

Abstract
Dystonia is a neurological disorder characterized by sustained or repetitive involuntary muscle contractions and abnormal postures. To understand the pathophysiology of dystonia, neurophysiological analyses were performed on hyperkinetic transgenic mice generated as a model of DYT1 dystonia. Abnormal muscle activity, such as coactivation of agonist and antagonist muscles and sustained muscle activation, was frequently observed in these mice. Recording of neuronal activity in the awake state revealed reduced spontaneous activity with bursts and pauses in both the external and internal segments of the globus pallidus. Motor cortical stimulation evoked responses composed of excitation and subsequent long-lasting inhibition in both pallidal segments, which were never observed in the normal mice. In addition, the somatotopic arrangements in both pallidal segments were disorganized. Long-lasting inhibition induced by cortical inputs in the internal pallidal segment may disinhibit thalamic and cortical activity, resulting in the motor hyperactivity observed in the transgenic mice.
Keywords: dystonia, transgenic mouse model, extracellular recording, globus pallidus, movement disorders, basal ganglia
Introduction
Dystonia is a neurological disorder characterized by sustained or repetitive involuntary muscle contractions and abnormal postures (Fahn, 1988; Fahn et al., 1998; Bressman, 2004). The pathophysiology of dystonia is poorly understood, and no consistent histopathological or biochemical changes associated with it have yet been detected. However, the internal (GPi) and external (GPe) segments of the globus pallidus have been discovered to exhibit decreased and bursting activity during stereotaxic surgery for deep brain stimulation (Lenz et al., 1998; Vitek et al., 1999; Vitek, 2002; Zhuang et al., 2004; Starr et al., 2005; Tang et al., 2007).
Early-onset torsion dystonia, the most common type of primary generalized dystonia, is inherited in an autosomal dominant manner with a penetrance of 30–40% (Kramer et al., 1988; Ozelius et al., 1997a). This dystonia is caused by a 3 bp (GAG) deletion in the DYT1 gene on chromosome 9q34, resulting in loss of a glutamic acid residue (ΔE) in the torsinA protein (Ozelius et al., 1997b). Recently, Shashidharan et al. (2005) generated a transgenic mouse model by overexpression of human ΔE-torsinA using a neuron-specific enolase promoter. These transgenic mice developed hyperkinesia and rapid bidirectional circling. They also exhibited abnormal involuntary movements with dystonic-appearing self-clasping of limbs and head-shaking. However, other mouse models have failed to exhibit such severe symptoms (Dang et al., 2005, 2006; Sharma et al., 2005; Grundmann et al., 2007; Zhao et al., 2008).
We therefore further examined the hyperkinesia and dystonic-like abnormal movements in this model by electrophysiological methods in the present study. We first tested whether the transgenic mice share electrophysiological features with human dystonia by recording of the electromyogram (EMG) and neuronal activity. We focused on activity in the GPi and GPe, two important nuclei in the basal ganglia circuitry, because activity change in these nuclei has been reported in human patients. Neuronal activity was recorded in the awake state to exclude effects of general anesthesia on neuronal firing rates and patterns (Bergstrom et al., 1984; Keane and Biziere, 1987; Löscher et al., 1995). We then investigated the responses of GPi and GPe neurons evoked by cortical stimulation to elucidate the neural mechanisms underlying the symptoms exhibited by the transgenic mice. Motor cortical stimulation typically induces triphasic responses composed of early excitation, inhibition, and late excitation in GPi and GPe neurons of normal monkeys and rodents (Nambu et al., 1990, 2000; Yoshida et al., 1993; Chiken and Tokuno, 2003). The origin of each component has been identified, with amplitudes and durations reflecting activity of the corresponding basal ganglia pathways and nuclei. Moreover, in voluntary movements, activity originating in the cortex is transmitted through the basal ganglia circuitry and finally reaches the output station of the basal ganglia (i.e., GPi). Cortical stimulation can mimic information processing through the basal ganglia circuitry (Nambu et al., 2002; Tachibana et al., 2008). We then discuss the pathophysiology of dystonia based on our electrophysiological findings.
