

Abstract
α-Synuclein (SYN) is the major component of Lewy bodies, the neuropathological hallmarks of Parkinson's disease (PD). Missense mutations and multiplications of the SYN gene cause autosomal dominant inherited PD. Thus, SYN is implicated in the pathogenesis of PD. However, the mechanism whereby SYN promotes neurodegeneration remains unclear. Familial PD with SYN gene mutations are rare because the majority of PD is sporadic and emerging evidence indicates that sporadic PD may result from genetic and environmental risk factors including neuroinflammation. Hence, we examined the relationship between SYN dysfunction and neuroinflammation in mediating dopaminergic neurodegeneration in mice and dopaminergic neuronal cultures derived from wild-type SYN and mutant A53T SYN transgenic mice in a murine SYN-null (SYNKO) background (M7KO and M83KO, respectively). Stereotaxic injection of an inflammagen, lipopolysaccharide, into substantia nigra of these SYN genetically engineered mice induced similar inflammatory reactions. In M7KO and M83KO, but not in SYNKO mice, the neuroinflammation was associated with dopaminergic neuronal death and the accumulation of insoluble aggregated SYN as cytoplasmic inclusions in nigral neurons. Nitrated/oxidized SYN was detected in these inclusions and abatement of microglia-derived nitric oxide and superoxide provided significant neuroprotection in neuron–glia cultures from M7KO mice. These data suggest that nitric oxide and superoxide released by activated microglia may be mediators that link inflammation and abnormal SYN in mechanisms of PD neurodegeneration. This study advances understanding of the role of neuroinflammation and abnormal SYN in the pathogenesis of PD and opens new avenues for the discovery of more effective therapies for PD.
Keywords: α-synuclein, dopamine, neuroinflammation, microglia, Parkinson's disease, neurodegeneration
Introduction
Parkinson's disease (PD) is characterized by progressive degeneration of the nigrostriatal dopaminergic pathway and formation of neuronal inclusions known as Lewy bodies (LBs) and Lewy neurites. Although most PD is sporadic, multiple genes harbor mutations or other abnormalities that are pathogenic for familial forms of PD (Cookson et al., 2005; Farrer, 2006), and pathological α-synuclein (SYN) is linked mechanistically to both familial and sporadic PD. Specifically, three missense mutations and multiplications of the SYN gene (SNCA) cause familial PD (FPD) in rare kindreds, and abnormal SYN filaments are the major components of LBs and Lewy neurites in both FPD and sporadic PD (Polymeropoulos et al., 1997; Spillantini et al., 1997; Krüger et al., 1998; Zarranz et al., 2004). In addition, age-associated increases of SYN in monkeys and humans are associated with nigrostriatal dopamine (DA) depletion (Chu and Kordower, 2007). However, how pathological SYN causes neurodegeneration in sporadic and familial PD remains unclear. Moreover, low rates of concordance for PD in monozygotic and dizygotic twins (Tanner, 2003; Wirdefeldt et al., 2004), the existence of asymptomatic carriers of genetic abnormalities pathogenic for FPD, and considerable phenotypic variations in FPD suggest the involvement of environmental factors in mechanisms underlying PD.
Indeed, neuroinflammation induced by a variety of toxins, brain insults, and proinflammagens has been implicated as an environmental risk factor for PD (Liu et al., 2003). For example, markers of inflammation such as glial alterations and elevated accumulation of proinflammatory factors have been observed in association with degenerating DA neurons in the substantia nigra (SN) of PD patients (McGeer et al., 1988), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-intoxicated patients (Langston et al., 1999), and animal models of PD created by MPTP, 6-OHDA, or rotenone (Liberatore et al., 1999; Cicchetti et al., 2002; Sherer et al., 2003). Additionally, the inflammagen lipopolysaccharide (LPS) can initiate dopaminergic neurodegeneration alone or in combination with other environmental toxins such as rotenone (Gao et al., 2002, 2003c; Ling et al., 2006). Moreover, suppressing neuroinflammation mitigates dopaminergic neurodegeneration in some of these animal models (Liu et al., 2000; Gao et al., 2003b; Choi et al., 2005). For example, blocking microglial activation with minocycline prevents dopaminergic neurodegeneration induced by MPTP (Du et al., 2001; Wu et al., 2002). Minocycline also appears to alleviate the clinical signs of early PD in a randomized, double-blind, phase II futility clinical trial (NINDS NET-PD Investigators, 2006). Additionally, epidemiological studies suggest a reduced risk for PD among long-term users of nonsteroidal anti-inflammatory drugs (Chen et al., 2005; Ton et al., 2006).
However, these previous studies were not conducted in SYN genetically engineered mice and the contribution of endogenous, human wild-type (WT) or mutant SYN in this pathogenesis process is unclear. SYN-null (SYNKO) mice and transgenic (Tg) mice overexpressing human WT or A53T mutant SYN have been generated (Giasson et al., 2002). In this study, we examined how pathological SYN and neuroinflammation interact and contribute to the degeneration of DA neurons in Tg mice engineered to overexpress mutant or WT human SYN in a murine SYN-null background.
Materials and Methods
Generation of human SYN Tg mice.
M7 and M83 lines were created on a C57BL/C3H background and expressed mouse endogenous SYN and human WT SYN (M7) or A53T mutant SYN (M83) driven by mouse prion protein promoter (Giasson et al., 2002). The SYN-null mutation was maintained on an inbred 129/Sv background (Abeliovich et al., 2000). Homozygous M7 or M83 mice were bred with homozygous SYN-null mice, and heterozygous mice were then bred to generate mice homozygous at both gene loci. Tg offspring were identified by Southern blot with a 32P-labeled oligonucleotide-primed SYN DNA probe and PCR analyses with primers specific for human SYN, mouse SYN, or the neomycin resistance gene using mouse tail DNA (Abeliovich et al., 2000; Giasson et al., 2002). Homozygous Tg lines were identified by quantitative Southern blot analysis and verified by backcrossing to nontransgenic (nTg) mice. Homozygous genetically engineered mice were maintained and bred to generate embryos for primary culture studies. The nTg mice used in this study were F1 mice produced by crossing C57BL with 129/Sv. Housing and breeding of the animals were performed in strict accordance with the National Institutes of Health guidelines.
Stereotaxic injection of LPS into mouse SN.
Twelve-month-old male nTg, SYN Tg, and SYN-null mice were anesthetized and LPS (Escherichia coli 0111:B4; Sigma) was stereotaxically injected into the right side of SN (5 μg in 2 μl of sterile saline) over a period of 2 min under the control of a motorized microinjection pump. The following coordinates were used for the injection: 3.0 mm posterior to the bregma, 1.3 mm lateral to the midline, and 4.7 mm ventral to the surface of the skull. Sterile saline alone was injected into the left side of SN as a control (Qin et al., 2004). None of the genetically engineered SYN mice used in this study presented an overt neurological impairment at the time of LPS injection. At desired time points, mice were anesthetized and transcardially perfused with PBS, followed by 70% ethanol/150 mm NaCl or PBS-buffered 4% paraformaldehyde.
Immunostaining and cell counting.
Immunostaining was performed with the following primary antibodies: anti-F4/80 antigen, anti-glial fibrillary acidic protein antiserum, antibodies against a neuron-specific nuclear protein (NeuN) and tyrosine hydroxylase (TH) (Gao et al., 2003c), anti-synaptophysin antibody (1:5000; Millipore), LB509, SYN211 and SYN505 (1:500; specific to human SYN), affinity-purified SNL1 (1:1200; recognizing both human and mouse SYN), nSYN14 (specific to nitrated α- and β-synuclein) (Giasson et al., 2000), and anti-SYN phosphorylated at Ser129 (Fujiwara et al., 2002). One set of TH-immunostaining brain sections was counterstained for 30 s with Nissl stain (0.1% cresyl violet in 2.5% acetic acid). Double-label immunofluorescence was performed by staining brain sections with SYN211 and anti-TH antibodies or SNL1 combined with anti-synaptophysin antibody, followed by incubation with Alexa 488 (green)- and Alexa 594 (red)-conjugated secondary antibodies (1:1000). For nitrated SYN immunostaining, 70% ethanol/150 mm NaCl-fixed, paraffin-embedded brain sections were used. Briefly, after deparaffinization and hydration, the brain sections were incubated with 5% H2O2/methanol at room temperature for 20 min followed by blocking with 5% horse serum/1% BSA at 37°C for 30 min and sequential incubation with neat nSYN14 for 90 min at 37°C.
To monitor dopaminergic neurodegeneration, the number of TH-immunoreactive (TH-IR) neurons in the SN pars compacta from the first (rostral) and every 10th section of a series of 80 paraffin-embedded sections of each brain, which cover the entire extent of the SN, was counted by two individuals blind to the treatment. Brain sections from six animals in each genotype were used. For visual enumeration of the immunostained cells in cultures, 10 representative areas per well were counted. The overall dendrite length for individual TH-IR neurons in the cultures was measured as described previously (Gao et al., 2002). For each experiment, three to four wells per treatment condition were used and results from three independent experiments were obtained.
Gel electrophoresis and Western blotting analysis.
The protein extracts from midbrains, whole brains, or cultured cells were homogenized in lysis buffer (2% SDS and 50 mm Tris-HCl, pH 7.5), sonicated, and heated to 100°C for 10 min. Protein concentrations were determined using the biocinchoninic acid assay (Pierce). Protein samples were resolved on SDS polyacrylamide gels and immunoblot analyses were performed using antibodies against inducible nitric oxide synthase (iNOS), F4/80, SNL1, LB509, or nSYN14. Monoclonal anti-actin antibody was included as an internal standard to monitor loading errors.
Sequential biochemical fractionation.
