetase/amidase from Escherichia coli. J Med Chem. 1997;40:3842–3850. doi: 10.1021/jm970414b. [DOI] [PubMed] [Google Scholar]
- 50.Chen S, Lin CH, Walsh CT, Coward JK. Novel inhibitors of trypanothione biosynthesis: synthesis and evaluation of a phosphinate analog of glutathionyl spermidine (GSP), a potent, slow-binding inhibitor of GSP synthetase. Bioorg Med Chem Lett. 1997;7:505–510. [Google Scholar]
- 51.Chen SJ, Coward JK. Investigations on new strategies for the facile synthesis of polyfunctionalized phosphinates: phosphinopeptide analogues of glutathionylspermidine. J Org Chem. 1998;63:502–509. doi: 10.1021/jo971318l. [DOI] [PubMed] [Google Scholar]
- 52.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-state Enzyme Systems. Wiley Classics Library Edition Published 1993 edn. New York, NY: Wiley; 1975. [Google Scholar]
- 53.Brekken DL, Phillips MA. Trypanosoma bruceiγ-glutamylcysteine synthetase – characterization of the kinetic mechanism and the role of Cys-319 in cystamine inactivation. J Biol Chem. 1998;273:26317–26322. doi: 10.1074/jbc.273.41.26317. [DOI] [PubMed] [Google Scholar]
- 54.Jez JM, Cahoon RE, Chen SX. Arabidopsis thaliana glutamate-cysteine ligase – functional properties, kinetic mechanism, and regulation of activity. J Biol Chem. 2004;279:33463–33470. doi: 10.1074/jbc.M405127200. [DOI] [PubMed] [Google Scholar]
- 55.Jez JM, Cahoon RE. Kinetic mechanism of glutathione synthetase from Arabidopsis thaliana. J Biol Chem. 2004;279:42726–42731. doi: 10.1074/jbc.M407961200. [DOI] [PubMed] [Google Scholar]
- 56.Broom AD. Rational design of enzyme-inhibitors – multisubstrate analog inhibitors. J Med Chem. 1989;32:2–7. doi: 10.1021/jm00121a001. [DOI] [PubMed] [Google Scholar]
- 57.Morrison JF. The slow-binding and slow, tight-binding inhibition of enzyme-catalyzed reactions. Trends Biochem Sci. 1982;7:102–105. [Google Scholar]
- 58.Copeland RA. Evaluation of Enzyme Inhbitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists. Hoboken, NJ: John Wiley & Sons; 2005. Tight binding inhibition; pp. 178–213. [PubMed] [Google Scholar]
- 59.Ariyanayagam MR, Fairlamb AH. Ovothiol and trypanothione as antioxidants in trypanosomatids. Mol Biochem Parasitol. 2001;115:189–198. doi: 10.1016/s0166-6851(01)00285-7. [DOI] [PubMed] [Google Scholar]
- 60.Amssoms K, Oza SL, Ravaschino E, Yamani A, Lambeir AM, Rajan P, Bal G, Rodriguez JB, Fairlamb AH, Augustyns K, et al. Glutathione-like tripeptides as inhibitors of glutathionylspermidine synthetase. Part 1: Substitution of the glycine carboxylic acid group. Bioorg Med Chem Lett. 2002;12:2553–2556. doi: 10.1016/s0960-894x(02)00489-4. [DOI] [PubMed] [Google Scholar]
- 61.Feng Y, Coward JK. Prodrug forms of N-[(4-deoxy-4-amino-10-methyl)pteroyl]glutamate-γ-[ψP(O)(OH)]-glutarate, a potent inhibitor of folylpoly-γ-glutamate synthetase: synthesis and hydrolytic stability. J Med Chem. 2006;49:770–788. doi: 10.1021/jm050871p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fyfe PK, Oza SL, Fairlamb AH, Hunter WN. Leishmania trypanothione synthetase–amidase structure reveals a basis for regulation of conflicting synthetic and hydrolytic activities. J Biol Chem. 2008;283:17672–17680. doi: 10.1074/jbc.M801850200. [DOI] [PMC free article] [PubMed] [Google Scholar]