Materials and Methods
Six transgenic mice (Shashidharan et al., 2005) from 5 to 28 weeks of age were used in the present study. Because ∼40% of the transgenic mice displayed dystonic-like behavior, only mice exhibiting such abnormalities on behavioral testing after genotype analyses were used in this study. We also used six age-matched wild-type mice as a control group. The experimental protocols were approved by the Animal Care and Use Committees of the Mount Sinai School of Medicine and the Okazaki Organization of National Institutes, and all experiments were conducted according to the guidelines of the National Institutes of Health Guide for Care and Use of Laboratory Animals. Before experiments, the mice were trained daily to acquaint them with handling by humans.
Surgery.
The head of the mouse was painlessly fixed in a stereotaxic apparatus while awake based on the method described by Yasoshima et al. (2005). Similar methods are usually used in recording neuronal activity from behaving monkeys. Each mouse was anesthetized with ketamine hydrochloride (100 mg/kg body weight, i.p.) and xylazine hydrochloride (4–5 mg/kg, i.p.) and fixed in a conventional stereotaxic apparatus. The skull was widely exposed, and 10 small stainless-steel screws (0.5 mm in diameter) were attached to the skull as anchors. The exposed skull and screws were completely covered with transparent acrylic resin, and then a small U-frame made of acetal resin for head fixation was mounted and fixed on the head of the mouse.
In three transgenic and three normal mice, bipolar wire electrodes (tip distance, 1–2 mm) made of 125-μm-diameter Teflon-coated multistranded stainless-steel wire (A-M Systems) were implanted in the triceps and biceps brachii muscles to record EMG activity. Wires were passed subcutaneously and connected to connectors attached to the U-frame. Antibiotics and analgesics were injected (intramuscularly) after surgery.
After recovery from the first surgery (2 or 3 d later), the mouse was positioned in a stereotaxic apparatus with its head restrained using the U-frame head holder under light anesthesia with ketamine hydrochloride (30–50 mg/kg, i.p.). A part of the skull in one hemisphere was removed to access the motor cortex, GPi, and GPe (classically termed the entopeduncular nucleus and globus pallidus in rodents, respectively). Large caudal and small rostral forelimb regions of the motor cortex have been reported in rats, corresponding to the primary and secondary motor cortices, respectively (Neafsey and Sievert, 1982; Neafsey et al., 1986; Liang et al., 1993; Rouiller et al., 1993). We inserted two pairs of bipolar stimulating electrodes (tip distance, 300–400 μm) made of 50-μm-diameter Teflon-coated tungsten wires into the motor cortex, one into the caudal forelimb region and the other into the orofacial region. These regions were confirmed by observation of body part movements evoked by intracortical microstimulation (ICMS) (train of 10 pulses at 333 Hz, 200 μs duration, up to 30 μΑ). Stimulating electrodes were then fixed therein using acrylic resin.
Recording of activity of pallidal neurons.
After full recovery from the second surgery, the mouse was positioned in a stereotaxic apparatus with its head restrained using the U-frame head holder in the awake state. For single-unit recording of GPi and GPe neurons, a glass-coated Elgiloy-alloy microelectrode (0.8–1.5 MΩ at 1 kHz) was inserted perpendicularly into the brain through the dura mater using a hydraulic microdrive (Narishige Scientific Instrument) with local application of lidocaine on the dura mater for anesthesia. The target area was 1.1–1.4 mm posterior and 1.4–2.2 mm lateral to bregma for the GPi, and 0.3–0.8 mm posterior and 1.6–2.4 mm lateral for the GPe (Paxinos and Franklin, 2001). Signals from the electrode were amplified and filtered (0.2–5 kHz). The GPi and GPe were identified by the depth profile of the microelectrode penetrations and their sustained spontaneous firing. Waveforms of the action potentials were continuously monitored with an oscilloscope. Unit activity was isolated and converted to digital pulses using a homemade time-amplitude window discriminator. Then the digital pulses were sampled at 2 kHz using a computer. Spontaneous discharges and responses to the cortical electrical stimulation (200 μs duration single pulse; 20–50 μA strength) through the electrode implanted in motor cortex were recorded. EMG activity was also amplified, filtered (0.25–1.5 kHz), and sampled at 20 kHz using a computer.
Data analysis.
Spontaneous discharge rates, interspike intervals (ISIs), and autocorrelograms (bin width of 0.5 ms) of the neurons were calculated from continuous digitized recordings for 30 s. Firing patterns of recorded neurons were classified into three types: regular, bursting, and irregular nonbursting.