Midbrain tissue samples were homogenized in 3 ml/g high-salt (HS) buffer [(in mm) 50 Tris-HCl, pH 7.5, 10 EGTA, 5 MgSO4, 750 NaCl, and 20 NaF] with protease inhibitor mixture and centrifuged at 100,000 × g for 30 min at 4°C. Pellets were re-extracted with HS buffer. The combined supernatant was used as the HS fraction. The pellets were sequentially extracted with HS buffer containing 1% Triton X-100 (HST fraction). The pellets were dissolved in radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 5 mm EDTA, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) and sedimented at 100,000 × g for 30 min at 4°C. The RIPA-soluble supernatant was used as RIPA fraction. The pellets were then re-extracted in 2% SDS buffer by sonication and heated to 100°C for 10 min. Protein samples were resolved on 12% SDS polyacrylamide gels and the distribution of SYN was determined by Western blotting using SNL1 and LB509 (Giasson et al., 2002).
Primary neuronal and glial cultures.
Neuron-enriched or neuron–glia cultures were prepared from the ventral midbrain of embryonic day 13 (E13) ± 0.5 mice (Gao et al., 2003a). Neuron–glia cultures were maintained in MEM supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 10% heat-inactivated horse serum, 1 g/L glucose, 2 mm l-glutamine, and 1 mm sodium pyruvate and 100 μm nonessential amino acids. Neuron-enriched cultures were first maintained in DMEM/F12 supplemented with 10% FBS, 6 g/L glucose, 1× B27. Then, 2 d later, the cultures were changed to serum-free neurobasal medium containing 1× B27, 0.5 mm l-glutamine, and 5 μm cytosine β-d-arabinofuranoside, which was used to suppress the proliferation of glia. Seven-day-old cultures were used for treatment. In either neuron-enriched cultures or neuron–glia cultures, the cell composition was not different among different genotypes.
Enriched glial cultures were prepared from whole brains of 1-d-old mice following the previous protocol (Gao et al., 2003d). After reaching confluence, microglia were isolated by shaking the flasks containing mixed glia for 5 h at 150 rpm. Enriched microglia were maintained in DMEM containing 10% FBS and 1 mm sodium pyruvate. To obtain primary astroglia, the mixed glial cultures, after the separation of microglia, were detached with trypsin-EDTA and seeded in the same culture medium used for microglia. After at least six consecutive passages, highly enriched astroglia were used for experiments.
Neuron–astroglia cocultures were prepared by coculturing fetal midbrain neurons with a confluent monolayer of astroglia (Gao et al., 2003d). Briefly, after six consecutive passages, nearly pure astroglia were seeded to 24-well plates. After reaching confluence, fetal midbrain cells from E13 ± 0.5 mice were added and maintained in neuron–glia culture medium. Reconstituted cell cultures were prepared as described previously (Gao et al., 2003a): midbrain neuron-enriched cultures were first prepared. Six days after the initial seeding, the cultures were changed to neuron–glia culture medium. Then highly enriched microglia (5 × 104) prepared from different mouse lines were added to neuron-enriched cultures. One day later, the reconstituted cell cultures were treated with LPS.
High-affinity [3H]dopamine uptake assay.
Uptake assays were determined by incubation of cultures for 15 min at 37°C with 1 μm [3H]DA (30 Ci/mmol; NEN) as described previously (Gao et al., 2003c). Nonspecific uptake was determined in the presence of 10 μm mazindol.
Measurement of superoxide, NO, and tumor necrosis factor-α.
The release of superoxide was determined by measuring the superoxide dismutase-inhibitable reduction of cytochrome c in microglia-enriched cultures treated for 60 min with normal saline (NS) or LPS. The production of NO and tumor necrosis factor-α (TNFα) was determined using Griess reagent and a mouse TNFα enzyme-linked immunosorbent assay kit, respectively (Gao et al., 2003a).
Immunoelectron microscopy.
Mice prepared for pre-embedding immunoelectron microscopy were perfused with 0.1 m PBS, pH 7.4, followed by 2% paraformaldehyde/0.5% glutaraldehyde in 0.1 m PBS. The tissues were further fixed for 12 h and cut into 50 μm sections. After reaction with fresh 0.1% sodium borohydrate in PBS for 10 min and extensive blocking, brain sections were labeled with antibody SYN211 and sequentially incubated with a biotinylated goat anti-mouse antibody and Vectastain ABC reagents. After the reaction with DAB, tissue sections were developed with silver methenamine. Sections were postfixed with 1.5% glutaraldehyde and 1% osmium tetraoxide, and, after dehydration in graded ethanol, they were embedded in Epon (Uryu et al., 2006).
Statistical analysis.
All values are expressed as the mean ± SEM. Differences among means were analyzed using one- or two-way ANOVA with treatment or genotype as the independent factors. When ANOVA showed significant differences, pairwise comparisons between means were tested by Newman–Keuls post hoc testing. In all analysis, the null hypothesis was rejected at the 0.05 level.
Results
Generation of human SYN Tg mice in a murine SYN-null background
To date, most of available human SYN Tg mice exhibit different phenotypes and fail to develop dopaminergic neurodegeneration. Although the reasons for this are unclear, the coexistence of human SYN and mouse SYN in these Tg mice is postulated to influence the phenotypes of human SYN Tg mice (Rochet et al., 2000; Cabin et al., 2005). Therefore, we produced a model whereby the effect of dysfunction of human SYN could be investigated without interference from endogenous mouse SYN. Two homozygous Tg lines, expressing human WT (M7) or A53T mutant SYN constructs (M83) and mouse endogenous SYN (Giasson et al., 2002), were bred with homozygous SYN-null mice (Abeliovich et al., 2000) and heterozygous mice were then intercrossed to generate mice homozygous at both gene loci. These novel Tg mice are referred to as M7KO and M83KO mice, respectively. Tg offspring were identified by Southern blot with a 32P-labeled oligonucleotide-primed SYN DNA probe and PCR analyses with primers specific for human SYN, mouse SYN, or the neomycin resistance gene using mouse tail DNA (Fig. 1A). The expression of SYN protein in the mouse brains was examined by Western blot analysis (Fig. 1B).
Figure 1.
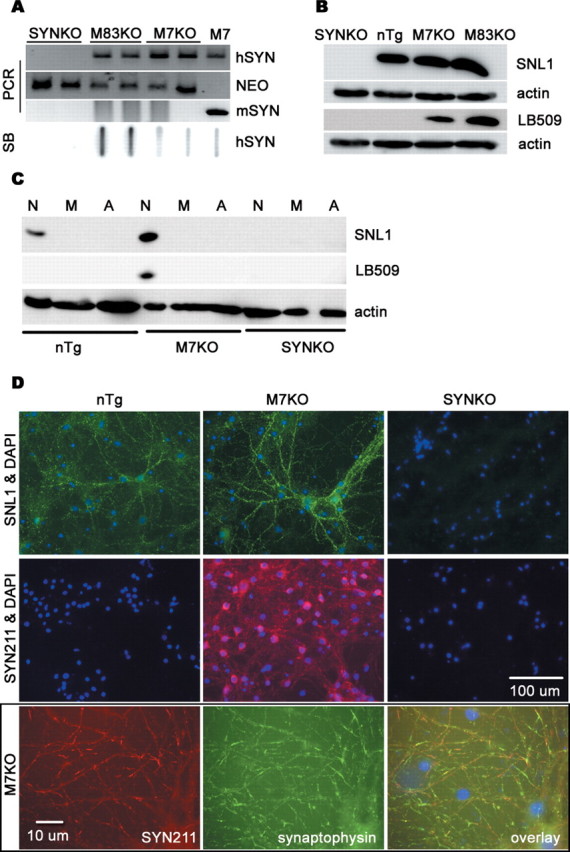
Expression of human SYN in Tg mice. A, Genotype analyses of tail DNA from homozygous mice by PCR amplification and slot blotting (SB). B, Western blot analysis of SYN expression in 3-month-old mice. The brain tissues were lysed in 2% SDS and 50 mm Tris-HCl, size fractionated by 12% SDS-PAGE gels, probed by Western blot for SNL1 (specific for mouse and human SYN), LB509 (specific for human SYN), or actin (as a loading control), and imaged by chemiluminescence. C, The cell-type specificity of SYN expression. The protein extracts from highly enriched neurons (N), microglia (M), or astroglia (A) were analyzed by Western blotting using anti-SYN antibodies. SYN was only expressed in neurons in both nTg and Tg mice. The level of SYN protein was undetectable in either microglia or astroglia even when lager amount of protein was loaded on the SDS-PAGE gel. D, Cellular localization of SYN. Immunofluorescence using antibodies against SYN and/or synaptophysin, a presynaptic protein, indicated that SYN was expressed predominantly at presynaptic terminals of cultured midbrain neurons in both nTg and SYN Tg mice. mSYN, Mouse SYN; hSYN, human SYN; NEO, neomycin.
Normally, SYN is expressed predominantly in neurons and particularly enriched at presynaptic terminals (Goedert, 1997; Norris et al., 2004). SYN is also developmentally expressed in cultured rat brain oligodendrocytes and is downregulated as the cultures mature (Richter-Landsberg et al., 2000). However, it is uncertain whether SYN proteins are expressed in microglia or astroglia under normal condition or after transgenic overexpression driven by the mouse prion protein promoter used to generate the M7 and M83 lines. To address this question, we generated highly enriched microglial or astroglial cultures from whole brains of 1-d-old SYN Tg mice and neuronal cultures from the cortex, hippocampus, and midbrain tissues of mouse embryos. Western blot analysis showed that SYN was only expressed in neurons in nTg and SYN Tg mice, whereas SYN proteins were undetectable in either microglia or astroglia (Fig. 1C). The same results were obtained when multiple different anti-SYN antibodies (SYN211, SYN204, and SYN208, specific for human SYN) were used (data not shown). Immunofluorescence staining using antibodies against SYN and synaptophysin, a presynaptic protein, indicated that SYN was expressed predominantly at presynaptic terminals of cultured neurons from both nTg and SYN Tg mice (Fig. 1D).
