

Abstract
Polymersomes are polymer-based vesicular shells that form upon hydration of amphiphilic block copolymers. These high molecular weight amphiphiles impart physicochemical properties that allow polymersomes to stably encapsulate or integrate a broad range of active molecules. This robustness together with recently described mechanisms for controlled breakdown of degradable polymersomes as well as escape from endolysosomes suggests that polymersomes might be usefully viewed as having structure/property/function relationships somewhere between lipid vesicles and viral capsids. Here we summarize the assembly and development of controlled release polymersomes to encapsulate therapeutics ranging from small molecule anti-cancer drugs to siRNA and therapeutic proteins.
Keywords: liposomes, amphiphile, block copolymers, nanoparticles, controlled release, siRNA, protein, encapsulation
INTRODUCTION
Nature has evolved many solutions to many problems of transport at the cellular and sub-cellular scale. Small lipid vesicles or liposomes bud from cell membranes and help traffic a wide range of biomolecules both within cells and outside cells. Fluidity, flexibility, and dynamics of these carriers largely arises from the fact that lipids are low molecular weight amphiphiles. In contrast, viral capsids self-assemble from virus-encoded polypeptides that are typically 1-2 orders of magnitude larger in molecular weight than lipids. Robust, solid-like capsid structures are genetically tailored to encapsulate, protect, and deliver the viral genome, often integrating mechanisms for targeting as well as controlled intracellular release. Viral vectors are indeed capable of high infection efficiency and sustained expression of foreign genes, but they are limited to delivery of nucleic acids and their polypeptides tend to be immunogenic.
Liposomes have been pursued as non-viral gene and drug delivery vehicles for several decades, but pure lipid vesicles are generally cleared within hours from the circulation. Addition of biocompatible poly(ethylene glycol) (PEG) to a small fraction of the lipids addresses this shortcoming. PEGylation is sometimes described as emulating the glycocalyx of cell membranes and is found commercially on chemotherapetuic liposomes such as DOXIL. However, vesicles composed of natural lipids also generally lack mechanisms for controlled release, and so additional synthetic schemes continue to be developed for lipids (Guo 2003).
Polymersomes are formed from high molecular weight (MW) amphiphilic block copolymers composed of distinct hydrophilic and hydrophobic blocks – somewhat like a scaled up lipid. Choices of polymer MW and chemistry impart polymersomes with a broad and tunable range of carrier properties. As reviewed here, polymersomes are capable of encapsulating a large range of therapeutically active molecules and biomolecules, with considerable work being done to engineer the release of those encapsulants at the desired place and time. The accelerated use of engineered polymer systems to create polymeric micellar structures for application in the field of drug delivery motivates this current review of the role of polymersomes in non-viral delivery.
POLYMERSOME STRUCTURE AND PROPERTIES
Self-Directed Assembly of Polymer Vesicles
The principles that govern the self-assembly of natural amphiphiles like lipids can be generalized to simple energetic and geometric arguments (Israelachvili 1991). At solution concentrations above a critical micelle concentration (CMC) – where CMC decreases exponentially with amphiphile molecular weight (i.e. CMC ~ exp(-MW)) – amphiphiles self-assemble to form super molecular weight aggregates. Aggregate geometry is dictated by the proportions of the hydrophilic and hydrophobic segments of the amphiphilic molecule. This idea is described by the molecular packing parameter p = v/alc, in which v is the volume of the densely packed hydrophobic segment, lc is the chain length of the hydrophobic block normal to the interface, and a is the effective cross-sectional area of the hydrophilic group. This simple idea of a packing parameter can then be used to predict whether the resultant morphology of these amphiphilic aggregates is spherical (p < 1/3), cylindrical (1/3 < p < 1/2), or a vesicle bilayer (1/2 < p < 1). It is this generality that has facilitated understanding of self-assembly to move beyond lipids and small molecular weight surfactants and on to high molecular weight amphiphiles like amphiphilic block copolymers, polypeptides, rod-coil polymers, and dendrimers (Discher 2002; Antonietti 2003).
Block copolymers can be designed with the same amphiphilic character as lipids but consist of polymer chains covalently linked as a series of two or more “blocks”. In the bulk polymer phase without organic solvent, block copolymers are known to display a wide range of ordered morphologies, including lamellar phases (with the same symmetry as a stack of paper). Hydration of these dry lamellae by aqueous solution results in a stable dispersion of amphiphilic block copolymer aggregates. Using the fully synthetic diblock copolymer poly(ethylene oxide)-polybutadiene (PEG-PBD, noting that PEO and PEG are the same), the role of the molecular packing factor in block copolymer aggregate morphology has been fully elucidated (Won 2002; Jain 2003; Jain 2004). By changing the packing factor of PEG-PBD through systematic changes in the hydrophilic block fraction f (≈ exp(-p/β); β≈ 0.66), the morphology of the resultant aggregates in aqueous solution can be tuned to form spherical micelles (f > 50%), worm-like micelles (40% < f < 50%) (Won 1999), or unilamellar polymer vesicles (25% < f < 40%) referred to as “polymersomes” (Discher 1999). The sensitivity of these morphological phase boundaries on f has not been systematically explored as a function of polymer chemistry or molecular weight, though they are consistent for the hydrogenated homolog of PEG-PBD, PEG-polyethylethylene (PEG-PEE).
Initial work with copolymers having values of f that consistently yield polymersomes and number-average molecular weights (MWs) ranging from ~2,000 to 20,000 Da has shown by cryogenic transmission electron microscopy (cryo-TEM) that the membrane core (d) increases with MW from d ~ 8 to 21 nm with the scaling d ~ Mhb (b ≈ 0.55) (Aranda-Espinoza 2001; Bermudez 2002). The scaling of membrane thickness with MW as well as the morphological dependence of PEG-PEE on f has been confirmed by novel coarse-grained molecular dynamics simulations (Figure 1a) (Srinivas 2004; Srinivas 2004; Ortiz 2005). Perhaps most revealing from simulation has been that only for low MW systems, the polymer bilayers exhibit a clear mid-plane of low density reminiscent of the “methyl trough” present in lipid bilayers. For high MW copolymers, the two leaflets of the bilayer interdigitate or “melt” together into a single thick and integrated shell of homogeneous density. Polymersome membranes thus present a novel opportunity to study membrane properties and performance in application as a function of membrane thickness.
Fig. 1. Polymersome assemblies, and Cellular uptake and release.

a) Coarse-grained molecular dynamics simulation of polymersomes of amphiphilic diblock copolymers in water demonstrates assembly of the vesicle bilayer. b) Schematic of cystolic drug delivery with degradable polymersomes illustrates the uptake and degradation of polymersomes within the endolysosomal pathway. Hydrolytic degradation of polymersomes results in a high concentration of spherical micelles that mimic detergents and lyse the endolysosomal membrane to allow the release of the therapeutic into the cytosol. c) Uptake of polymersomes labeled with a green fluorescent membrane probe and loaded with the red fluorescent anti-cancer drug Doxorubicin is demonstrated in vitro using a human cancer cell line. Non-degradable polymersomes show colocalization of the polymersome label and Doxorubicin near the nucleus as expected for endolysosomal cargo. With degradable polymersomes the green membrane probe remains outside the nucleus while the red Doxorubicin localizes to the nucleus.
Physiochemical Characterization of Polymersomes
The formation of giant, micron-sized vesicles allows for detailed characterization of membrane properties by single-vesicle micromanipulation methods (Discher 1999). Measurements of lateral diffusivity (Lee 2002) as well as apparent membrane viscosity (Dimova 2002) have shown that membrane fluidity generally decreases with increasing MW, and the fluidity decreases most drastically when the chains are sufficiently long to entangle. Area elasticity measurements for PEG-PBD membranes provide an indirect measure of the monomer’s effective interaction energy (εh) as interfacial tension (γ ~ 30 mN/m), which appears independent of MW. Opposition to interface dilation is expected to be dictated by polymer chemistry and solvent. Electromechanical stability also increases with membrane thickness up to a limit set by γ(Aranda-Espinoza 2001). Phospholipid membranes rupture well below such limits as their thin membrane core increases susceptibility to catastrophic defects. Even when intact, permeation of water through the thick polymersome membranes is considerably reduced in comparison to lipid membranes (Battaglia 2006). The trends with MW generalize early measurements on liposomes (Bangham 1993).
In parallel with experimental studies, coarse grain molecular dynamics has also been used to study the effects of block length on the area elastic modulus and lateral chain mobility of the bilayer (Srinivas 2004). Simulation has indicated that the area elastic modulus is indeed independent of block length. Experimental diffusivities of the diblocks compared with simulation data for the extent of overlap and entanglement between the two leaflets of the bilayer suggest a crossover in diffusivity from the linear Rouse regime at low MW to a more viscoelastic regime dominated by entanglements at high MW. What appears most suggestive from the physicochemical studies to date is that lipid membranes have evolved in nature to be optimized more for fluidity than for stability. In contrast, robustness and solidity are hallmarks of viral capsids.
Evolution of Polymersome Design
Polyeletrolyte-based Polymersomes
Although positively charged liposomes tend to be cleared quickly from the circulation (Liu 1995), addition of charge to polymer amphiphiles presents additional opportunities for controlled release in response to external stimuli, such as pH (eg. poly(acrylic acid) (PAA)) mediated endolysosomal rupture (Jones 2003)). The shape and structure of charged diblock copolymer aggregates in solution are governed by a surprisingly delicate balance of forces. Interfacial tension between the core and bulk solvent orients, confines, and stretches the chains, but the interaction between solvated corona blocks can be repulsive through sterics and electrostatics as well as attractive if multivalent ions are involved (Israelachvili 1991).