Whether a firing pattern was regular was judged by autocorrelograms. The mean and SD of autocorrelation coefficients between 0.9 and 1.0 s (200 bins), which were sufficiently far after time 0, were calculated as control values because the autocorrelogram was flat during this period. Peaks and troughs of autocorrelation were judged significant if the coefficient during at least two consecutive bins (1 ms) exceeded the confidence limits (p < 0.005, one-tailed t test) (Abeles, 1982; Karmon and Bergman, 1993; Tachibana et al., 2008). The firing pattern of neurons was judged to be regular if at least two cycles, consisting of a pair of the significant peak and trough, were observed.
Bursts were detected by the “Poisson surprise” method (Legéndy and Salcman, 1985; Aldridge and Gilman, 1991; Wichmann and Soares, 2006). It was assumed that spikes in a certain interval followed a Poisson distribution. This method calculated the probability (P) that a given spike train would be found. First, three spikes trains shorter than the mean ISIs in the normal mice (i.e., GPi, 19.8 ms; GPe, 18.3 ms) were detected. The Poisson surprise value, which is defined as −log10 P, was maximized by adding additional intervals after the initial interval or deleting first intervals from the initial interval. The spike train was judged to be a burst if it included at least three spikes and if its surprise value was >3.0. We judged the firing pattern of a neuron to be bursting when at least two bursts were observed in 30 s of data. Distribution of numbers of bursts is shown in supplemental Figure 1, A and C (available at www.jneurosci.org as supplemental material), and the cutoff value of two bursts in 30 s was arbitrarily determined. The firing pattern of the neurons was judged to be irregular nonbursting if firing was neither regular nor bursting.
Pauses of spikes were detected using the method of Elias et al. (2007), whose principles are similar to the algorithm for surprise burst detection described above (Legéndy and Salcman, 1985). First, ISIs >250 ms were detected in the spike train. The Poisson surprise value was maximized by adding additional intervals (less than five) before or after the initial interval. The final interval was considered a pause if its interval was >300 ms and if its surprise value was >3.0. Finally, two adjacent pauses were merged into one continuous pause if they were separated by one spike. We judged a neuron to be a neuron with pauses if at least one pause was observed in 30 s of the data. Distribution of numbers of pauses is shown in supplemental Figure 1, B and D (available at www.jneurosci.org as supplemental material), and the cutoff value of one pause in 30 s was arbitrarily determined.
Responses to cortical electrical stimulation were examined by constructing peristimulus time histograms (PSTHs) (bin width of 1 ms) for 100 stimulus trials. The mean value and SD of the discharge rate during the 100 ms period preceding onset of stimulation were calculated for each PSTH and considered the baseline discharge rate. Changes in neuronal activity in response to cortical stimulation (i.e., excitation and inhibition) were judged significant if the discharge rate during at least two consecutive bins reached a significance level of p < 0.05 (one-tailed t test) (Nambu et al., 2000; Kita et al., 2004, 2005, 2006; Tachibana et al., 2008). The latency of each response was defined as the time at which the discharge rate first exceeded this level.
Histology.
In the final experiment, several sites of neuronal recording were marked by passing cathodal DC current (15 μA for 15 s) through the recording electrodes. The mice were anesthetized deeply with sodium pentobarbital (80 mg/kg, i.p.), and perfused transcardially with 0.1 m phosphate buffer (PB), pH 7.3, followed by 10% formalin in 0.1 m PB, and then 0.1 m PB containing 10% sucrose. The brains were removed immediately and saturated with the same buffer containing 30% sucrose. They were cut into frontal 50-μm-thick sections on a freezing microtome. The sections were mounted onto gelatin-coated glass slides, stained with 0.7% neutral red, dehydrated, and coverslipped. The sections were observed under a light microscope, and the recording sites were reconstructed according to the lesions made by current injection and traces of electrode tracks. The sites of stimulation in motor cortex were also examined histologically.
Results
EMG activity
The activities of the triceps and biceps brachii muscles, the extensor and flexor muscles of the forelimb, were simultaneously recorded in three normal and three transgenic mice (Fig. 1). Patterns of EMG activity during voluntary forelimb movements in the transgenic mice did not appear to differ from those in the normal mice during most of the time of recording (Fig. 1A,B). In all three transgenic mice, however, the triceps and biceps muscles were sometimes coactivated during forelimb movements (Fig. 1C). Sharp EMG activities in the triceps and biceps muscles in these mice were synchronized with each other (Fig. 1C); such synchronization was never observed in the normal mice. Moreover, sustained muscle activity that lasted >10 s was frequently observed when the transgenic mice stopped movement (Fig. 1D). The sustained muscle activity was sometimes accompanied by coactivation of the triceps and biceps muscles (Fig. 1D).