Differential vulnerability of DA neurons to inflammation-mediated neurotoxicity
Because SYN pathology and neuroinflammation are implicated in PD, we stereotaxically injected LPS (5 μg in 2 μl of sterile saline) unilaterally into the SN of the right side of the midbrain of SYN Tg and SYNKO mice to induce local inflammation. NS alone was injected into the left SN as a control. This strategy enables us to investigate whether the presence or absence of SYN affected the sensitivity of DA neurons to LPS-induced inflammation because LPS is one of the most common inflammagens used to investigate how inflammation contributes to the pathogenesis of a variety of diseases (Hauss-Wegrzyniak et al., 2000). Moreover, LPS does not directly affect neurons because its neurotoxicity is dependent on the activation of glia (Gao et al., 2002).
LPS-induced inflammation caused a significant loss of TH-IR neurons in the SN in nTg and SYN Tg mice 4 weeks after LPS injection (Fig. 2A). The loss of DA neurons after LPS injection was more prominent in M83KO mice than that in M7KO mice (Fig. 2B). The destruction of nigral neurons in SN was confirmed by Nissl counterstaining. In contrast, no significant loss of TH-IR and Nissl-positive neurons was detected in SYNKO mice after LPS injection (Fig. 2A,B). Quantification of the number of NeuN-IR neurons in the LPS-injected SN revealed an insignificant decrease in M7KO mice and a 31% decrease in M83KO mice (Fig. 2C). The decrease in the number of NeuN-IR neurons in M83KO mice could be attributable to the dramatic loss of DA neurons and a slight loss of non-DA neurons. The relative selectivity of nigral neurodegeneration was further characterized by double-label immunofluorescent analysis for TH and NeuN (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In the LPS-injected SN, the number of DA neurons labeled for both cytoplasmic TH (green) and nuclear NeuN (red) was markedly reduced compared with the corresponding region of the NS-injected side in nTg and SYN Tg mice, whereas non-DA neurons (red nuclear staining only) in the SN were not significantly affected. These results indicate that LPS-induced inflammation results in a preferential destruction of nigral DA neurons in the presence of neuronal SYN.
Figure 2.
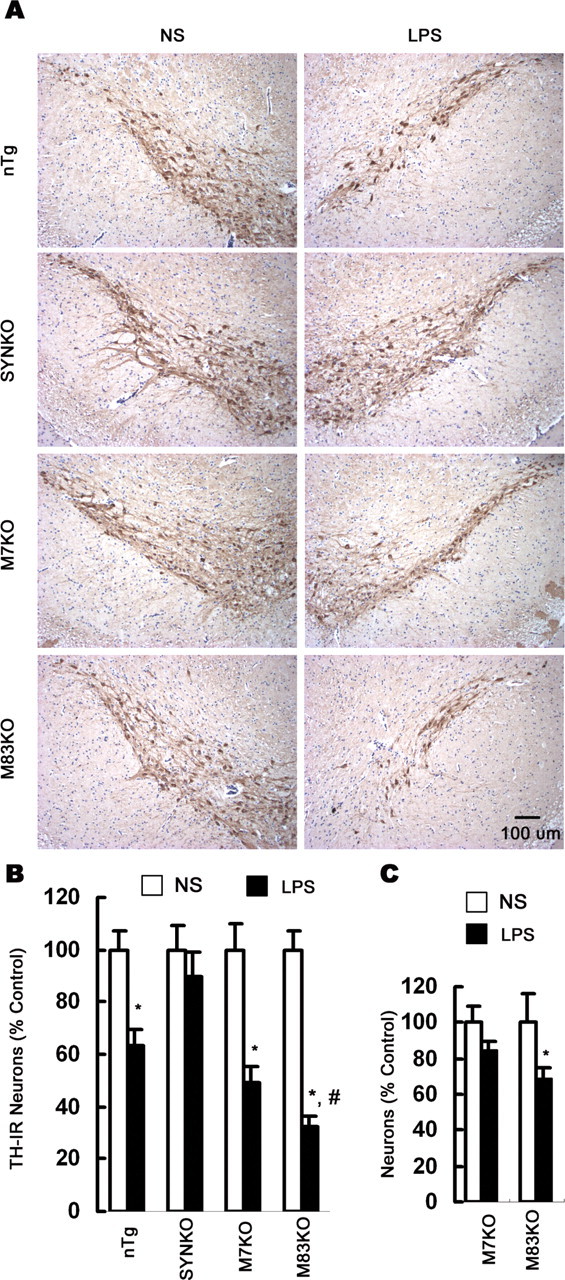
Differential vulnerability of nigral DA neurons to LPS-mediated neurotoxicity. A, Mouse brain sections were immunolabeled with antibody against TH and counterstained with Nissl. Representative images indicate the SN lesion in the LPS-injected side in nTg, M7KO, and M83KO mice 4 weeks after LPS injection, whereas in SYNKO mice, no significant damage of DA neurons was observed in the LPS-injected side compared with the NS-injected side. The loss of nigral DA neurons was most prominent in M83KO mice. B, Comparison of the number of nigral TH-IR neurons between the LPS-injected and the NS-injected side indicated that no significant loss of TH-IR neurons was detected in SYNKO mice, whereas significant loss of DA neurons was found in LPS-injected SN in nTg and SYN Tg mice 4 weeks after the injection. C, Quantification of NeuN-IR neurons in the LPS-injected SN revealed a significant decrease in M83KO mice but not in M7KO mice. Results are expressed as a percentage of the NS-injected side of the corresponding genotype. Error bars indicate mean ± SEM. *p < 0.01, compared with the corresponding NS-injected control side; #p < 0.01, compared with the LPS-injected side of M7KO mice.
To further examine the mechanisms of inflammation-mediated SN degeneration in the presence of SYN, we generated various primary cell cultures from genetically engineered SYN Tg and SYNKO mice. As shown in Figure 3, the in vitro studies confirmed that DA neurons are more sensitive to LPS-mediated neurotoxicity in the presence of human SYN, but this requires the presence of glia. In primary midbrain neuron–glia cultures prepared from M7KO mice, application of 5 or 10 ng/ml LPS resulted in a significant loss of DA neurons, shown by the significant reduction in the number of TH-IR neurons, [3H]DA uptake, and the overall length of dendrites of TH-IR neurons. In contrast, in neuron–glia cultures prepared from SYNKO mice, the same concentrations of LPS failed to induce significant neuronal loss (Fig. 3). Additionally, in the neuron-enriched cultures, 7 d after the administration of LPS, there was no neuron loss regardless of the origin of the cultures (M7KO or SYNKO mice), indicating that the LPS-mediated neurotoxicity is dependent on the presence of glia in the neuron–glia mixed cultures (Fig. 3A). Moreover, quantification of the number of NeuN-IR neurons in midbrain neuron–glia cultures from M7KO mice indicated that 7 d after the LPS treatment, there was no significant loss of neurons in general (Fig. 3D), supporting the relative selectivity of inflammation-mediated neurodegeneration in the presence of SYN.
Figure 3.
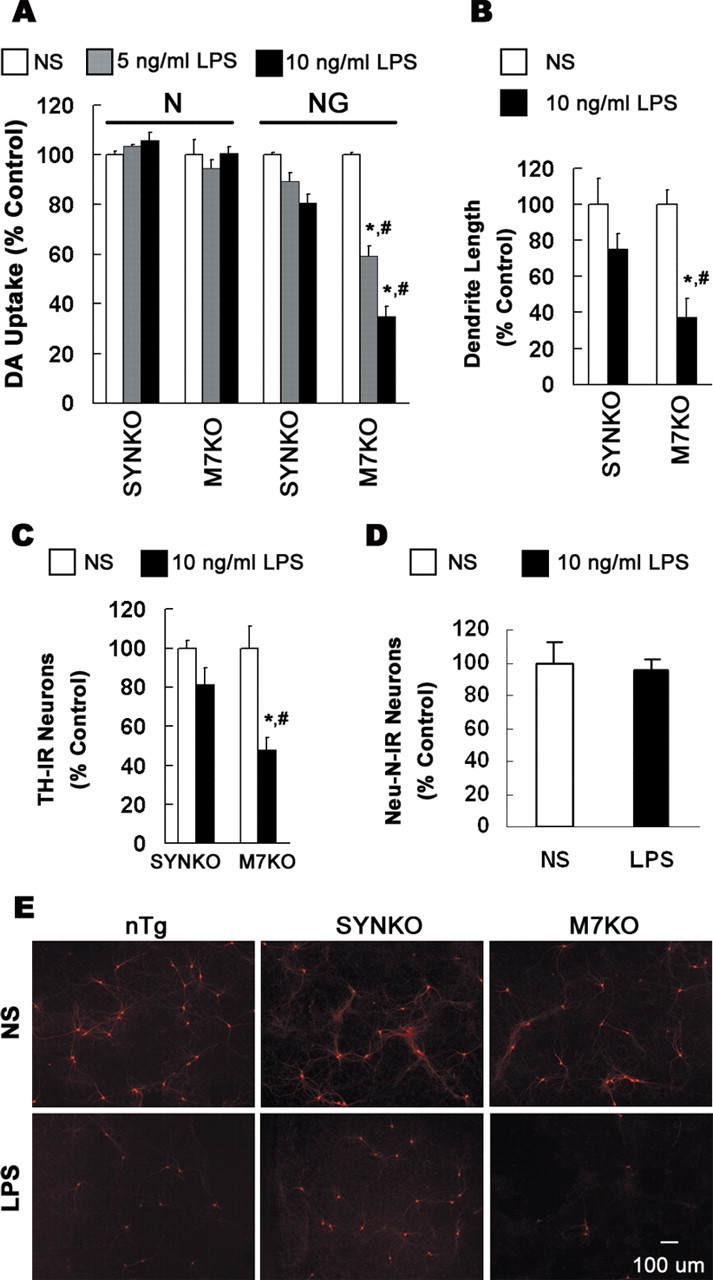
Overexpression of human SYN in neurons renders DA neurons more sensitive to inflammation. Midbrain neuron-enriched (N) or neuron–glia cultures (NG) from SYNKO or M7KO mice were treated for 7 d with NS or 5–10 ng/ml LPS. DA neurons were labeled with anti-TH antibody. A–C, Dopaminergic neurodegeneration was evaluated with [3H]DA uptake assay (A), measurement of dendrite length of individual TH-IR neurons (B), or quantification of TH-IR neurons in NG cultures (C). D, Quantification of the number of NeuN-IR neurons in midbrain NG cultures from M7KO mice 7 d after the treatment with 10 ng/ml LPS. The results are expressed as percentage of the control cultures and are the mean ± SEM of three to four experiments performed in triplicate. *p < 0.01, compared with the corresponding NS-treated NG cultures; #p < 0.01, compared with the corresponding LPS-treated NG cultures from SYNKO mice. E, Midbrain NG cultures were treated for 7 d with NS or LPS. Cultures then were immunostained for TH.