The first morphological phase diagram of charged diblock copolymers in dilute solution was mapped out by Eisenberg and coworkers for “crew-cut” PAA-PS glassy copolymers (Zhang 1996). The rich phase behavior and dynamics of a relatively symmetric and fluid diblock of PAA-PBD have now also been mapped out in water (Geng 2005). Depending on the concentration of added salt as well as pH, these diblocks are seen to self-assemble in water into stable vesicles, wormlike micelles (‘worms’), and spherical micelles (‘spheres’). Furthermore, using fluorescence microscopy, transitions of vesicles into worms and spheres within minutes were visualized with a sudden jump in pH or chelation of salt. Liu and Eisenberg have likewise shown rapid pH-triggered inversion of biamphiphilic triblock copolymer vesicles of PAA-PS-poly(4-vinyl-pyridine) (P4VP) in organic/aqueous mixtures (Liu 2003). With increasing bulk pH, the aggregate morphology changes from vesicles (with P4VP outside) to solid aggregates, and then inverts back into a vesicle assembly (with PAA on the outside). Some of these design principles seem likely to make their way into biocompatible polymersomes, and certainly inform us about the dramatic effects of charge in polymer systems.
Peptide-Based Vesicles
A rapidly emerging approach to polymer-based vesicles involves a return to biology with peptide-based assemblies. In contrast to viral capsids that exploit precise lock-and-key interactions within remarkable geodesic assemblies, the peptide-based vesicle assemblies are based more simply again on amphiphilicity. Most polypeptide-based amphiphilic copolymers have been composed of a hydrophilic polypeptide segment covalently coupled to a synthetic hydrophobic block (Checot 2002; Kukula 2002; Rong 2003; Dong 2004; Babin 2005). The first examples of this approach used PBD as the synthetic hydrophobic block and a form of poly(L-glutamic acid) as the peptide-based hydrophilic block (Checot 2002; Kukula 2002) to form self-assembled vesicles or “peptosomes” (Kukula 2002) where the polypeptide forms a well-defined α-helical structure. In aqueous solution, these copolymers form either spherical micelles or larger vesicular aggregates and exhibit pH and ionic strength dependent changes in the hydrodynamic radius and in coil-helix transitions.
More recently, Koide and co-workers have synthesized polypeptide-based copolymers that have replaced the synthetic hydrophobic block with a hydrophilic block of PEG and coupled it to a block of charged polypeptide that is either cationic or anionic (Koide 2006). These block copolymers do not rely on amphiphilicity for assembly, rather simple mixing of a pair of opposite and equally charged block copolymers results in a stable polyion complex (PIC) membrane that forms vesicles or “PICsomes”. Both anionic and cationic copolymers are synthesized from the same precursor, PEG-poly(β-benzyl-L-aspartate) (PEG-PBLA), where the anionic PEG-poly(α,β-aspartic acid) (PEG-P(Asp)) is obtained from alkali hydrolysis and the cationic PEG-poly([5-aminopentyl]-αβ-aspartamide) (PEG-P(Asp-AP)) is achieved by aminolysis with the diamine 1,5-diaminopentane. Because these PICsome membranes do not have a hydrophobic core, they have an increased permeability to hydrophilic compounds with MWs as high as ~450 Da and can be made without the use of organic solvent, making them amenable carriers for sensitive hydrophilic compounds like nucleic acids or proteins.
The formation of purely polypeptide polymer vesicles was first demonstrated with the amphiphilic diblock co-polypeptide [poly(Nε-2-(2-(2-methoxyethoxy)ethoxy)acetyl-L-lysine]x-(poly-L-leucine)y (KxLy) (Bellomo 2004) where the size and structure of these vesicles are dictated primarily by the ordered conformation of the peptide segments. Further work with KxLy has demonstrated the tunability of the aggregates where the fluidity of the vesicle membrane allows for control of vesicle size and the morphology of aggregates can be tuned with changes in the chain lengths of polylysine and polyleucine (Holowka 2005). By replacing the poly-lysine block of the KxLy co-polypeptide with poly-arginine (RxLy), it is possible to impart the enhanced cellular delivery properties of other arginine-rich protein-transduction domains (i.e. HIV-1 Tat peptide) to co-polypeptide vesicles (Holowka 2007). Studies with R60L20 vesicles demonstrate the ability of these arginine-based vesicles to encapsulate hydrophilic compounds and successfully deliver them to cells in vitro with high efficacy.
In order to mimic pH-responsive synthetic block copolymer vesicles developed by Liu and Eisenberg (Liu 2003), Lecommandoux and co-workers have expanded work with co-polypeptide vesicles. By synthesizing a zwitterionic block co-polypeptide composed poly(L-glutamic acid)-b-poly(L-lysine) (PGA-b-PLys), the structure of the vesicle in aqueous solution could be tuned via pH (Rodriguez-Hernandez 2005). In acidic conditions (pH < 4), the PGA is neutralized and forms a hydrophobic α-helical structure resulting in a vesicle with a positively charged PLys coil brush and PGA membrane core. As pH is increased to basic conditions (pH > 10), a similar vesicle structure is formed but with an α-helical PLys membrane core and a negatively charged PGA coil brush.
In addition to forming amphiphilic co-polypeptides for vesicle assembly, polypeptides can be used as a backbone to form synthetic block copolymers. The synthesis of amino acid (AA) chains composed of poly-L-lysine of poly-L-ornithine provides a linear building block that allows for the covalent attachment of different polymers through the primary amines of lysine or ornithine. By this method, Brown and co-workers successfully created an amphiphilic block copolymer chain via the addition of the hydrophilic methoxy-poly(ethylene glycol) (mPEG) and the hydrophobic palmitic acid (Brown 2000). These recent advances in polypeptide-containing vesicles from enhanced cellular delivery to pH-sensitive assembly suggest potential for therapeutic application, although immunogenicity of peptides is more likely than with synthetic polymers – which is why virus based gene therapy is problematic and limiting to gene therapy.
CONTROLLED RELEASE FROM POLYMERSOMES
Viruses have evolved to deliver cargos effectively to a target cell, and to achieve this, viruses disassemble only when triggered by the appropriate cellular cues. Polymersomes can circulate in vivo for extended periods, probably because of the 100% PEGylation which scales the circulation half-life with PEG MW (MP) as τ1/2 ~ MPa (a ≈ 0.43) (Photos 2003), but release mechanisms – triggered externally or by intracellular cues – have just begun to be engineered into polymersomes for controlled delivery. External cues include temperature changes in the tissue resulting from the application of heating or cooling devices to specific regions of the body. Since some polymersomes have been found to enter cellular endolysosomes via ‘pinocytosis’ pathways as sketched in Figure 1b (Ahmed 2006), intracellular stimuli can be environmental changes that occur in the endolysosomes, particularly a pH decrease or a change in redox condition. While some work in this area has been done with liposomes and spherical micelles, we focus here on the incorporation of release mechanisms in polymersomes.
The most common mechanism for release of encapsulated therapeutics from polymersomes is via hydrolytic degradation of hydrophobic polyester blocks such as polylactic acid (PLA) or polycaprolactone (PCL). This release mechanism takes advantage of the molecular shape-dictated morphology of block copolymer aggregates. As the chain-end hydrolyzes (Geng 2005), the hydrophobic block systematically changes the molecular shape by increasing f. PEG-polyester copolymer molecules that begin cylindrically shaped (f ~35% ± 10%) and form bilayer vesicles transition to a wedge and finally to a cone-shaped molecule. This degradation is accelerated at acidic pH as found within the endolysosomal pathway of cells (Ahmed 2004), and at the very high copolymer concentrations (~100 mg/ml) achieved by taking up a single polymersome within an endolysosome the micelles can porate endolysosomal membranes and provide a means for drug escape into cytoplasm (Ahmed 2006). Eventually – upon complete hydrolysis – single PEG chains can be secreted from cells and excreted through the kidney.
Polyester-based degradable polymersomes have thus far been formed from PEG-PLA, PEG-PCL, and PEG-PMCL (Ahmed 2003; Ahmed 2004; Meng 2005; Ghoroghchian 2006; Zupancich 2006; Du 2007). The blending of an inert diblock such as PEG-PBD (but slowly degrading polymers could also be used) with degradable copolymers has been shown to result in homogeneously mixed polymersome membranes as verified by fluorescence microscopy (Ahmed 2003), and this blending exploited to control polymersome degradation kinetics as the release rates of encapsulated molecules vary linearly with the mole ratio of PEG-PLA (Ahmed 2004). The release of these encapsulants as well as other physical measurements (fluorescence and electron microscopy images (Ahmed 2006)) makes evident that the degradation-induced molecular shape transitions of the PEG-PLA copolymer play a significant role in the morphology taken by the blended micellar structure. While there has been initial work on these systems, the crystallinity or glassiness of pure polyester blocks at temperatures below 50°C can complicate the morphological phase diagram, membrane integrity at physiological temperatures, and the ability to make nano-sized polymersomes suitable for drug delivery purposes without otherwise fluidizing the aggregates (Ghoroghchian 2006). Over the last five or so years, various laboratories have described both vesicles and worm-like micelles made from PEO-polyester diblocks, and recent work has begun to elaborate the morphological phase diagram of PEO-PCL as a function of f (Geng 2005; Du 2007).
In contrast to the gradual mechanism of release incorporated by hydrolytic degradation of the hydrophobic block of the copolymer, other release mechanisms relying on the systematic change in hydrophobic block polarity in response to physiological or external cues have also been incorportated into the polymersome membrane (Rijcken 2007). Two examples of external cues that change copolymer polarity include temperature and UV light. UV-dependent release from polymersomes has been explored in two different systems: PEG-poly(methylphenylsilane) (Kros 2002) and PEG-(Malachite Green-CN) (Jiang 2007). Studies with PEG-poly(methylphenylsilane) have shown that polymersomes can be formed to encapsulate hydrophilic molecules. The release of these encapsulants is achieved as UV irradiation results in the cleavage of the Si-Si bonds in the poly(methylphenylsilane) that makes up the hydrophobic core of the polymersome bilayer. Upon Si-Si bond cleavage, the polarity of the bilayer core increases, resulting in a loss of membrane integrity, polymersome disintegration, and a release of hydrophilic encapsulants. Similarly, PEG-(Malachite Green-CN) (PEG-MG-CN) has been shown by electron microscopy and dynamic light scattering to form nano-sized polymersomes in aqueous solution (Jiang 2007). Exposure of these polymersomes to UV irradiation induces the loss of the cyanide anion from MG, resulting in an ionized MG molecule. This ionization significantly increases the polarity of the MG that makes up the hydrophobic core of the polymersome membrane which results in PEG-MG polymersome disassembly and release of any hydrophilic encapsulants. UV-sensitive release mechanisms are more likely to be used for in vitro delivery of encapsulants, as UV exposure in vivo has many technical challenges.