Figure 1.
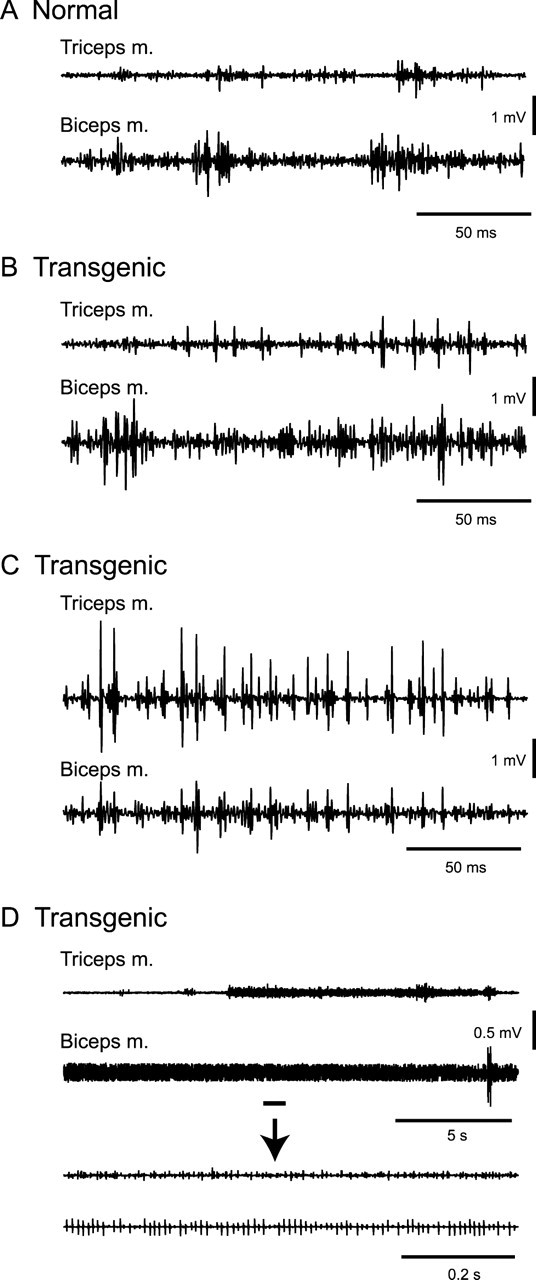
EMG activity of triceps and biceps brachii muscles. A, EMG activity in a normal mouse during voluntary forelimb movements. B–D, EMG activity in a transgenic mouse. EMG activity during voluntary forelimb movements did not appear to differ from that in the normal mice during most recording time (B). However, sharp EMG activities in the triceps and biceps muscles were sometimes observed, synchronized with each other (C). Sustained muscle activity that lasted >10 s was frequently observed (D). The underlined portions of the top two traces are expanded in the bottom two traces.
Locations of stimulating electrodes in motor cortex
The caudal forelimb and orofacial regions were successfully identified in all normal and transgenic mice by ICMS. The caudal forelimb region was found 0.3–1.3 mm anterior and 1.3–1.8 mm lateral to bregma in both normal and transgenic mice. Stimulation applied to more medial sites induced movements of the vibrissae. The orofacial region including jaw, lip, and tongue representations was located more rostrally and laterally than the forelimb region, at 1.7–2.6 mm anterior and 1.8–2.5 mm lateral to bregma in both normal and transgenic mice. In some cases, fingers or wrist movements were evoked by stimulation applied to more medial sites, corresponding to the rostral forelimb region. The threshold for evocation of movement by ICMS in the caudal forelimb and orofacial regions of the transgenic mice was 6–30 μA, and similar to that in the normal mice. These somatotopic arrangements were consistent with those previously reported in rats (Donoghue and Wise, 1982; Neafsey et al., 1986). These observations indicate that the somatotopic arrangement and excitability of motor cortex in the transgenic mice did not differ from those in the normal mice. Movements evoked by single-pulse stimulation (up to 70 μA) through the implanted stimulating electrodes were restricted to the forelimb (with stimulation in the forelimb region) or face (with stimulation in the orofacial region), suggesting that current spread to neighboring regions was negligible in daily experimental sessions.