Neuronal SYN sensitizes DA neurons to neuroinflammation
To further examine the basis for these differences in dopaminergic neurodegeneration, we next compared the morphologic alterations in glial cells and the expression levels of F4/80 antigen (a marker of activated mouse microglia and macrophages) after LPS injection in all mouse lines. We found that the LPS-induced inflammatory reaction in different mouse lines was not different (Fig. 4A,B, supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Although these in vivo data and in vitro measurements of proinflammatory factors in cultures after LPS stimulation (Fig. 5A–E) implied that there were no differences in the LPS-induced inflammatory reactions of glia in mice with different genotypes, it is possible that other inflammatory factors not measured here may differ and thereby contribute to neurodegeneration. To rule out this possibility, we generated reconstituted cell cultures and added highly enriched microglia prepared from either M7KO or SYNKO mice to midbrain neuron-enriched cultures prepared from either of these mouse lines. One day after the supplement of microglia, the cultures were treated with 10 ng/ml LPS. Dopaminergic neurodegeneration was monitored by [3H]DA uptake 7 d after the treatment. The results showed that it is the presence of human SYN in neurons that dictated the differential sensitivity of DA neurons to inflammation regardless of the origin of the microglia used as supplement to the neuronal cultures (Fig. 4C). Specifically, the loss of DA neurons depended on the presence of SYN rather than the source of the microglia added to the neuronal cultures. Additionally, in neuron–astroglia cocultures, the activation of astroglia provided similar neuroprotective effect in cultures from SYNKO and M7KO mice (Fig. 4D). These data confirm that it is neuronal SYN, but not differences in inflammatory reactions that renders DA neurons more vulnerable to inflammation-mediated neurotoxicity.
Figure 4.
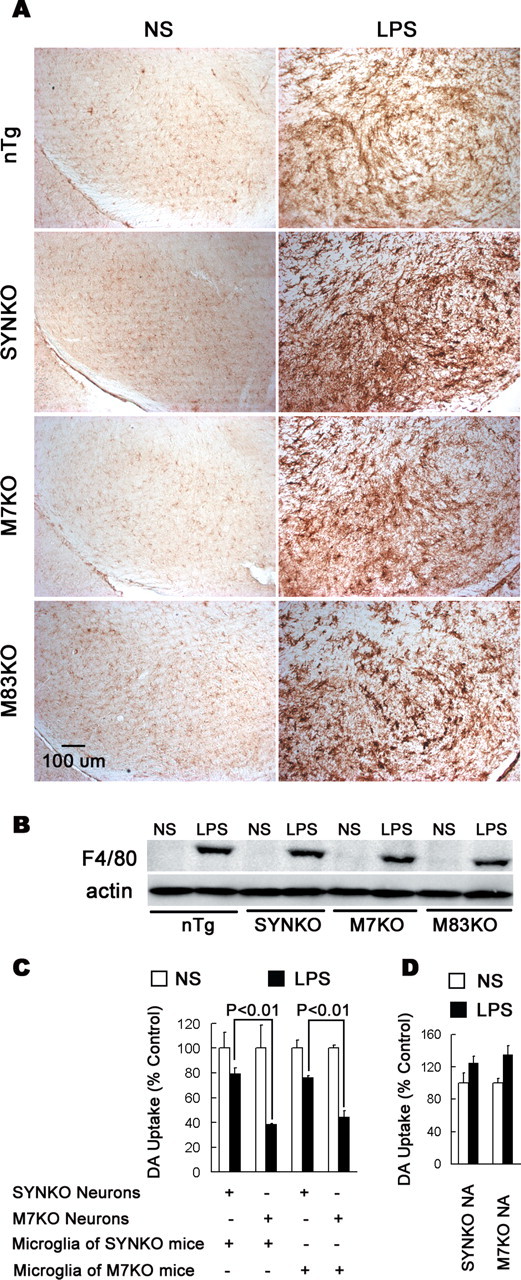
The differential degeneration of DA neurons is not attributable to different inflammatory reactions. A, LPS-induced activation of microglia in the mouse SN. One week after the injection, brain sections were immunostained with an antibody against F4/80 to determine the activation status of the microglia. In the NS-injected SN, microglia exhibited the typical ramified morphology of resting microglia. In the LPS-injected SN, microglia were highly activated with the characteristics of larger size, irregular shape, and dramatically increased expression of the F4/80 antigen. Note that the activation status of microglia appeared similar in all genotypes. B, Western blot analysis showed strikingly increased expression of F4/80 in the mouse midbrain 1 week after LPS injection. The levels of F4/80 expression were not different among different mouse genotypes. C, Midbrain neuron-enriched cultures from SYNKO or M7KO mice were supplemented with 5 × 104 microglia per well prepared from either SYNKO or M7KO mice. After 24 h, the cultures were treated with NS or 10 ng/ml LPS, and [3H]DA uptake was determined 7 d after the treatment. p < 0.01 was considered statistically significant. Note that in these reconstituted cultures, the presence of human SYN in neurons rendered DA neurons more sensitive to inflammation, regardless of the origin of supplemented microglia. D, The neuron–astroglia cocultures (NA) were treated with NS or LPS, and [3H]DA uptake was determined 7 d after the treatment. The activation of astroglia after LPS treatment provided similar neuroprotection in cultures from SYNKO and M7KO mice. Error bars indicate SEM.
Figure 5.
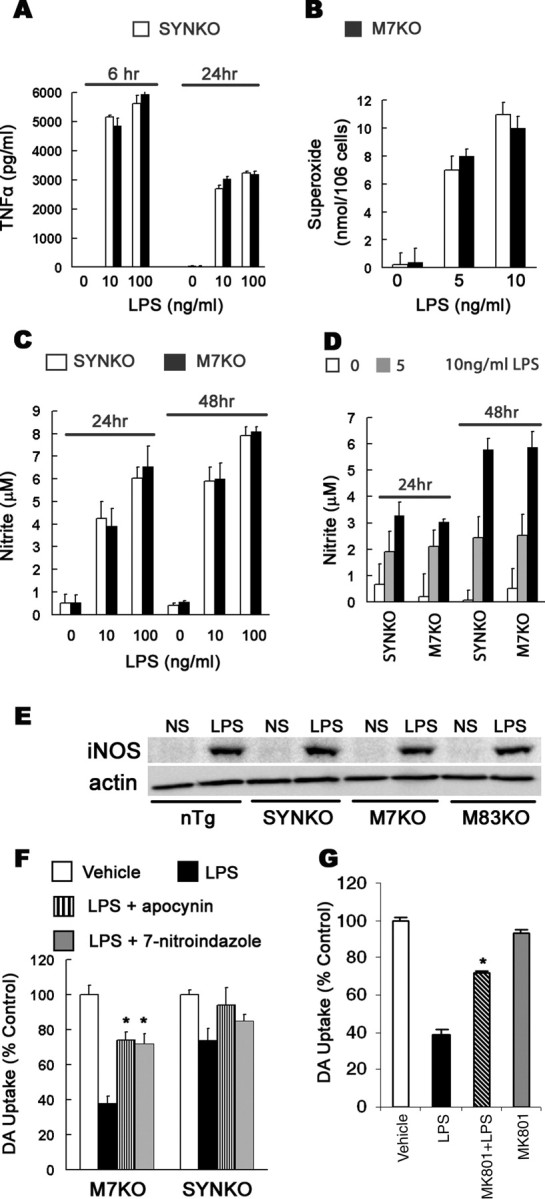
Microglia-derived superoxide and NO are major mediators of dopaminergic neurodegeneration. A–D, Neuron–glia cultures (A, D) and microglia-enriched cultures (B, C) were treated with NS or LPS. At the indicated time point, the production of TNFα (A), superoxide (B), and nitrite (an indicator of NO production) (C, D) was measured. The levels of these factors were dramatically increased with LPS stimulation, but they were not significantly different between two genotypes. E, In vivo iNOS expression was similarly increased 1 week after LPS injection in mouse midbrains. F, Abatement of NO and superoxide mitigated LPS-induced degeneration of DA neurons. Neuron–glia cultures were pretreated for 30 min with the iNOS inhibitor 7-nitroindazole or NADPH oxidase inhibitor apocynin followed with the treatment with NS or 10 ng/ml LPS for 7 d. Cultures were then assayed for DA uptake. Both 7-nitroindazole and apocynin provided significant neuroprotection in the cultures from M7KO mice. G, NMDA glutamate receptor antagonist MK801 attenuated LPS neurotoxicity in neuron–glia cultures prepared from M7KO mice. The cultures were pretreated for 30 min with 10 μm MK801 and the DA uptake assay was performed 7 d after the treatment with NS or 10 ng/ml LPS. The results are the mean ± SEM of two to three experiments performed in triplicate. *p < 0.01 compared with the corresponding LPS-treated cultures.