In contrast to UV-dependent release, temperature-dependent mechanisms are more feasible for in vivo delivery and are starting to be explored in polymer systems by making use of the polymer poly(N-isopropylacrylamide) (PNIPAm) (Li 2006; Qin 2006). The polarity of PNIPAm is dependent on temperature: for T ≥ 40°C, the block is hydrophobic. Therefore, by covalently attaching PNIPAm to a hydrophilic block like PEG or poly(N-(3-aminopropyl)methyl-acrylamide hydrochloride) (PAMPA), the resulting diblock copolymer is amphiphilic at high temperatures and self-assembles to form micellar structures like polymersomes. At these higher temperatures it has been shown by fluorescence microscopy (Qin 2006) and electron microscopy (Li 2006) that PNIPAm-based diblocks can indeed self-assemble to form polymersomes capable of encapsulating hydrophilic therapeutics such as doxorubicin. As predicted, decreasing the temperature of the polymersome solution results in the disassembly of polymersome structures and the transition to single copolymer molecules suspended in aqueous solution (Li 2006; Qin 2006). Initial studies have shown that this disassembly allows for the release of encapsulated therapeutics (Qin 2006), inviting the application of such a carrier in conjunction with localized hypothermic therapy for targeted drug delivery.
Polymersomes with release mechanisms that respond to external cues including temperature and UV light are limited by the accessibility of the target tissue to these cues. In contrast, the effective delivery from polymersomes with release mechanisms dependent on intracellular cues is significantly less limited. Such intracellular cues include changes in pH and redox conditions in the endolysosomal pathway. In the endolysosomal pathway, pH drops from 7.4 (as present in blood plasma) to ~5. Using polymersomes whose structure is sensitive in this pH range will allow for the intracellular release of polymersome encapsulants. While the rate of polyester hydrolysis increases with decreasing pH (Ahmed 2004; Ahmed 2006), such degrading polymersomes nonspecifically release encapsulant prior to pH changes. Alternatively, polymers which change polarity in response to pH will maintain structural integrity and result in localized burst release. To this point, work with two different diblock copolymers – poly(2-vinylpyridine)-PEO (P2VP-PEO) (Borchert 2006) and poly(2-(methacryloyloxy)ethyl phosphorylcholine)-b-poly(2-(diisopropylamino)ethyl methacrylate) (PMPC-b-PDPA) (Du 2005) – has demonstrated the pH-sensitive formation of polymersomes by light or electron microscopy and encapsulation experiments. In both systems, one block of the copolymer is composed of a polybase – P2VP or PDPA – that is hydrophobic at pH values above their respective pKa values. If these polymersomes are endocytosed, the pH in the endolysosomes would progressively decrease to pH ~ 4.5 – below the pKa for both P2VP and PDPA. Below the pKa, each polybase deprotonates to become a cationic polyelectrolyte block which results in the disassembly of the polymersome membrane. Time lapse microscopy captures the process of polymersome membrane failure as well as the release of hydrophilic encapsulants upon exposure to an acidic environment (Borchert 2006).
Instead of relying on changes in pH to shift the polarity of the hydrophobic block, Hubbell and coworkers have developed release mechanisms in polymersome systems that are either oxidation (Napoli 2004; Napoli 2004) or reduction responsive (Cerritelli 2007). To incorporate an oxidation-responsive mechanism that can be triggered in the oxidizing environment at sites of inflammation or in the endolysosomal pathway, they used an amphiphilic triblock copolymer to form stable polymersomes composed of PEO-poly(propylene-sulphide)-PEO. Upon exposure to an oxidizing agent like hydrogen peroxide (H2O2), the hydrophobic poly(propylene-sulfide) transitions to more polar poly(sulfoxides) and poly(sulfones). This increase in polarity in the hydrophobic core thus destabilizes the polymersome membrane to release encapsulated materials. By incorporating a disulfide linkage between the hydrophilic PEO and hydrophobic PPS blocks of the diblock copolymer (PEO-SS-PPS), polymersomes can be made to remain intact in oxidizing environments (i.e. blood plasma) and to disassemble in reducing environments like the cytosol where concentrations of reducing agents like glutathione increase by an order of magnitude. This reducing environment results in a bursting rupture of polymersomes due to the cleavage of the disulfide bond between the PEO and PPS blocks. Studies with these reduction-responsive polymersomes have demonstrates the release of hydrophilic encapsulants like calcein in solution as well as within cells in vitro.(Cerritelli 2007) Both oxidation and reduction responsive polymersomes help to broaden the possibilities for selective release of encapsulated therapeutics using natural cues in vivo.
ENCAPSULATION OF SMALL MOLECULE THERAPEUTICS
For polymersomes to achieve their potential as effective delivery vehicles, they must efficiently encapsulate therapeutic agents. The ability of polymersomes to encapsulate small molecules into either the aqueous lumen of the vesicle (eg. doxorubicin) or the hydrophobic core of the membrane (eg. taxol) has been thoroughly studied. Early studies used fluorescent or light-absorbing molecules both to provide evidence of encapsulation in both the lumen and membrane core and to allow for the tracking of polymersome fate in vivo.(Discher 1999; Photos 2003) Polymersome encapsulation of small molecules has advanced to include anti-cancer therapeutics (Choucair 2005; Ahmed 2006; Ahmed 2006; Li 2007) as well as near-infrared fluorophores that are designed to be used for deep tissue imaging (Lin 1994) or cell-tracking in vivo (Christian 2007).
Efficient loading of the anti-cancer drug doxorubicin into the lumen of preformed polymersomes can be achieved by the pH-gradient entrapment method developed originally for liposomes (Ahmed 2004; Choucair 2005). Polymersomes are also capable of stably loading significant quantities of the hydrophobic anti-cancer drug Taxol into the hydrophobic core of the membrane – a distinct advantage over lipid-based carriers (Ahmed 2006; Ahmed 2006; Li 2007). By loading both doxorubicin and taxol simultaneously into degradable polymersomes, an effective, dual drug anti-cancer therapeutic device has been developed (Ahmed 2006; Ahmed 2006). In studying the uptake of these anti-cancer degradable polymersomes by cultured cells in vitro, delivery of doxorubicin (which is naturally fluorescent) to the nucleus indicated these degradable polymersomes were capable of efficiently escaping the endolysosomal pathway. As mentioned above, degradable polymersomes break down with the pH decrease in the endolysosomal pathway: they transition to spherical micelles which are membrane-lytic at the very high polymer to lipid ratios of endolysosomes (Ahmed 2006). In fact, the circulating concentrations of copolymer are very low (mg/mL) and sub-lytic, but lytic concentrations of ~100 mg/mL are achieved in the small endolysosomes after uptake and the accelerated degradation only promotes the release of drug into the cytosol (eg. Figure 1b).
The long-circulating properties of these 100% PEG-ylated degradable polymersomes allow passive accumulation in tumor sites by the Enhanced Permeability and Retention (EPR) effect (Maeda 2000). By combining passive tumor site accumulation, endolytic escape and the capacity to increase tolerable doses of two anti-cancer drugs in the same carrier, anti-cancer polymersomes are able to effectively shrink tumors in vivo (Ahmed 2006; Ahmed 2006). These in vivo studies tracked the size of human-derived tumors in nude mice over time. Mice were treated with saline, empty polymersomes, free drug, or anti-cancer polymersomes. Treatments with saline and empty polymersomes showed substantial tumor growth while free drug prevented further tumor growth. In contrast, treatments with anti-cancer polymersomes resulted in tumor shrinkage over time. Analysis of drug accumulation in the tumor and tumor cell death versus the different treatment methods all corroborate the results. Successful treatment of tumors in vivo with these anti-cancer polymersomes suggests significant great opportunity for the use of polymersomes in the effective delivery of other therapeutic molecules.
POLYMERSOMES AS NON-VIRAL GENE/OLIGO VECTORS
With the effectiveness of gene therapy proven in many different disease models, the development of vectors to safely and effectively administer these materials in vivo has been an area of intense study for 10-20 years. The optimal vectors should be capable of delivering genetic material to a wide variety of cells and tissues, and considerable work has gone into re-engineering viruses. Viral vectors are certainly capable of high transfection efficiency and sustained expression of a foreign gene, but they can face problems with production scale-up (including avoidance of empty capsids) and safety concerns with an immune response. The substantial challenges facing viral vectors have led to increased efforts to synthesize effective non-viral gene delivery vectors. While such carriers do not pose the same risks as viral vectors, they are not without their disadvantages. Two of the major challenges facing work with non-viral vectors are 1) low transfection efficiency caused by the many barriers between administering the vector and delivering viable genes to the nucleus, and 2) transient expression of the foreign gene. Non-viral vectors are frequently composed of the negatively charged, genetic material (DNA, RNA, etc.) complexed with a cationic lipid carrier system referred to as lipoplexes. Lipoplexes like Lipofectamine have commonly been used to effectively deliver genetic material in vitro. The composition of these complexes has been expanded to include cationic polymers (polyplex), cationic polymers encapsulated by cationic liposomes (lipopolyplex), and now both charged and neutral polymersomes.
Mixtures of cationic and anionic surfactants at different ratios were shown by Kaler and coworkers to yield different aggregate morphologies from spheres to cylinders to planar bilayers that result in catanionic surfactant vesicles (Kaler 1989). Though surfactant vesicles are more reminiscent of low MW lipid structures than polymersomes, the initial work with DNA encapsulation in non-lipid vesicle structures merits discussion. Drawing from work with lipoplexes, these vesicles were investigated for use as gene therapy vectors, and initial studies of DNA interaction with different catanionic vesicle systems showed DNA compaction to the vesicle surface that could be released upon the addition of anionic amphiphiles (Mel’nikov 1999; Bonincontro 2007). Investigation of the ability of these vesicles to encapsulate DNA for gene delivery revealed that the complexation of anionic DNA with catanionic vesicles resulted in multibilayer lamellar stacks intercalated with DNA (Dias 2002; Dias 2002). Despite the ability of these vesicles to encapsulate DNA by complexation, they were not a viable vector due to the cytotoxic nature of the positively charged surfactant cetyltrimethylammonium bromide (CTAB). Therefore, Lindman and coworkers replaced CTAB with arginine-N-lauroyl amide dihydrochloride (ALA) as a cationic surfactant (Rosa 2007; Rosa 2007). Catanionic vesicles composed of ALA and the anionic surfactant sodium octylsulfate (SOS) show similar DNA intercalation as with CTAB/SOS systems and the release of DNA from DNA/ALA/SOS complexes was shown to result in DNA/ALA complexes. While these catanionic vesicles demonstrate the ability to encapsulate DNA, complexation and subsequent intercalation between bilayers makes controlled release difficult to achieve. In addition, any exposed cation is problematic for sustained circulation in vivo, in part because cell surfaces are negatively charged.