Microglia-derived NO and superoxide mediate SYN pathology and dopaminergic neurodegeneration
Because iNOS-derived NO and NADPH oxidase-derived superoxide were major players in inflammation-mediated neurodegeneration (Gao et al., 2002; Liu et al., 2002), we measured the production of NO, superoxide, and a representative proinflammatory cytokine TNFα. All of these factors were dramatically increased after LPS stimulation in neuron–glia cultures and microglia-enriched cultures from M7KO or SYNKO mice (Fig. 5A–D). We also found that 1 week after LPS injection into the SN, iNOS expression significantly increased in midbrain extracts, whereas there was no detectable iNOS expression in NS-injected midbrain by Western blot analysis (Fig. 5E). Notably, the production of each inflammatory factor in cultures and the level of iNOS expression in vivo were not different among the mouse genotypes, supporting the view that LPS induced similar inflammatory reactions in all mouse lines.
To more rigorously evaluate which factor plays a major and detrimental role in mediating the death of DA neurons and the dysfunction of SYN, we next analyzed the effects of each factor on the survival of DA neurons. Inhibition of NO production using iNOS inhibitor 7-nitroindazole and blockade of superoxide release using NADPH oxidase inhibitor apocynin from activated microglia attenuated degeneration of DA neurons in neuron–glia cultures from M7KO mice. In contrast, neither 7-nitroindazole nor apocynin showed significant neuroprotection in neuron–glia cultures generated from SYNKO mice (Fig. 5F). These results suggest that both superoxide and NO play important roles in SYN alterations and the degeneration of DA neurons. In addition to causing direct oxidative and nitrative damage to neurons, superoxide, NO, and/or peroxynitrite produced by activated microglia might also participate in excitotoxicity to neurons by triggering glutamate release and consequent glutamate receptor activation. Indeed, reduction of superoxide production by NADPH oxidase inhibitors, diphenylene iodonium chloride and apocynin, blocked microglial glutamate release triggered by LPS (Barger et al., 2007). Additionally, glutamate excitotoxicity mediated by the NMDA subtype glutamate receptor has been reported to contribute to neuronal damage during neuroinflammation in a variety of neurodegenerative diseases. Therefore, the ability of glutamate receptor antagonist to prevent LPS-activated, microglia-dependent toxicity to DA neurons was assessed. We found that NMDA receptor blocker (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK801; 10 μm) significantly, although not completely, prevented LPS toxicity in neuron–glia cultures from M7KO mice (Fig. 5G). Thus, glutamate excitotoxicity seems to be an important mechanism in oxidative stress-induced neuronal damage during neuroinflammation.
Accumulation of insoluble and aggregated SYN
To examine interactions of SYN pathology and inflammation as well as oxidative stress, we characterized abnormal SYN in the context of inflammation and oxidative stress. Double-label immunofluorescence using antibodies specific for TH and human SYN showed the formation of SYN-positive aggregates in the LPS-injected SN but not in NS-injected SN. Some SYN aggregates colocalized with TH, but others were localized in non-DA cells (Fig. 6A). In neuron–glia cultures, SYN pathology mainly appeared in perinuclear locations and formed aggregates 7 d after LPS treatment. In addition, SYN505, an antibody raised to oxidized human SYN that preferentially recognizes pathological SYN in human brain tissues from PD patients (Giasson et al., 2000), positively stained these SYN aggregates (Fig. 6B).
Figure 6.
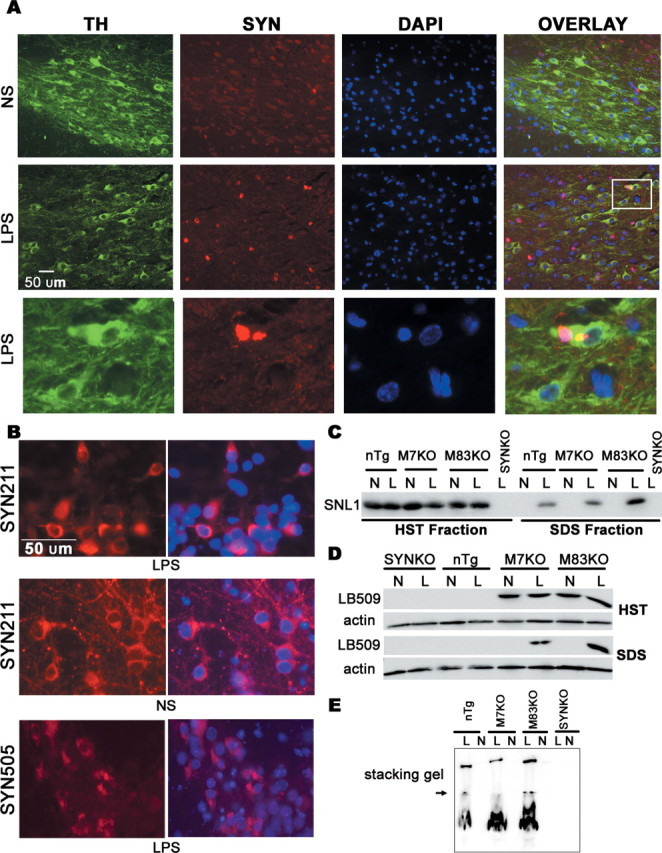
Accumulation of insoluble and aggregated SYN in mouse midbrain after LPS injection. A, Mouse brain sections were double labeled with anti-TH (green) and SYN211 (specific to human SYN, red) antibodies. SYN211 staining revealed that some of TH-expressing cells in the LPS-injected SN contained SYN aggregates. The inset and its magnified photomicrograph (bottom) display SYN aggregates in TH-expressing neurons in the LPS-injected SN. B, In neuron–glia cultures, SYN appeared mainly in perinuclear locations and formed aggregation 7 d after the LPS treatment. SYN505, antibody raised to oxidized human SYN, positively stained these aggregates. C, D, Midbrain tissues were sequentially extracted and size fractionated by 12% SDS-PAGE gels, followed by Western blot analysis using antibody SNL1 or LB509. Insoluble SYN was detected only in extracts of LPS-injected midbrains, but not in NS-injected midbrains. E, HST-insoluble fraction (dissolved in RIPA buffer) was size fractionated on a nondenaturing 12% polyacrylamide gel and probed by Western blotting for SNL1. The aggregated SYN was seen in the stacking gel in LPS-injected midbrain extracts, but not in NS-injected midbrain extracts. The arrow indicates the resolving and stacking gel interface. N, NS; L, LPS; DAPI, 4′,6′-diamidino-2-phenylindole dihydrochloride.
Next, we sequentially extracted midbrain tissue samples 4 weeks after LPS/NS injection using buffers of increasing solubilization strength. In extracts from NS-injected midbrains, SYN was detected only in HS and HST soluble fractions, but not in HST insoluble fractions. However, nTg and SYN Tg mice developed an accumulation of insoluble and aggregated SYN in LPS-injected midbrains, indicating that the neuroinflammation altered the solubility of SYN (Fig. 6C,D). To further confirm the formation of SYN aggregates, we then analyzed the HST insoluble proteins (dissolved in RIPA buffer) using nondenaturing polyacrylamide gels. The aggregated SYN appeared in the stacking gel in the LPS-injected midbrain extracts, whereas no similar staining was detected in the NS-injected midbrain extracts (Fig. 6E). The abnormal accumulation of insoluble and aggregated SYN positively correlated with the loss of DA neurons (Figs. 3, 6).
Immunoelectron microscopy for human SYN was used to further characterize SYN abnormalities and examine the subcellular location of this pathology in the LPS-injected SN of SYN Tg mice. The specific labeling of SYN was detected within neuronal cell bodies (Fig. 7). SYN-positive silver grains appeared to be associated with the lysosomes, Golgi apparatus, and multilamellar bodies. Occasionally, mitochondria also showed immunoreactivity for human SYN and they appeared morphologically abnormal. Additionally, ultrastructural assays show neuronal damages in the LPS-injected nigral neurons of M7KO mice, as shown by the myelin pathology and axonal degeneration. In contrast, in the NS-injected SN, SYN immunoreactivity was scant and not associated with mitochondria, and no neuronal damage was seen.
Figure 7.
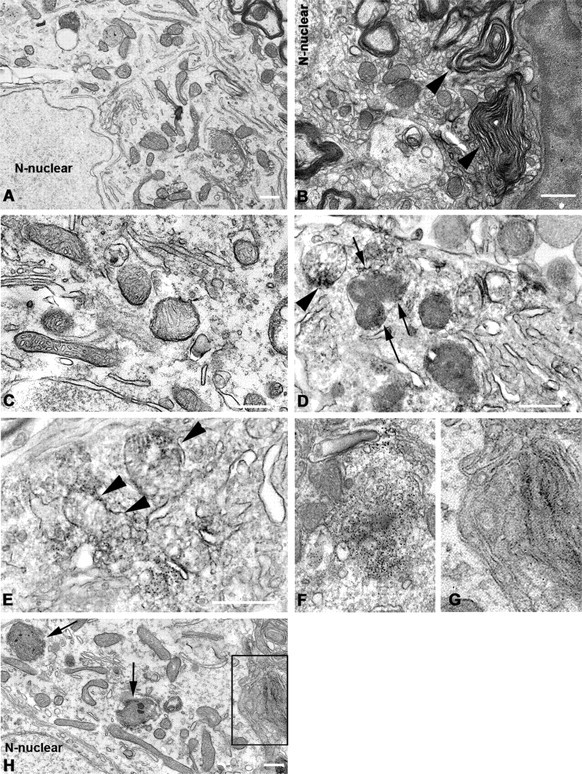
Neuronal degeneration associated with SYN pathology in the LPS-injected SN of M7KO mice. A, C, Unaffected neuron reveals normal morphology of mitochondria and other organelles. B, D, Myelin disruption and axonal degeneration in the LPS-injected nigral neurons. Myelin is fragmented and disorganized, and the axons within the myelin sheaths are severely atrophic (arrowheads). D, Interestingly, there is an abnormal membrane structure that appears in the nucleus of a damaged neuron (arrow). E–H, Anti-SYN antibody SYN211 staining associated with membrane structures showed damaged mitochondria (arrowheads) in affected neurons. SYN-positive silver grains also appeared to be associated with the lysosomes (arrow), Golgi apparatus (F), and multilamellar bodies (G). G, Magnified photomicrograph from boxed area (defined by solid black rectangles) in H shows the specifically labeled silver grains in the multilamellar bodies. Scale bars, 500 nm.
Pathogical modifications of SYN
To detect pathological modifications of SYN under inflammation and oxidative stress conditions, we performed double-label immunocytochemistry. The brain sections were first stained with the anti-NeuN antibody using DAB as a chromogen and nickel sulfate as an intensifying agent (dark blue) followed with nSYN14 antibody (specific for nitrated human SYN) or pSER129 SYN antibody (specific for Ser129-phosphorylated SYN) using DAB as a chromogen (brown). nSYN14-positive cytoplasmic inclusions were observed in neurons in LPS-injected SN, but not in NS-injected SN, in M7KO and M83KO mice. Western blot analyses using nSYN14 showed that nitrated SYN appeared in LPS-injected midbrain extracts from both M7KO and M83KO mice but not in NS-injected midbrain samples (Fig. 8A,B). These results indicated that glia-derived free radicals resulted in the nitration of SYN, which, in turn, promotes formation of intracytoplasmic SYN inclusions. Interestingly, the treatment of neuron–glia cultures prepared from M7KO mice with MK801 for 7 d led to substantial amelioration of the nitration of SYN induced by LPS challenge as detected by Western blot (Fig. 8C).
Figure 8.

Pathogenic modifications of SYN in cytoplasmic inclusions. A, D, Brain sections were first stained with an anti-NeuN antibody using DAB as a chromophore and nickel sulfate as an intensifying agent (dark blue) followed by (A) nSYN14 antibody (specific for nitrated human SYN) or (D) pSER129 SYN antibody (specific for Ser129-phosphorylated SYN) using DAB as a chromophore (brown). nSYN14-positive cytoplasmic inclusions were observed in neurons in LPS-injected SN, but not in NS-injected SN in M7KO and M83KO mice. B, Western blot analyses using nSYN14 shows that nitrated SYN appeared in LPS-injected (L) midbrain extracts from both M7KO and M83KO mice, but not in NS-injected (N) midbrain samples. C, Blockade of NMDA glutamate receptors using MK801 (M) significantly reduced the nitration of SYN. Neuron–glia cultures prepared from M7KO mice were pretreated for 30 min with MK801 followed with the treatment with NS or 10 ng/ml LPS for 7 d. Cultures were then extracted with RIPA buffer and Western blot analysis using nSYN14 was used to detect the nitration of SYN. D, Phosphorylated SYN at Ser129 appeared in the neuronal inclusions in the LPS-injected SN of M7KO mice. E, Midbrain tissues were sequentially extracted 4 weeks after LPS/NS injection, and analyzed by Western blotting using LB509 or an antibody specific for Ser129-phosphorylated (pSER129) SYN. Ser129-phosphorylated SYN appeared in HST-insoluble fractions in LPS-injected midbrains, but not in NS-injected midbrains. In sequential extracts of midbrain from a PD patient, Ser129-phosphorylated SYN only could be detected in insoluble fractions (SDS fractions). C, Control; M+L, MK801 plus LPS.
SYN deposited in neuronal inclusions in the LPS-injected SN of SYN Tg mice was phosphorylated at Ser129, as shown by positive staining for Ser129-phosphorylated SYN (Fig. 8D). Western blot analysis of sequential extracts of the human PD midbrain revealed that Ser129-phosphorylated SYN only could be detected in the insoluble SDS fraction (Fig. 8E). Similarly, 4 weeks after LPS injection, Ser129-phosphorylated SYN appeared in HST-insoluble fractions in LPS-injected midbrains but not in NS-injected midbrains. Different from PD patients, in the SYN Tg mice, SYN is relatively highly phosphorylated at Ser129 in HST-soluble fractions (Fig. 8E). Additionally, Ser129-phosphorylated SYN was also present in normal mouse brains (data not shown), which is consistent with a previous report (Hirai et al., 2004). Although evidence suggests that Ser129 phosphorylation may mediate SYN neurotoxicity and inclusion formation, this remains controversial.
Discussion
Although growing evidence suggests that PD may result from interactions among multiple factors, including exposure to environmental toxins, the occurrence of inflammation in the brain, and genetic predisposition, how these processes interact and contribute to the onset and progression of PD is poorly understood. Although toxins (MPTP, paraquat, and rotenone) have been shown to interact with SYN, it is not clear how this results in SYN pathology and neurodegeneration (Manning-Bog et al., 2003). Using genetically engineered SYN mice and primary cell cultures generated from these mice, we demonstrate here that pathological SYN and neuroinflammation synergistically contribute to dopaminergic neurodegeneration. Transgenic neuronal overexpression of human SYN rendered DA neurons more vulnerable to LPS-induced inflammation, and this inflammatory reaction led to the accumulation of insoluble SYN aggregates and the formation of cytoplasmic SYN inclusions in nigral neurons.
Recent advances in molecular genetics have revealed an important genetic influence underlying the development of PD. It is especially of interest that SNCA gene multiplications cause FPD, and disease severity is gene copy number- or dose-dependent (Singleton et al., 2003; Chartier-Harlin et al., 2004; Farrer et al., 2004; Ibáñez et al., 2004; Nishioka et al., 2006). In addition, age-associated increases of SYN in monkeys and humans were reported to be associated with nigrostriatal DA depletion (Chu and Kordower, 2007). These findings suggest that overproduction and/or accumulation of WT SYN may contribute to the development of familial as well as sporadic PD. To explore how SYN promotes neurodegeneration, various genetically engineered SYN models have been created, and SYN ablation leads to a subtle electrophysiological phenotype but no neurodegenerative pathology (Perez and Hastings, 2004). The relationship between SYN overexpression, accumulation/aggregation, and DA neuron dysfunction is not straightforward. Several Tg animals overexpressing either WT or mutant human SYN variably recapitulate some features of PD and other human neurodegenerative synucleinopathies (Farrer, 2006). SYN Tg flies (Feany and Bender, 2000) and SYN virus-infected mice, rats, and primates (Klein et al., 2002; Lo Bianco et al., 2002; Kirik et al., 2003; Lauwers et al., 2003; Yamada et al., 2004) developed loss of DA neurons and LB-like inclusions. However, of many different SYN Tg mouse models, a few showed alterations in the nigrostriatal DA system (Masliah et al., 2000; Richfield et al., 2002; Rockenstein et al., 2002; Tofaris et al., 2006), and only very few of them developed loss of DA neurons in SN, where human WT or doubly mutated SYN (A53T and A30P) was expressed using rat TH promoter (Thiruchelvam et al., 2004). In contrast, it was reported that even high levels of expression and accumulation of SYN (WT, A30P, or A53T) in DA neurons were achieved by using rat TH promoter, but the Tg mice did not display dopaminergic deficits (Matsuoka et al., 2001). In fact, it is the demise of DA neurons that correlates with motor impairments in PD and other forms of parkinsonism lacking LBs and Lewy neurites, the signature lesions of classic PD. The lack of nigral DA neuron loss in most SYN Tg mouse models of PD suggests that other factors participate in the development of PD and that better models are needed to better investigate the pathogenesis of PD, as well as for PD drug discovery efforts.
By using our two-hit model (neuroinflammation and SYN overexpression), we found that the expression of SYN in neurons was important for the inflammation-mediated dopaminergic neurodegeneration; genetic ablation of SYN made DA neurons more resistant to the inflammation. To rule out potential strain effects on LPS-induced inflammatory response and consequent SYN dysfunction as well as neurodegeneration, we used complementary approaches to demonstrate that there was no difference in inflammatory reaction in different mouse lines. More importantly, taking advantage of reconstituted cultures, where we added microglia from either SYNKO mice or SYN Tg mice to neuron-enriched cultures, we showed that it is the presence of human SYN in neurons that rendered DA neurons more sensitive to inflammation-mediated neurotoxicity. These findings suggest a prominent role of pathological SYN in PD neurodegeneration, which is also supported by the following evidence: (1) SYN knock-out mice are more resistant to MPTP neurotoxicity (Dauer et al., 2002; Drolet et al., 2004; Klivenyi et al., 2006); (2) overexpression of SYN enhances the sensitivity of cells to the neurotoxins MPTP (Richfield et al., 2002; Nieto et al., 2005), paraquat (Thiruchelvam et al., 2004), rotenone (Orth et al., 2003; Ved et al., 2005), or a combination of maneb and paraquat (Norris et al., 2007); (3) most importantly, WT SYN gene multiplications cause FPD and disease severity correlates with gene copy number (Singleton et al., 2003; Chartier-Harlin et al., 2004; Farrer et al., 2004); (4) age-associated increases of SYN in humans and monkeys are related to nigrostriatal DA depletion (Chu and Kordower, 2007).