To create an effective gene therapy vector based on a vesicle architecture, controlled release must be incorporated for the encapsulated gene. By encapsulating DNA in the aqueous lumen of vesicles in addition to complexation with a cationic polymer, Korobko and coworkers were able to effectively release and deliver genes in vitro (Korobko 2005; Korobko 2006). The cationic amphiphilic diblock copolymer poly-(butadiene-b-N-methyl-4-vinyl pyridinium) (PBD-P4VPQI) was dissolved in toluene and added to an aqueous solution containing DNA to form cationic polymersomes encapsulating DNA in the vesicle lumen as well as intercalated into the P4VPQI brush as visualized by fluorescence microscopy. DNA intercalation provides the stability necessary for these polymersomes to be purified in air and redissolved in an aqueous PEG solution. To demonstrate that these polymersomes are not limited to small DNA molecules as are viral vectors, the DNA plasmid pUC18 was successfully encapsulated in the lumen and P4VPQI brush as shown by fluorescence microscopy (Korobko 2006). Because DNA is suspended in the lumen of these vesicles, release is simply achieved by lysing the polymersomes with changes in osmotic pressure or ionic strength. The cationic polymersomes were shown effective in in vitro transfection: the DNA-containing vesicles stuck to a glass surface, and HeLa cells cultured on the glass took up the vesicles, resulting in successful transfections after two hours (Korobko 2006). While these cationic polymersomes allow for the controlled release of DNA by encapsulation in the lumen of the vesicle, the cationic surface charge results in a non-viable in vivo delivery system due to nonspecific uptake and challenges to vesicle and DNA stability.
Encapsulation and delivery of DNA has also been investigated with poly(amino acid) (poly(AA)) based polymer vesicles. Brown and coworkers have created an amphiphilic triblock copolymer by attaching methoxy-poly(ethylene glycol) (mPEG) and the hydrophobic chain palmitic acid in block segments along a poly-L-lysine (PLL) or poly-L-ornithine (PO) backbone, leaving a block segment of bare, cationic PLL or PO (Brown 2000). This triblock self-assembles to form vesicle structures capable of intercalating with DNA, where the surface charge of the vesicles can be tuned by the polymer:DNA ratio. Incubation of these DNA-containing vesicles with two different cell lines shows improved DNA transfection versus poly(AA) + DNA or DNA alone. Injection of these DNA-encapsulating poly(AA) vesicles in vivo, where the surface charge is slightly anionic, has shown that gene transfer can be achieved in the lungs and liver (Brown 2003). However, measurements of cytotoxicity in cell culture show cell death at vesicle concentrations as low as 0.1 mg/mL of polymer, demonstrating the need for a more biocompatible polymer gene delivery vector.
As demonstrated by studies with anti-cancer polymersomes, the in vivo advantages of a charge-neutral, 100% PEGylated surface – i.e. long circulation in vivo, biocompatibility – make it desirable to create a PEG-based polymersome vector that is capable of effectively encapsulating and delivering genes or oligonucleotides. Initial work with degradable PEG-PLA (OLA) polymersomes has shown the encapsulation and delivery of siRNA in vitro is comparable to levels achieved with the commonly used lipoplex of siRNA and Lipofectamine 2000 (LF2K) (Kim 2008). Encapsulation of an siRNA that works to silence the gene responsible for the nuclear proteins lamin A and lamin C (lamin A/C) was achieved by an emulsion technique wherein OLA dissolved in dimethyl sulfoxide (DMSO) was added to an aqueous solution containing fluorescently-labeled siRNA. Nano-sized polymersomes with a hydrodynamic radius (Rh) of 93 ± 7 nm were subsequently formed, encapsulating siRNA in the aqueous lumen where Rh was measured by dynamic light scattering (DLS). Organic solvent and unencapsulated siRNA were removed by dialysis, and the colocalization in fluorescence micrographs of the hydrophobic membrane labeling dye (PKH26) and FITC-labeled siRNA indicate the successful loading of siRNA into polymersomes (Figure 2a).
Fig. 2. Delivery of siRNA by polymersomes.
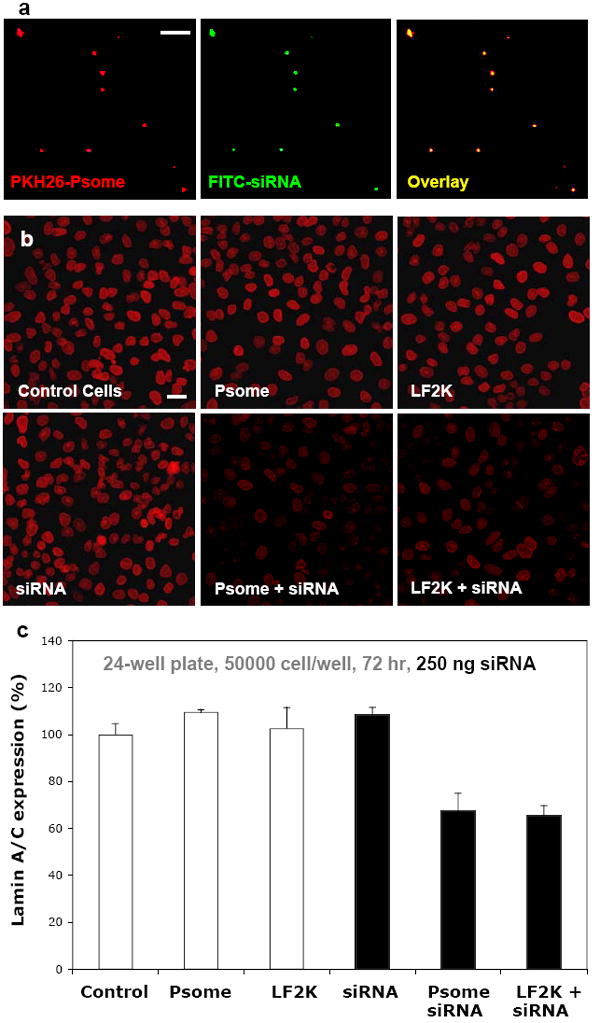
a) Fluorescent micrographs of fluorescent PKH26 labeling the membrane of nano-sized OLA polymersomes (Psome) and FITC-labeled siRNA for lamin A/C. The precise overlay of these two images indicates the encapsulation of siRNA in the aqueous lumen of OLA polymersomes. Scale bar = 10 μm. b) Representative images of A549 cells fixed and stained for lamin A/C at 72 hours display the knockdown of Lamin A/C in cultures incubated with LF2K-encapsulated siRNA and OLA-Psome encapsulated siRNA when compared to controls. Scale bar = 40 μm. c) Quantification of lamin A/C immunofluorescence in A549 cells indicates a 33-35% knockdown of lamin A/C by LF2K-encapsulated siRNA and OLA-Psome encapsulated siRNA.
Size, loading efficiency, and surface charge of these OLA polymersomes (OLA-Psomes) with siRNA was similar to LF2K + siRNA lipoplexes. The ability of siRNA-containing OLA polymersomes to knockdown lamin A/C in the A549 cell line in vitro was then compared to LF2K + siRNA. After incubating with OLA-Psomes, LF2K, siRNA, LF2K + siRNA, or OLA-Psomes + siRNA, cells were examined at 72 hours to determine protein knockdown. Lamin A/C knockdown was measured by comparing immunostain fluorescence intensity to that of control cells that were not exposed to any carrier or siRNA. Representative fluorescent micrographs of lamin A/C knockdown at 72 hours display similar decreases in fluorescence intensity in cultures exposed to LF2K + siRNA and OLA + siRNA compared to controls (Figure 2b). Analysis of lamin A/C knockdown (Figure 2c) indicates that bare siRNA had no knockdown effect while both LF2K + siRNA and OLA-Psome + siRNA show about one-third decreases in lamin A/C expression. Viral based methods show greater knockdown (about 60-70%; Kim 2008), but such virus strategies are not especially viable in vivo as discussed above. The advantage of the polymersome system compared to LF2K is that its neutral 100% PEGylated surface is already known to be effective for in vivo delivery of small molecules.
POLYMERSOME ENCAPSULATION OF PROTEINS
Therapeutic proteins are generally potent but short-lived, which has motivated recent work on direct modification of proteins with poly(ethylene glycol) (PEG) (Perez 2002; Harris 2003; Parveen 2006). Encapsulation is another route and has been pursued with several different carrier systems, including liposomes and (micro/nano)particles. While both approaches have been shown to increase the protein plasma half-life, both have disadvantages. Liposomes have been shown to be leaky and unstable over time, and – without modification – are cleared quickly (t1/2 < 5 hours) by the body (Lasic 1998; Photos 2003). The process of forming (micro/nano)particles to encapsulate therapeutics can cause denaturation and thus inactivation of proteins. During particle formation, proteins are exposed to hydrophobic interfaces (i.e. organic solvent, air) and subsequently denature to form inactive protein conformations (Perez 2002). The covalent attachment of PEG molecules to proteins (PEGylation) has been shown to be effective in increasing the effectiveness of protein therapies. PEGylation increases the half-life of the protein in plasma as well as decreases the immune response from the body (Harris 2003; Parveen 2006). The drawback of PEGylation is that the non-specific covalent attachment of a large PEG molecule to the protein can lead to changes in the protein structure and thus activity (Harris 2003). In contrast to these encapsulation methods, polymersomes formed by film rehydration have been shown to incorporate transmembrane proteins in the bilayer as well as encapsulate proteins in the aqueous lumen without loss of their functional conformation.