Neurons are highly susceptible to oxidative stress, and oxidative stress appears to provide a critical link between environmental factors and genetic risk factors in the pathogenic mechanisms of PD neurodegeneration. We showed in this study that nitrated and possibly oxidized SYN occurs in neuronal cytoplasmic inclusions of the LPS-injected SN. The neuroprotective effects of abatement of microglia-derived NO and superoxide in the presence of SYN (Fig. 5) suggest that NO and superoxide may contribute to SYN pathology and dopaminergic neurodegeneration. Thus, neuroinflammation and consequent oxidative/nitrative modification of SYN may be critical for SYN dysfunction and dopaminergic neurodegeneration. It is well known that neurons are extraordinarily susceptible to excitotoxicity and there is cross talk between glutamate excitotoxicity and oxidative/nitrative damage during neurodegeneration. Indeed, it is known that activation of NOS or application of certain NO donors stimulates glutamate release (Bal-Price and Brown, 2001). That the neurotoxicity associated with NO and superoxide exposure during LPS-induced neuroinflammation was abrogated by the NMDA receptor antagonist MK801 (Fig. 5) implies that the secondary excitotoxicity involving NMDA receptor could be one of critical mechanisms underlying neuroinflammation-mediated neurodegeneration. However, the findings that SYN nitration was positively correlated to LPS-induced DA neuronal death and both events could be ameliorated effectively by MK801 (Figs. 5, 8) indicate that glutamate excitotoxicity might be involved in the production of NO, the nitration of SYN, and subsequent neuron demise. In fact, NO has been suggested to mediate neurotoxicity in response to glutamate–NMDA receptor overactivation (Boehning and Snyder, 2003). Together, neuroinflammation appears to drive a positive feedback toxic cycle in which activated microglia release a variety of inflammatory and neurotoxic mediators including superoxide, NO, and glutamate. These factors can work in concert to initiate or exacerbate neuronal injury, whereas neuronal injury will further stimulate inflammatory reaction. The self-propagating cycle of neuroinflammation will cause a progressive SYN pathology and neuronal death, where glutamate excitotoxicity could be an important mediator, bridging inflammation and neurodegeneration in Parkinson's disease.
In addition to oxidation and nitration, SYN is also phosphorylated at Ser129 within neuronal inclusions in SN after LPS injection. SYN in LBs is hyperphosphorylated at Ser129 in PD patients (Fujiwara et al., 2002; Arawaka et al., 2006). SYN phosphorylation has been reported to modulate neurotoxicity and inclusion formation in a Drosophila model of PD and in SH-SY5Y cells (Chen and Feany, 2005; Smith et al., 2005), suggesting a pathogenic role for the phosphorylation of SYN in PD. However, how phosphorylated SYN affects inclusion formation and neurodegeneration remains to be further determined.
Microglia are readily activated during most neuropathological conditions, including PD (Rock et al., 2004); microglial activation and consequent oxidative stress induce a selective loss of DA neurons in response to multiple stimuli (Block et al., 2004; Zhang et al., 2005), and the midbrain shows a high density of microglia (Lawson et al., 1990; Kim et al., 2000), which might explain why DA neurons are vulnerable to neuroinflammation and oxidative stress in PD. This study shows that microglial activation caused modification and aggregation of SYN through releasing free radicals. However, multiple stressors can cause aggregation of neuronal SYN, which, in turn, would activate microglia if the aggregated SYN interacts with microglia after its release or leak from dead and dying neurons. It has been reported that exogenous aggregated SYN causes microglial activation in the cell culture system (Zhang et al., 2005), monomeric as well as aggregated SYN can be released from cultured cells, and this release is elevated with proteasomal and mitochondrial dysfunction (Lee et al., 2005). Additionally, a recent study shows aggregated SYN induced microglial reactive oxygen species production and is attenuated by blockade of ion channels (Thomas et al., 2007).
This study demonstrates that SYN pathology and neuroinflammation interact to synergistically lead to PD neurodegeneration, although microglia-derived NO and superoxide also appear to contribute to these events and mediate these neurodegenerative mechanisms in PD. Thus, our data provide insight into how neuroinflammation and SYN pathology are linked mechanistically to the onset and progression of PD, and additional understanding of these processes may augment efforts to discover more effective disease-modifying therapeutic agents for PD.
Footnotes
This work was supported by National Institutes of Health–National Institute on Aging Grants AG-P01-09215 and AG-T32-00255. V.M.-Y.L. is the John H. Ware III professor in Alzheimer's disease research. J.Q.T. is the William Maul Measey-Truman G. Schnabel Jr Chair of Geriatric Medicine and Gerontology.
References
- Abeliovich et al., 2000.Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Arawaka et al., 2006.Arawaka S, Wada M, Goto S, Karube H, Sakamoto M, Ren CH, Koyama S, Nagasawa H, Kimura H, Kawanami T, Kurita K, Tajima K, Daimon M, Baba M, Kido T, Saino S, Goto K, Asao H, Kitanaka C, Takashita E, et al. The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson's disease. J Neurosci. 2006;26:9227–9238. doi: 10.1523/JNEUROSCI.0341-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal-Price and Brown, 2001.Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21:6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger et al., 2007.Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block et al., 2004.Block ML, Wu X, Pei Z, Li G, Wang T, Qin L, Wilson B, Yang J, Hong JS, Veronesi B. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 2004;18:1618–1620. doi: 10.1096/fj.04-1945fje. [DOI] [PubMed] [Google Scholar]
- Boehning and Snyder, 2003.Boehning D, Snyder SH. Novel neural modulators. Annu Rev Neurosci. 2003;26:105–131. doi: 10.1146/annurev.neuro.26.041002.131047. [DOI] [PubMed] [Google Scholar]
- Cabin et al., 2005.Cabin DE, Gispert-Sanchez S, Murphy D, Auburger G, Myers RR, Nussbaum RL. Exacerbated synucleinopathy in mice expressing A53T SNCA on a Snca null background. Neurobiol Aging. 2005;26:25–35. doi: 10.1016/j.neurobiolaging.2004.02.026. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin et al., 2004.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Chen et al., 2005.Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, Ascherio A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease. Ann Neurol. 2005;58:963–967. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- Chen and Feany, 2005.Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Choi et al., 2005.Choi DK, Pennathur S, Perier C, Tieu K, Teismann P, Wu DC, Jackson-Lewis V, Vila M, Vonsattel JP, Heinecke JW, Przedborski S. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson's disease in mice. J Neurosci. 2005;25:6594–6600. doi: 10.1523/JNEUROSCI.0970-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu and Kordower, 2007.Chu Y, Kordower JH. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson's disease? Neurobiol Dis. 2007;25:134–149. doi: 10.1016/j.nbd.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Cicchetti et al., 2002.Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci. 2002;15:991–998. doi: 10.1046/j.1460-9568.2002.01938.x. [DOI] [PubMed] [Google Scholar]
- Cookson et al., 2005.Cookson MR, Xiromerisiou G, Singleton A. How genetics research in Parkinson's disease is enhancing understanding of the common idiopathic forms of the disease. Curr Opin Neurol. 2005;18:706–711. doi: 10.1097/01.wco.0000186841.43505.e6. [DOI] [PubMed] [Google Scholar]
- Dauer et al., 2002.Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson-Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R. Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drolet et al., 2004.Drolet RE, Behrouz B, Lookingland KJ, Goudreau JL. Mice lacking alpha-synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology. 2004;25:761–769. doi: 10.1016/j.neuro.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Du et al., 2001.Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer et al., 2004.Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Farrer, 2006.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- Feany and Bender, 2000.Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Fujiwara et al., 2002.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Gao et al., 2002.Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem. 2002;81:1285–1297. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
- Gao et al., 2003a.Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2003a;23:6181–6187. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao et al., 2003b.Gao HM, Liu B, Zhang W, Hong JS. Novel anti-inflammatory therapy for Parkinson's disease. Trends Pharmacol Sci. 2003b;24:395–401. doi: 10.1016/S0165-6147(03)00176-7. [DOI] [PubMed] [Google Scholar]
- Gao et al., 2003c.Gao HM, Hong JS, Zhang W, Liu B. Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson's disease. J Neurosci. 2003c;23:1228–1236. doi: 10.1523/JNEUROSCI.23-04-01228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao et al., 2003d.Gao HM, Liu B, Zhang W, Hong JS. Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson's disease. FASEB J. 2003d;17:1954–1956. doi: 10.1096/fj.03-0109fje. [DOI] [PubMed] [Google Scholar]
- Giasson et al., 2000.Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Giasson et al., 2002.Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Goedert, 1997.Goedert M. Familial Parkinson's disease. The awakening of alpha-synuclein. Nature. 1997;388:232–233. doi: 10.1038/40767. [DOI] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak et al., 2000.Hauss-Wegrzyniak B, Vraniak PD, Wenk GL. LPS-induced neuroinflammatory effects do not recover with time. Neuroreport. 2000;11:1759–1763. doi: 10.1097/00001756-200006050-00032. [DOI] [PubMed] [Google Scholar]
- Hirai et al., 2004.Hirai Y, Fujita SC, Iwatsubo T, Hasegawa M. Phosphorylated alpha-synuclein in normal mouse brain. FEBS Lett. 2004;572:227–232. doi: 10.1016/j.febslet.2004.07.046. [DOI] [PubMed] [Google Scholar]
- Ibáñez et al., 2004.Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Kim et al., 2000.Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik et al., 2003.Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Björklund A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson's disease. Proc Natl Acad Sci U S A. 2003;100:2884–2889. doi: 10.1073/pnas.0536383100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein et al., 2002.Klein RL, King MA, Hamby ME, Meyer EM. Dopaminergic cell loss induced by human A30P alpha-synuclein gene transfer to the rat substantia nigra. Hum Gene Ther. 2002;13:605–612. doi: 10.1089/10430340252837206. [DOI] [PubMed] [Google Scholar]
- Klivenyi et al., 2006.Klivenyi P, Siwek D, Gardian G, Yang L, Starkov A, Cleren C, Ferrante RJ, Kowall NW, Abeliovich A, Beal MF. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol Dis. 2006;21:541–548. doi: 10.1016/j.nbd.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Krüger et al., 1998.Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Langston et al., 1999.Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Lauwers et al., 2003.Lauwers E, Debyser Z, Van Dorpe J, De Strooper B, Nuttin B, Baekelandt V. Neuropathology and neurodegeneration in rodent brain induced by lentiviral vector-mediated overexpression of alpha-synuclein. Brain Pathol. 2003;13:364–372. doi: 10.1111/j.1750-3639.2003.tb00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson et al., 1990.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Lee et al., 2005.Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6048. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberatore et al., 1999.Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Ling et al., 2006.Ling Z, Zhu Y, Tong C, Snyder JA, Lipton JW, Carvey PM. Progressive dopamine neuron loss following supra-nigral lipopolysaccharide (LPS) infusion into rats exposed to LPS prenatally. Exp Neurol. 2006;199:499–512. doi: 10.1016/j.expneurol.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Liu et al., 2000.Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000;293:607–617. [PubMed] [Google Scholar]
- Liu et al., 2002.Liu B, Gao HM, Wang JY, Jeohn GH, Cooper CL, Hong JS. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann N Y Acad Sci. 2002;962:318–331. doi: 10.1111/j.1749-6632.2002.tb04077.x. [DOI] [PubMed] [Google Scholar]
- Liu et al., 2003.Liu B, Gao HM, Hong JS. Parkinson's disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: role of neuroinflammation. Environ Health Perspect. 2003;111:1065–1073. doi: 10.1289/ehp.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco et al., 2002.Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. alpha-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc Natl Acad Sci U S A. 2002;99:10813–10818. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning-Bog et al., 2003.Manning-Bog AB, McCormack AL, Purisai MG, Bolin LM, Di Monte DA. Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J Neurosci. 2003;23:3095–3099. doi: 10.1523/JNEUROSCI.23-08-03095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah et al., 2000.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Matsuoka et al., 2001.Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- McGeer et al., 1988.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- Nieto et al., 2005.Nieto M, Gil-Bea FJ, Dalfo E, Cuadrado M, Cabodevilla F, Sanchez B, Catena S, Sesma T, Ribe E, Ferrer I, Ramirez MJ, Gomez-Isla T. Increased sensitivity to MPTP in human alpha-synuclein A30P transgenic mice. Neurobiol Aging. 2005;27:848–856. doi: 10.1016/j.neurobiolaging.2005.04.010. [DOI] [PubMed] [Google Scholar]
- NINDS NET-PD Investigators, 2006.NINDS NET-PD Investigators. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology. 2006;66:664–671. doi: 10.1212/01.wnl.0000201252.57661.e1. [DOI] [PubMed] [Google Scholar]
- Nishioka et al., 2006.Nishioka K, Hayashi S, Farrer MJ, Singleton AB, Yoshino H, Imai H, Kitami T, Sato K, Kuroda R, Tomiyama H, Mizoguchi K, Murata M, Toda T, Imoto I, Inazawa J, Mizuno Y, Hattori N. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson's disease. Ann Neurol. 2006;59:298–309. doi: 10.1002/ana.20753. [DOI] [PubMed] [Google Scholar]
- Norris et al., 2004.Norris EH, Giasson BI, Lee VM. Alpha-synuclein: normal function and role in neurodegenerative diseases. Curr Top Dev Biol. 2004;60:17–54. doi: 10.1016/S0070-2153(04)60002-0. [DOI] [PubMed] [Google Scholar]
- Norris et al., 2007.Norris EH, Uryu K, Leight S, Giasson BI, Trojanowski JQ, Lee VM. Pesticide exposure excerates alpha-synucleinopathy in an A53T transgenic mouse model. Am J Pathol. 2007;170:658–666. doi: 10.2353/ajpath.2007.060359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth et al., 2003.Orth M, Tabrizi SJ, Schapira AH, Cooper JM. Alpha-synuclein expression in HEK293 cells enhances the mitochondrial sensitivity to rotenone. Neurosci Lett. 2003;351:29–32. doi: 10.1016/s0304-3940(03)00941-8. [DOI] [PubMed] [Google Scholar]
- Perez and Hastings, 2004.Perez RG, Hastings TG. Could a loss of alpha-synuclein function put dopaminergic neurons at risk? J Neurochem. 2004;89:1318–1324. doi: 10.1111/j.1471-4159.2004.02423.x. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos et al., 1997.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Qin et al., 2004.Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- Richfield et al., 2002.Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG, Di Monte DA, Federoff HJ. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol. 2002;175:35–48. doi: 10.1006/exnr.2002.7882. [DOI] [PubMed] [Google Scholar]
- Richter-Landsberg et al., 2000.Richter-Landsberg C, Gorath M, Trojanowski JQ, Lee VM. alpha-synuclein is developmentally expressed in cultured rat brain oligodendrocytes. J Neurosci Res. 2000;62:9–14. doi: 10.1002/1097-4547(20001001)62:1<9::AID-JNR2>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Rochet et al., 2000.Rochet JC, Conway KA, Lansbury PT., Jr Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse alpha-synuclein. Biochemistry. 2000;39:10619–10626. doi: 10.1021/bi001315u. [DOI] [PubMed] [Google Scholar]
- Rock et al., 2004.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein et al., 2002.Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Sherer et al., 2003.Sherer TB, Betarbet R, Kim JH, Greenamyre JT. Selective microglial activation in the rat rotenone model of Parkinson's disease. Neurosci Lett. 2003;341:87–90. doi: 10.1016/s0304-3940(03)00172-1. [DOI] [PubMed] [Google Scholar]
- Singleton et al., 2003.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Smith et al., 2005.Smith WW, Margolis RL, Li X, Troncoso JC, Lee MK, Dawson VL, Dawson TM, Iwatsubo T, Ross CA. Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J Neurosci. 2005;25:5544–5552. doi: 10.1523/JNEUROSCI.0482-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini et al., 1997.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Tanner, 2003.Tanner CM. Is the cause of Parkinson's disease environmental or hereditary? Evidence from twin studies. Adv Neurol. 2003;91:133–142. [PubMed] [Google Scholar]
- Thiruchelvam et al., 2004.Thiruchelvam MJ, Powers JM, Cory-Slechta DA, Richfield EK. Risk factors for dopaminergic neuron loss in human alpha-synuclein transgenic mice. Eur J Neurosci. 2004;19:845–854. doi: 10.1111/j.0953-816x.2004.03139.x. [DOI] [PubMed] [Google Scholar]
- Thomas et al., 2007.Thomas MP, Chartrand K, Reynolds A, Vitvitsky V, Banerjee R, Gendelman HE. Ion channel blockade attenuated aggregated alpha synuclein induction of microglial reactive oxygen species: relevance for the pathogenesis of Parkinson's disease. J Neurochem. 2007;100:503–519. doi: 10.1111/j.1471-4159.2006.04315.x. [DOI] [PubMed] [Google Scholar]
- Tofaris et al., 2006.Tofaris GK, Garcia Reitböck P, Humby T, Lambourne SL, O'Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, Spillantini MG. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1–120): implications for Lewy body disorders. J Neurosci. 2006;26:3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton et al., 2006.Ton TG, Heckbert SR, Longstreth WT, Jr, Rossing MA, Kukull WA, Franklin GM, Swanson PD, Smith-Weller T, Checkoway H. Nonsteroidal anti-inflammatory drugs and risk of Parkinson's disease. Mov Disord. 2006;21:964–969. doi: 10.1002/mds.20856. [DOI] [PubMed] [Google Scholar]
- Uryu et al., 2006.Uryu K, Richter-Landsberg C, Welch W, Sun E, Goldbaum O, Norris EH, Pham CT, Yazawa I, Hilburger K, Micsenyi M, Giasson BI, Bonini NM, Lee VM, Trojanowski JQ. Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. Am J Pathol. 2006;168:947–961. doi: 10.2353/ajpath.2006.050770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ved et al., 2005.Ved R, Saha S, Westlund B, Perier C, Burnam L, Sluder A, Hoener M, Rodrigues CM, Alfonso A, Steer C, Liu L, Przedborski S, Wolozin B. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J Biol Chem. 2005;280:42655–42668. doi: 10.1074/jbc.M505910200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirdefeldt et al., 2004.Wirdefeldt K, Gatz M, Schalling M, Pedersen NL. No evidence for heritability of Parkinson disease in Swedish twins. Neurology. 2004;63:305–311. doi: 10.1212/01.wnl.0000129841.30587.9d. [DOI] [PubMed] [Google Scholar]
- Wu et al., 2002.Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada et al., 2004.Yamada M, Iwatsubo T, Mizuno Y, Mochizuki H. Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: resemblance to pathogenetic changes in Parkinson's disease. J Neurochem. 2004;91:451–461. doi: 10.1111/j.1471-4159.2004.02728.x. [DOI] [PubMed] [Google Scholar]
- Zarranz et al., 2004.Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2005.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]