Incorporation of proteins into polymer bilayers began by integrating membrane proteins into black block copolymer membranes – two-dimensional bilayer membranes stretched across an interface between two aqueous solutions (Meier 2000). This work proved proteins could be incorporated into hyperthick triblock copolymer membranes while maintaining their functionality as measured by membrane conductance. Incorporation of proteins in “black films” od block copolymer has been expanded for applications in sensors (Wong 2006) and protein-driven energy transduction (Ho 2004) across polymeric biomembranes. By expanding their work to integrate selective protein channels into polymersome membranes, Meier and coworkers were able to form polymersome nano-reactors (Nardin 2000; Nardin 2000) as well as load DNA into polymersomes by (a very impressive) viral injection (Graff 2002). These polymersomes were formed from an ABA triblock copolymer composed of a flexible poly(dimethylsiloxane) (PDMS) hydrophobic block and two water soluble poly(2-methyloxazoline) (PMOXA) side blocks. Creation of nano-reactors is achieved by the incorporation of the channel protein OmpF which selectively allows the passage of the antibiotic ampicillin into the lumen of the vesicle where it can be hydrolyzed by the encapsulated enzyme β-lactamase (Nardin 2000; Nardin 2001). Similarly, by incorporating the channel protein LamB – a bacterial channel protein that serves as a receptor for the λ-phage virus – into the polymersome membrane, the λ-phage virus recognizes its receptor, binds to the polymersome and injects its DNA as visualized by incredible electron microscopy micrographs (Graff 2002). To better understand how membrane proteins can be incorporated into the hyperthick polymersome membrane and maintain functionality, the insertion of pore forming membrane into a polymer bilayer was simulated using coarse-grained molecular dynamics (Srinivas 2005). Simulations suggest that polymer chain flexibility permits the integration of proteins with small membrane-spanning domains, but the flexibility of the hydrophilic chains can partially block the functional pore of the membrane protein, resulting in decreased functionality compared to when the proteins are incorporated in more natural lipid membranes.
Encapsulation of proteins within the aqueous lumen of polymersomes can in principle take advantage of the extended circulation kinetics and controlled release properties of polymersomes. To test the ability of neutral diblock or charged triblock polymersomes to encapsulate larger, globular proteins, initial studies were performed with FITC-labeled bovine serum albumin (FITC-BSA) as well as Myoglobin and Hemoglobin (Lee 2001; Wittemann 2007). Fluorescent micrographs of either giant or fluorescently-labeled, nano-sized polymersomes provide visual proof that FITC-BSA is indeed encapsulated in the aqueous lumen of the vesicles. By encapsulating different enzymes and metalloproteins in polymersomes, different groups have proven that proteins remain functional after encapsulation (Nardin 2000; Nardin 2001; Napoli 2004; Arifin 2005; Kishimura 2007). As previously discussed, Meier and coworkers were able to create polymersome nano-reactors by incorporating functional proteins into the membrane while also encapsulating the enzyme β-lactamase in the aqueous lumen of the vesicle (Nardin 2000; Nardin 2001). The activity of β-lactamase was demonstrated through the hydrolysis of ampicillin to form ampicillinoic acid. Also discussed previously was the incorporation of an oxidation-responsive release mechanism from PEG-PPS-PEG polymersomes (Napoli 2004). While this mechanism can be cued in vivo (i.e. sites of inflammation, endolysosomal pathway), release can also be triggered via the encapsulation of oxidizing enzyme. Glucose oxidase (GOx) works to break down glucose to form gluconolactone by oxidation and in the process reduces oxygen to form hydrogen peroxide. Because GOx that was encapsulated in PEG-PPS-PEG polymersomes remained functional, the addition of glucose to the surrounding medium resulted in glucose crossing the polymersome membrane where it was subsequently being oxidized, resulting in the formation of hydrogen peroxide (Napoli 2004). Hydrogen peroxide – a strong oxidizing compound – then worked to break down the PEG-PPS-PEG polymersomes (as measured by optical density at 600nm) by oxidizing PPS to its more polar byproducts poly(sulfoxide) and poly(sulfone). In these studies it was shown that GOx not only maintained its functionality after encapsulation, but its functionality was extended compared to GOx in solution.
The synthetic mimicry of cellular structures was the initial inspiration for the study of micellar polymer structures like polymersomes and worm-like micelles (Dalhaimer 2004). Red blood cells (RBCs) are perhaps the easiest functional cells to mimic because they lack intracellular structures – nucleus, organelles – that make all other cells so very complex. By encapsulating the metalloproteins hemoglobin (Arifin 2005) and myoglobin (Kishimura 2007) in polymersomes, an RBC ‘mimic’ has been created. Because oxygen is capable of diffusing across the polymersome membrane at rates similar to lipid bilayers, polymersomes encapsulating hemoglobin have been shown to have oxygen affinities similar to those of human RBCs (Arifin 2005). Similarly, Kishimura and coworkers successfully encapsulated myoglobin (Mb) in PICsomes as shown by fluorescent micrographs of fluorescently-labeled Mb (Kishimura 2007). Functionality of encapsulated Mb was determined by oxygen binding which could be monitored by absorbance at 434 nm. Though these studies have shown that encapsulation of functional proteins is possible with polymersomes, little work has been done to encapsulate therapeutic proteins.
The encapsulation of recombinant insulin in polymersomes provides a good model for the encapsulation of therapeutic proteins. Enhanced circulation kinetics and controllable release of insulin in vivo is desirable to increase patient compliance and decrease the need for intravenous injections. While insulin is a low molecular weight protein, it has been shown to aggregate to dimers, hexamers, and eventually fibrils when the structure is disrupted by exposure to agitation or hydrophobic interfaces. Therefore, insulin was a challenging first test of therapeutic protein encapsulation in PEG-based polymersomes. Briefly, by a film rehydration method, thin films of PEG-PBD (OB2, 1.2 – 2.4kD) blended with the hydrophobic membrane label PKH26 were hydrated at 60°C with phosphate buffered aqueous solutions (pH = 7.4) containing either fluorescein-labeled bovine insulin or recombinant human zinc-insulin at 1.5 mg/mL. As expected, film rehydration resulted in giant, membrane-labeled OB2 polymersomes encapsulating insulin in their aqueous lumen. The encapsulation of fluorescein-insulin provided visual proof of insulin encapsulation by fluorescence microscopy as micrographs of the red fluorescent membrane label overlayed perfectly with micrographs of fluorescein-insulin in the aqueous lumen (Figure 3a).
Fig. 3. Therapeutic protein encapsulation in Polymersomes.
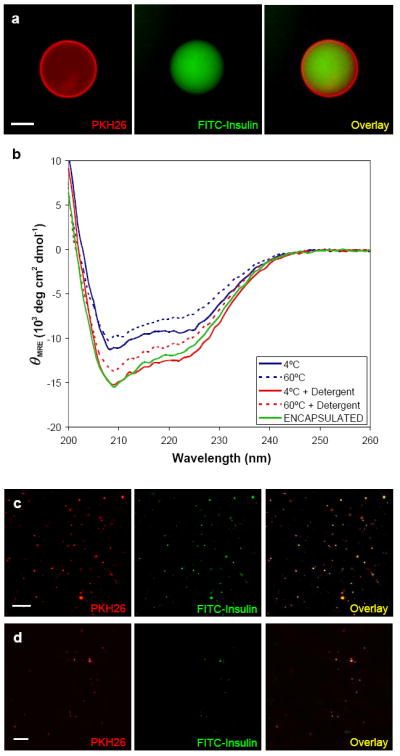
a) Fluorescent micrographs of fluorescent PKH26 labeling the membrane of a giant OB2 polymersome and fluorescein-labeled bovine insulin (diluted 100X). Overlay of these images display the encapsulation of fluorescein-insulin in OB2 polymersomes. Scale bar = 10μm. b) Circular dichroism (CD) results for recombinant human Zn-insulin after incubation for 8 hours at 4°C and 60°C indicates that the film rehydration technique does not result in the loss of insulin secondary structure. The addition of the nonionic detergent Tween80 further stabilizes the secondary structure at both 4°C and 60°C. Most importantly, CD of insulin encapsulated in OB2 polymersomes in the presence of Tween80 does not indicate any loss of protein secondary structure. c) Fluorescent micrographs of PKH26 labeling the membrane of nano-sized OB2 polymersomes and fluorescein-insulin (diluted 0X). The precise overlay of these two images shows that polymersomes can stably encapsulate insulin through sizing down and separation from unencapsulated insulin. Scale bar = 10μm. d) Fluorescent micrographs of PKH26 labeling the membrane of nano-sized OB2 polymersomes encapsulating fluorescein-insulin after 10X dilution in whole blood. Fluorescence overlay indicates stable encapsulation in a biologically relevant medium up to 8 hours. Scale bar = 10μm.
In previous work with growth factors similar to insulin, the nonionic detergent Tween80 was used to prevent protein denaturation and aggregation (Katakam 1995). Similarly, the addition of Tween80 (0.05% v/v) to solutions of zinc-insulin was shown by circular dichroism to enhance the stability of insulin’s secondary structure during hydration (Figure 3b). Solutions of zinc-insulin alone were incubated either at 4°C or 60°C for eight hours with or without Tween80. Circular dichroism was performed at constant temperatures and the final protein concentrations were measured to calculate the mean residue ellipticity (θMRE). Promising circular dichroism results also indicated that the film rehydration technique allowed for the encapsulation of insulin in OB2 polymersomes without significant loss of secondary structure.
For polymersomes to be used as viable delivery vehicles, they must be sized down to a radius of approximately 100 nm. To prevent denaturation of insulin, polymersomes were sized down by repeated extrusion through sequentially smaller pore sizes without prior sonication or freeze-thaw cycles. The Rh of these nano-polymersomes was then measured to be 97 ± 6 nm using dynamic light scattering (data not shown). Fluorescein-insulin loaded, nano-polymersomes were then separated from unencapsulated fluorescein-insulin by size exclusion chromatography through a Sephacryl S-500 column, collecting fractions every 500μl. Separation of the polymersomes from unencapsulated fluorescein-insulin was confirmed by fluorescence absorbance measurements of each elution fraction using a Fluoroskan microplate fluorimeter. After sizing down and separation, these polymersomes were shown to maintain their encapsulants for months (at 4°C) by fluorescence microscopy (Figure 3c). While these structures were too small to visualize the presence of fluorescein-insulin in the aqueous lumen as done with giant polymersomes, the precise colocalization of fluorescent signals from the fluorescent membrane label and fluorescein-insulin confirms encapsulation. To prove these polymersomes provide a stable carrier system for therapeutic proteins, fluorescein-insulin loaded nano-polymersomes were incubated for hours in whole blood in vitro at 37°C and shown to stably maintain their encapsulant (Figure 3d). Encapsulation of insulin in neutral PEG-PBD polymersomes provides a promising method to increase therapeutic efficiency by maintaining protein structure – and in a carrier system that has been shown to be stable in biological environments.
ACTIVE TARGETING OF POLYMERSOMES
Although few if any drug carriers in clinical use today are targeted, viruses often show preferential – although not exclusive – interactions with particular cell types and cell entry pathways. Given that polymersomes exhibit minimal nonspecific adhesion to cells (initially), that they can circulate for many tens of hours, and that they can integrate controlled release mechanisms, targeted polymersomes should add an additional level of functionality – although there is an added cost for the complexity. With PEG-based assemblies, much work has been focused on attaching ligands or antibodies to the hydroxyl end-group (Torchilin 2001; Discher 2002; Christian 2007; Nam 2007). Biotinylated nondegradable block copolymer assemblies have been shown to attach to surfaces coated with the biotin receptor avidin (Dalhaimer 2004; Lin 2004; Nam 2007; Nam 2007) as well as to cells where they successfully delivered the hydrophobic drug Taxol (Dalhaimer 2004). Similar chemistry has been used to attach either an antihuman IgG, or antihuman serum to PEG-carbonate- or PEG-polyester-assembled polymer vesicles.(Meng 2005; Lin 2006) The attachment of HIV-derived Tat peptide – a cationic peptide shown to enhance cellular delivery of nanoparticles (Gupta 2005) – to PEG-PBD polymersomes has been shown to increase dendritic cell uptake in vitro (Christian 2007). Broz and coworkers (Broz 2005) have modified their triblock copolymer polymersomes with polyguancylic acid to target a macrophage scavenger receptor SRA1. This scavenger receptor is a pattern-recognition antigen upregulated only in activated tissue macrophages, not in monocytes or monocyte precursor cells. These in vitro cell targeting studies have not yet been translated to in vivo conditions, where complications may arise from opsonization by serum components and competitive interactions with other cells (Discher 2002). Though promising, chemically attached targeting moieties do not act as a ‘homing beacon’ to the desired in vivo target. Instead, targeted carriers must contact the desired target either by convective or diffusive collisions; only then can targeted adhesion increase the likelihood of cellular uptake or localized release. Therefore, future decisions to increase carrier complexity with targeting ligands should be carefully weighed against the advantages of active targeting versus passive targeting via the EPR effect.
CONCLUSIONS
The tunability of polymersome structure and properties has expanded considerably with the recent advances in block copolymer chemistries. The ability to specifically tailor polymersome formulation methods, physicochemical properties, release mechanisms, and even targeting chemistries makes polymersomes an ideal platform for the encapsulation of a broad range of therapeutic molecules. As demonstrated by their effective treatment of tumor models in vivo and stable encapsulation of other actives (i.e. dyes, nucleic acids, proteins), polymersomes show great promise in moving from design and onto therapeutic application.
Lastly, one might conclude from our initial descriptions of copolymer assemblies that a library of any given block copolymer (eg. 10% < f < 90%) could be extremely useful to develop and study, even if the initial focus were on polymersomes. Indeed, the anticancer performance of polymersomes loaded with hydrophobic drugs such as taxol can be readily compared to that of worm-like micelles and spherical micelles of very similar composition. This has already proven an interesting direction of study with the demonstration that very long worm-like micelles appear to circulate longer than any synthetic carrier described to date and also that worm-like ‘filomicelles’ more effectively shrink tumors than the smaller carriers (Geng 2007).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed F, Discher DE. Self-porating polymersomes of PEG-PLA and PEG-PCL: hydrolysis-triggered controlled release vesicles. Journal of Controlled Release. 2004;96(1):37–53. doi: 10.1016/j.jconrel.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Ahmed F, Hategan A, Discher DE, Discher BM. Block copolymer assemblies with cross-link stabilization: From single-component monolayers to bilayer blends with PEO-PLA. Langmuir. 2003;19(16):6505–6511. [Google Scholar]
- Ahmed F, Pakunlu RI, Brannan A, Bates F, Minko T, Discher DE. Biodegradable polymersomes loaded with both paclitaxel and doxorubicin permeate and shrink tumors, inducing apoptosis in proportion to accumulated drug. Journal of Controlled Release. 2006;116(2):150–158. doi: 10.1016/j.jconrel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Ahmed F, Pakunlu RI, Srinivas G, Brannan A, Bates F, Klein ML, Minko T, Discher DE. Shrinkage of a Rapidly Growing Tumor by Drug-Loaded Polymersomes: pH-Triggered Release through Copolymer Degradation. Mol Pharmaceutics. 2006;3(3):340–350. doi: 10.1021/mp050103u. [DOI] [PubMed] [Google Scholar]
- Antonietti M, Forster S. Vesicles and liposomes: A self-assembly principle beyond lipids. Advanced Materials. 2003;15(16):1323–1333. [Google Scholar]
- Aranda-Espinoza H, Bermudez H, Bates FS, Discher DE. Electromechanical limits of polymersomes. Physical Review Letters. 2001;8720(20) doi: 10.1103/PhysRevLett.87.208301. [DOI] [PubMed] [Google Scholar]
- Arifin DR, Palmer AF. Polymersome encapsulated hemoglobin: A novel type of oxygen carrier. Biomacromolecules. 2005;6(4):2172–2181. doi: 10.1021/bm0501454. [DOI] [PubMed] [Google Scholar]
- Babin J, Rodriguez-Hernandez J, Lecommandoux S, Klok HA, Achard MF. Self-assembled nanostructures from peptide-synthetic hybrid block copolymers: Complex, stimuli-responsive rod-coil architectures. Faraday Discussions. 2005;128:179–192. doi: 10.1039/b403203a. [DOI] [PubMed] [Google Scholar]
- Bangham AD. Liposomes - the Babraham Connection. Chemistry and Physics of Lipids. 1993;64(13):275–285. doi: 10.1016/0009-3084(93)90071-a. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Ryan AJ, Tomas S. Polymeric vesicle permeability: A facile chemical assay. Langmuir. 2006;22(11):4910–4913. doi: 10.1021/la060354p. [DOI] [PubMed] [Google Scholar]
- Bellomo EG, Wyrsta MD, Pakstis L, Pochan DJ, Deming TJ. Stimuli-responsive polypeptide vesicles by conformation-specific assembly. Nature Materials. 2004;3(4):244–248. doi: 10.1038/nmat1093. [DOI] [PubMed] [Google Scholar]
- Bermudez H, Brannan AK, Hammer DA, Bates FS, Discher DE. Molecular weight dependence of polymersome membrane structure, elasticity, and stability. Macromolecules. 2002;35(21):8203–8208. [Google Scholar]
- Bonincontro A, La Mesa C, Proietti C, Risuleo G. A biophysical investigation on the binding and controlled DNA release in a cetyltrimethylammonium bromide-sodium octyl sulfate cat-anionic vesicle system. Biomacromolecules. 2007;8(6):1824–1829. doi: 10.1021/bm0612079. [DOI] [PubMed] [Google Scholar]
- Borchert U, Lipprandt U, Bilang M, Kimpfler A, Rank A, Peschka-Suss R, Schubert R, Lindner P, Forster S. pH-induced release from P2VP-PEO block copolymer vesicles. Langmuir. 2006;22(13):5843–5847. doi: 10.1021/la060227t. [DOI] [PubMed] [Google Scholar]
- Brown MD, Gray AI, Tetley L, Santovena A, Rene J, Schatzlein AG, Uchegbu IF. In vitro and in vivo gene transfer with poly(amino acid) vesicles. Journal of Controlled Release. 2003;93(2):193–211. doi: 10.1016/j.jconrel.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Brown MD, Schatzlein A, Brownlie A, Jack V, Wang W, Tetley L, Gray AI, Uchegbu IF. Preliminary characterization of novel amino acid based polymeric vesicles as gene and drug delivery agents. Bioconjugate Chemistry. 2000;11(6):880–891. doi: 10.1021/bc000052d. [DOI] [PubMed] [Google Scholar]
- Broz P, Benito SM, Saw C, Burger P, Heider H, Pfisterer M, Marsch S, Meier W, Hunziker P. Cell targeting by a generic receptor-targeted polymer nanocontainer platform. Journal of Controlled Release. 2005;102(2):475–488. doi: 10.1016/j.jconrel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Cerritelli S, Velluto D, Hubbell JA. PEG-SS-PPS: Reduction-sensitive disulfide block copolymer vesicles for intracellular drug delivery. Biomacromolecules. 2007;8(6):1966–1972. doi: 10.1021/bm070085x. [DOI] [PubMed] [Google Scholar]
- Checot F, Lecommandoux S, Gnanou Y, Klok HA. Water-soluble stimuli-responsive vesicles from peptide-based diblock copolymers. Angewandte Chemie-International Edition. 2002;41(8):1339–1343. doi: 10.1002/1521-3773(20020415)41:8<1339::aid-anie1339>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Choucair A, Soo PL, Eisenberg A. Active loading and tunable release of doxorubicin from block copolymer vesicles. Langmuir. 2005;21(20):9308–9313. doi: 10.1021/la050710o. [DOI] [PubMed] [Google Scholar]
- Christian NA, Milone MC, Ranka SS, Li GZ, Frail PR, Davis KP, Bates FS, Therien MJ, Ghoroghchian PP, June CH, Hammer DA. Tat-functionalized near-infrared emissive polymersomes for dendritic cell labeling. Bioconjugate Chemistry. 2007;18(1):31–40. doi: 10.1021/bc0601267. [DOI] [PubMed] [Google Scholar]
- Dalhaimer P, Bermudez H, Discher DE. Biopolymer mimicry with polymeric wormlike micelles: Molecular weight scaled flexibility, locked-in curvature, and coexisting microphases. Journal of Polymer Science Part B-Polymer Physics. 2004;42(1):168–176. [Google Scholar]
- Dalhaimer P, Engler AJ, Parthasarathy R, Discher DE. Targeted worm micelles. Biomacromolecules. 2004;5(5):1714–1719. doi: 10.1021/bm049884v. [DOI] [PubMed] [Google Scholar]
- Dias RS, Lindman B, Miguel MG. Compaction and decompaction of DNA in the presence of catanionic amphiphile mixtures. Journal of Physical Chemistry B. 2002;106(48):12608–12612. [Google Scholar]
- Dias RS, Lindman B, Miguel MG. DNA interaction with catanionic vesicles. Journal of Physical Chemistry B. 2002;106(48):12600–12607. [Google Scholar]
- Dimova R, Seifert U, Pouligny B, Forster S, Dobereiner HG. Hyperviscous diblock copolymer vesicles. European Physical Journal E. 2002;7(3):241–250. [Google Scholar]
- Discher BM, Won YY, Ege DS, Lee JCM, Bates FS, Discher DE, Hammer DA. Polymersomes: Tough vesicles made from diblock copolymers. Science. 1999;284(5417):1143–1146. doi: 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]
- Discher DE, Eisenberg A. Polymer vesicles. Science. 2002;297(5583):967–973. doi: 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
- Discher DE, Photos PJ, Ahmed F, Parthasarathy R, Bates FS. Polymersomes: a new platform for drug targeting. In: Muzykantov VR, Torchilin VP, editors. Biomedical Aspects of Drug Targeting. Boston: Kluwer Academic Pub.; 2002. pp. 459–471. [Google Scholar]
- Dong CM, Sun XL, Faucher KM, Apkarian RP, Chaikof EL. Synthesis and characterization of glycopolymer-polypeptide triblock copolymers. Biomacromolecules. 2004;5(1):224–231. doi: 10.1021/bm0343500. [DOI] [PubMed] [Google Scholar]
- Du JZ, Tang YP, Lewis AL, Armes SP. pH-sensitive vesicles based on a biocompatible zwitterionic diblock copolymer. Journal of the American Chemical Society. 2005;127(51):17982–17983. doi: 10.1021/ja056514l. [DOI] [PubMed] [Google Scholar]
- Du ZX, Xu JT, Fan ZQ. Micellar morphologies of poly(epsilon-caprolactone)-b-poly(ethylene oxide) block copolymers in water with a crystalline core. Macromolecules. 2007;40(21):7633–7637. [Google Scholar]
- Geng Y, Ahmed F, Bhasin N, Discher DE. Visualizing worm micelle dynamics and phase transitions of a charged diblock copolymer in water. Journal of Physical Chemistry B. 2005;109(9):3772–3779. doi: 10.1021/jp0459559. [DOI] [PubMed] [Google Scholar]
- Geng Y, Discher DE. Hydrolytic degradation of poly(ethylene oxide)-block-polycaprolactone worm micelles. Journal of the American Chemical Society. 2005;127(37):12780–12781. doi: 10.1021/ja053902e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Dalhaimer P, Cai SS, Tsai R, Tewari M, Minko T, Discher DE. Shape effects of filaments versus spherical particles in flow and drug delivery. Nature Nanotechnology. 2007;2(4):249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoroghchian PP, Li GZ, Levine DH, Davis KP, Bates FS, Hammer DA, Therien MJ. Bioresorbable vesicles formed through spontaneous self-assembly of amphiphilic poly(ethylene oxide)-block-polycaprolactone. Macromolecules. 2006;39(5):1673–1675. doi: 10.1021/ma0519009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff A, Sauer M, Van Gelder P, Meier W. Virus-assisted loading of polymer nanocontainer. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(8):5064–5068. doi: 10.1073/pnas.062654499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Szoka FC. Chemical approaches to triggerable lipid vesicles for drug and gene delivery. Accounts of Chemical Research. 2003;36(5):335–341. doi: 10.1021/ar9703241. [DOI] [PubMed] [Google Scholar]
- Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Advanced Drug Delivery Reviews. 2005;57(4):637–651. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nature Reviews Drug Discovery. 2003;2(3):214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- Ho D, Chu B, Lee H, Montemagno CD. Protein-driven energy transduction across polymeric biomembranes. Nanotechnology. 2004;15(8):1084–1094. [Google Scholar]
- Holowka EP, Pochan DJ, Deming TJ. Charged polypeptide vesicles with controllable diameter. Journal of the American Chemical Society. 2005;127(35):12423–12428. doi: 10.1021/ja053557t. [DOI] [PubMed] [Google Scholar]
- Holowka EP, Sun VZ, Kamei DT, Deming TJ. Polyarginine segments in block copolypeptides drive both vesicular assembly and intracellular delivery. Nature Materials. 2007;6(1):52–57. doi: 10.1038/nmat1794. [DOI] [PubMed] [Google Scholar]
- Israelachvili JN. Intermolecular and surface forces. London; San Diego: Academic Press; 1991. [Google Scholar]
- Jain S, Bates FS. On the origins of morphological complexity in block copolymer surfactants. Science. 2003;300(5618):460–464. doi: 10.1126/science.1082193. [DOI] [PubMed] [Google Scholar]
- Jain S, Bates FS. Consequences of nonergodicity in aqueous binary PEO-PB micellar dispersions. Macromolecules. 2004;37(4):1511–1523. [Google Scholar]
- Jiang YG, Wang YP, Ma N, Wang ZQ, Smet M, Zhang X. Reversible self-organization of a UV-responsive PEG-terminated malachite green derivative: Vesicle formation and photoinduced disassembly. Langmuir. 2007;23(7):4029–4034. doi: 10.1021/la063305l. [DOI] [PubMed] [Google Scholar]
- Jones RA, Cheung CY, Black FE, Zia JK, Stayton PS, Hoffman AS, Wilson MR. Poly(2-alkylacrylic acid) polymers deliver molecules to the cytosol by pH-sensitive disruption of endosomal vesicles. Biochemical Journal. 2003;372:65–75. doi: 10.1042/BJ20021945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler EW, Murthy AK, Rodriguez BE, Zasadzinski JAN. Spontaneous Vesicle Formation in Aqueous Mixtures of Single-Tailed Surfactants. Science. 1989;245(4924):1371–1374. doi: 10.1126/science.2781283. [DOI] [PubMed] [Google Scholar]
- Katakam M, Bell LN, Banga AK. Effect of Surfactants on the Physical Stability of Recombinant Human Growth-Hormone. Journal of Pharmaceutical Sciences. 1995;84(6):713–716. doi: 10.1002/jps.2600840609. [DOI] [PubMed] [Google Scholar]
- Kim Y, Tewari M, Pajerowski JD, Cai SS, Sen S, Williams J, Sirsi S, Lutz G, Discher DE. Polymersome delivery of RNAi and Antisense Oligonucleotides. 2008 doi: 10.1016/j.jconrel.2008.10.020. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimura A, Koide A, Osada K, Yamasaki Y, Kataoka K. Encapsulation of myoglobin in PEGylated polyion complex vesicles made from a pair of oppositely charged block lonomers: A physiologically available oxygen carrier. Angewandte Chemie-International Edition. 2007;46(32):6085–6088. doi: 10.1002/anie.200701776. [DOI] [PubMed] [Google Scholar]
- Koide A, Kishimura A, Osada K, Jang WD, Yamasaki Y, Kataoka K. Semipermeable polymer vesicle (PICsome) self-assembled in aqueous medium from a pair of oppositely charged block copolymers: Physiologically stable micro-/nanocontainers of water-soluble macromolecules. Journal of the American Chemical Society. 2006;128(18):5988–5989. doi: 10.1021/ja057993r. [DOI] [PubMed] [Google Scholar]
- Korobko AV, Backendorf C, van der Maarel JRC. Plasmid DNA encapsulation within cationic diblock copolymer vesicles for gene delivery. Journal of Physical Chemistry B. 2006;110(30):14550–14556. doi: 10.1021/jp057363b. [DOI] [PubMed] [Google Scholar]
- Korobko AV, Jesse W, van der Maarel JRC. Encapsulation of DNA by cationic diblock copolymer vesicles. Langmuir. 2005;21(1):34–42. doi: 10.1021/la047967r. [DOI] [PubMed] [Google Scholar]
- Kros A, Jansen JA, Holder SJ, Nolte RJM, Sommerdijk N. Silane-based hybrids for biomedical applications. Journal of Adhesion Science and Technology. 2002;16(2):143–155. [Google Scholar]
- Kukula H, Schlaad H, Antonietti M, Forster S. The formation of polymer vesicles or “peptosomes” by polybutadiene-block-poly(L-glutamate)s in dilute aqueous solution. Journal of the American Chemical Society. 2002;124(8):1658–1663. doi: 10.1021/ja012091l. [DOI] [PubMed] [Google Scholar]
- Lasic DD, Papahadjopoulos D. Medical applications of liposomes. Amsterdam; New York: Elsevier; 1998. [Google Scholar]
- Lee JCM, Bermudez H, Discher BM, Sheehan MA, Won YY, Bates FS, Discher DE. Preparation, stability, and in vitro performance of vesicles made with diblock copolymers. Biotechnology and Bioengineering. 2001;73(2):135–145. doi: 10.1002/bit.1045. [DOI] [PubMed] [Google Scholar]
- Lee JCM, Santore M, Bates FS, Discher DE. From membranes to melts, rouse to reptation: Diffusion in polymersome versus lipid bilayers. Macromolecules. 2002;35(2):323–326. [Google Scholar]
- Li SL, Byrne B, Welsh J, Palmer AF. Self-assembled poly(butadiene)-b-poly(ethylene oxide) polymersomes as paclitaxel carriers. Biotechnology Progress. 2007;23(1):278–285. doi: 10.1021/bp060208+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lokitz BS, McCormick CL. Thermally responsive vesicles and their structural “locking” through polyelectrolyte complex formation. Angewandte Chemie-International Edition. 2006;45(35):5792–5795. doi: 10.1002/anie.200602168. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Ghoroghchian P, Zhang Y, Hammer DA. Adhesion of antibody-functionalized polymersomes. Langmuir. 2006;22(9):3975–3979. doi: 10.1021/la052445c. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Silas JA, Bermudez H, Milam VT, Bates FS, Hammer DA. The effect of polymer chain length and surface density on the adhesiveness of functionalized polymersomes. Langmuir. 2004;20(13):5493–5500. doi: 10.1021/la036417a. [DOI] [PubMed] [Google Scholar]
- Lin VSY, Dimagno SG, Therien MJ. Highly Conjugated, Acetylenyl Bridged Porphyrins - New Models for Light-Harvesting Antenna Systems. Science. 1994;264(5162):1105–1111. doi: 10.1126/science.8178169. [DOI] [PubMed] [Google Scholar]
- Liu DX, Liu F, Song YK. Recognition and Clearance of Liposomes Containing Phosphatidylserine Are Mediated by Serum Opsonin. Biochimica Et Biophysica Acta-Biomembranes. 1995;1235(1):140–146. doi: 10.1016/0005-2736(95)00005-n. [DOI] [PubMed] [Google Scholar]
- Liu FT, Eisenberg A. Preparation and pH triggered inversion of vesicles from poly(acrylic acid)-block-polystyrene-block-poly(4-vinyl pyridine) Journal of the American Chemical Society. 2003;125(49):15059–15064. doi: 10.1021/ja038142r. [DOI] [PubMed] [Google Scholar]
- Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. Journal of Controlled Release. 2000;65(12):271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- Meier W, Nardin C, Winterhalter M. Reconstitution of channel proteins in (polymerized) ABA triblock copolymer membranes. Angewandte Chemie-International Edition. 2000;39(24):4599–+. [PubMed] [Google Scholar]
- Mel’nikov SM, Dias R, Mel’nikova YS, Marques EF, Miguel MG, Lindman B. DNA conformational dynamics in the presence of catanionic mixtures. Febs Letters. 1999;453(12):113–118. doi: 10.1016/s0014-5793(99)00699-7. [DOI] [PubMed] [Google Scholar]
- Meng FH, Engbers GHM, Feijen J. Biodegradable polymersomes as a basis for artificial cells: encapsulation, release and targeting. Journal of Controlled Release. 2005;101(13):187–198. doi: 10.1016/j.jconrel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Nam J, Santore MM. Adhesion plaque formation dynamics between polymer vesicles in the limit of highly concentrated binding sites. Langmuir. 2007;23(13):7216–7224. doi: 10.1021/la700542t. [DOI] [PubMed] [Google Scholar]
- Nam J, Santore MM. The adhesion kinetics of sticky vesicles in tension: The distinction between spreading and receptor binding. Langmuir. 2007;23(21):10650–10660. doi: 10.1021/la7017709. [DOI] [PubMed] [Google Scholar]
- Napoli A, Boerakker MJ, Tirelli N, Nolte RJM, Sommerdijk N, Hubbell JA. Glucose-oxidase based self-destructing polymeric vesicles. Langmuir. 2004;20(9):3487–3491. doi: 10.1021/la0357054. [DOI] [PubMed] [Google Scholar]
- Napoli A, Valentini M, Tirelli N, Muller M, Hubbell JA. Oxidation-responsive polymeric vesicles. Nature Materials. 2004;3(3):183–189. doi: 10.1038/nmat1081. [DOI] [PubMed] [Google Scholar]
- Nardin C, Hirt T, Leukel J, Meier W. Polymerized ABA triblock copolymer vesicles. Langmuir. 2000;16(3):1035–1041. [Google Scholar]
- Nardin C, Thoeni S, Widmer J, Winterhalter M, Meier W. Nanoreactors based on (polymerized) ABA-triblock copolymer vesicles. Chemical Communications. 2000;(15):1433–1434. [Google Scholar]
- Nardin C, Widmer J, Winterhalter M, Meier W. Amphiphilic block copolymer nanocontainers as bioreactors. European Physical Journal E. 2001;4(4):403–410. [Google Scholar]
- Ortiz V, Nielsen SO, Discher DE, Klein ML, Lipowsky R, Shillcock J. Dissipative particle dynamics simulations of polymersomes. Journal of Physical Chemistry B. 2005;109(37):17708–17714. doi: 10.1021/jp0512762. [DOI] [PubMed] [Google Scholar]
- Parveen S, Sahoo SK. Nanomedicine - Clinical applications of polyethylene glycol conjugated proteins and drugs. Clinical Pharmacokinetics. 2006;45(10):965–988. doi: 10.2165/00003088-200645100-00002. [DOI] [PubMed] [Google Scholar]
- Perez C, Castellanos IJ, Costantino HR, Al-Azzam W, Griebenow K. Recent trends in stabilizing protein structure upon encapsulation and release from bioerodible polymers. Journal of Pharmacy and Pharmacology. 2002;54(3):301–313. doi: 10.1211/0022357021778448. [DOI] [PubMed] [Google Scholar]
- Photos PJ, Bacakova L, Discher B, Bates FS, Discher DE. Polymer vesicles in vivo: correlations with PEG molecular weight. Journal of Controlled Release. 2003;90(3):323–334. doi: 10.1016/s0168-3659(03)00201-3. [DOI] [PubMed] [Google Scholar]
- Qin SH, Geng Y, Discher DE, Yang S. Temperature-controlled assembly and release from polymer vesicles of poly(ethylene oxide)-block-poly(N-isopropylacrylamide) Advanced Materials. 2006;18(21):2905–+. [Google Scholar]
- Rijcken CJF, Soga O, Hennink WE, van Nostrum CF. Triggered destabilisation of polymeric micelles and vesicles by changing polymers polarity: An attractive tool for drug delivery. Journal of Controlled Release. 2007;120(3):131–148. doi: 10.1016/j.jconrel.2007.03.023. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Hernandez J, Lecommandoux S. Reversible inside-out micellization of pH-responsive and water-soluble vesicles based on polypeptide diblock copolymers. Journal of the American Chemical Society. 2005;127(7):2026–2027. doi: 10.1021/ja043920g. [DOI] [PubMed] [Google Scholar]
- Rong GZ, Deng MX, Deng C, Tang ZH, Piao LH, Chen XS, Jing XB. Synthesis of poly(epsilon-caprolactone)-b-poly(gamma-benzyl-L-glutamic acid) block copolymer using amino organic calcium catalyst. Biomacromolecules. 2003;4(6):1800–1804. doi: 10.1021/bm034208z. [DOI] [PubMed] [Google Scholar]
- Rosa M, Miguel MD, Lindman B. DNA encapsulation by biocompatible catanionic vesicles. Journal of Colloid and Interface Science. 2007;312(1):87–97. doi: 10.1016/j.jcis.2006.07.084. [DOI] [PubMed] [Google Scholar]
- Rosa M, Moran MD, Miguel MD, Lindman B. The association of DNA and stable catanionic amino acid-based vesicles. Colloids and Surfaces a-Physicochemical and Engineering Aspects. 2007;301(13):361–375. [Google Scholar]
- Srinivas G, Discher DE, Klein ML. Self-assembly and properties of diblock copolymers by coarse-grain molecular dynamics. Nature Materials. 2004;3(9):638–644. doi: 10.1038/nmat1185. [DOI] [PubMed] [Google Scholar]
- Srinivas G, Discher DE, Klein ML. Key roles for chain flexibility in block copolymer membranes that contain pores or make tubes. Nano Letters. 2005;5(12):2343–2349. doi: 10.1021/nl051515x. [DOI] [PubMed] [Google Scholar]
- Srinivas G, Shelley JC, Nielsen SO, Discher DE, Klein ML. Simulation of diblock copolymer self-assembly, using a coarse-grain model. Journal of Physical Chemistry B. 2004;108(24):8153–8160. [Google Scholar]
- Torchilin VP, Levchenko TS, Lukyanov AN, Khaw BA, Klibanov AL, Rammohan R, Samokhin GP, Whiteman KR. p-nitrophenylcarbonyl-PEG-PE-liposomes: fast and simple attachment of specific ligands, including monoclonal antibodies, to distal ends of PEG chains via p-nitrophenylcarbonyl groups. Biochimica Et Biophysica Acta-Biomembranes. 2001;1511(2):397–411. doi: 10.1016/s0005-2728(01)00165-7. [DOI] [PubMed] [Google Scholar]
- Wittemann A, Azzam T, Eisenberg A. Biocompatible polymer vesicles from biamphiphilic triblock copolymers and their interaction with bovine serum albumin. Langmuir. 2007;23(4):2224–2230. doi: 10.1021/la062805b. [DOI] [PubMed] [Google Scholar]
- Won YY, Brannan AK, Davis HT, Bates FS. Cryogenic transmission electron microscopy (cryo-TEM) of micelles and vesicles formed in water by polyethylene oxide)-based block copolymers. Journal of Physical Chemistry B. 2002;106(13):3354–3364. [Google Scholar]
- Won YY, Davis HT, Bates FS. Giant wormlike rubber micelles. Science. 1999;283(5404):960–963. doi: 10.1126/science.283.5404.960. [DOI] [PubMed] [Google Scholar]
- Wong D, Jeon TJ, Schmidt J. Single molecule measurements of channel proteins incorporated into biomimetic polymer membranes. Nanotechnology. 2006;17(15):3710–3717. [Google Scholar]
- Zhang LF, Yu K, Eisenberg A. Ion-induced morphological changes in “crew-cut ” aggregates of amphiphilic block copolymers. Science. 1996;272(5269):1777–1779. doi: 10.1126/science.272.5269.1777. [DOI] [PubMed] [Google Scholar]
- Zupancich JA, Bates FS, Hillmyer MA. Aqueous dispersions of poly(ethylene oxide)-b-poly(gamma-methyl-epsilon-caprolactone) block copolymers. Macromolecules. 2006;39(13):4286–4288. [Google Scholar]