Spontaneous activity of GPi and GPe neurons
The spontaneous activity of 94 GPi and 70 GPe neurons in six normal mice and 90 GPi and 204 GPe neurons in six transgenic mice was recorded. GPi and GPe neurons in the normal mice fired continuously at high discharge rate >50 Hz, as shown in traces of digitized spikes (Fig. 2A1,B1). Autocorrelograms indicated that these neurons fired irregularly (Fig. 2A2,B2). However, GPi and GPe neurons in the transgenic mice fired at low frequency with bursts and pauses (Fig. 2A3,B3). The mean discharge rates of GPi and GPe neurons in the transgenic mice were significantly lower than those in the normal mice (Table 1; Fig. 2A5,B5) (p < 0.001, t test). Discharge patterns also differed in the transgenic mice. Bursts (indicated by thick black lines in Fig. 2A3,B3) and pauses (indicated by thick white lines) were frequently observed in GPi and GPe neurons of the transgenic mice. The major firing pattern of GPi (63%) and GPe (77%) neurons in the normal mice was irregular nonbusting, whereas other firing patterns, such as regular (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and bursting neurons were observed (Fig. 2A6,B6). However, numbers of bursting neurons in the GPi (57%) and GPe (43%) were significantly increased in the transgenic mice (p < 0.001, χ2 test), and numbers of regular and irregular nonbursting neurons decreased. Spike shapes of bursting and irregular nonbursting neurons in the transgenic mice were compared. Both neurons showed positive–negative deflection with similar peak-to-peak amplitude (mean ± SD in microvolts, average of 10 neurons; bursting GPi, 86 ± 17; irregular nonbursting GPi, 79 ± 17; bursting GPe, 75 ± 14; irregular nonbursting GPe, 72 ± 21) and similar duration (in milliseconds, average of 10 neurons; bursting GPi, 1.6 ± 0.2; irregular nonbursting GPi, 1.6 ± 0.2; bursting GPe, 1.6 ± 0.2; irregular nonbursting GPe, 1.6 ± 0.2), suggesting that both neurons may belong to a single neuronal group. Bursts in the normal and transgenic mice are quantitatively compared in Table 2 and supplemental Figure 1, A and C (available at www.jneurosci.org as supplemental material). Number of bursts in the GPe and total lengths of bursts in the GPi and GPe were increased in the transgenic mice, although the duration of each burst in the GPe was decreased. More GPi (28%) and GPe (41%) neurons exhibited pauses in the transgenic mice than in the normal mice (Fig. 2A7,B7) (p < 0.001, χ2 test). Pauses in the normal and transgenic mice are compared in Table 3 and supplemental Figure 1, B and D (available at www.jneurosci.org as supplemental material). The duration of each pause and total lengths of pauses in the GPi and GPe were increased in the transgenic mice.
Figure 2.

Spontaneous activity of neurons in the internal (GPi) (A) and external (GPe) (B) segments of the globus pallidus. 1, 3, Spikes are shown as digital signals in the normal (1) and transgenic mice (3). Bursts and pauses in the digital signals are indicated by horizontal black thick lines and horizontal white thick lines, respectively. Neither bursts nor pauses are observed in traces of the normal mice exemplified in A1 and B1. 2, 4, Autocorrelograms with long and short (insets) timescales are shown in the normal (2) and transgenic mice (4). 5, Box plots of firing rates in the normal (left) and transgenic mice (right). The boxes are constructed with the top line bounding the first-quartile and the bottom line bounding the third quartile. Median and mean values are indicated by a thick horizontal line and an open circle in the box, respectively. The short horizontal lines show the largest and smallest values that are not outliers. Outliers are shown as small closed circles. *Significantly different (p < 0.001, t test). 6, Proportions of neurons classified by firing pattern (i.e., regular, bursting, or irregular nonbursting) in the normal (top) and transgenic (bottom) mice. 7, Proportions of neurons with or without pauses in the normal (top) and transgenic (bottom) mice.
Table 1.
Firing rates of GPi and GPe neurons in normal and transgenic mice
| GPi |
GPe |
|||
|---|---|---|---|---|
| Normal | Transgenic | Normal | Transgenic | |
| Total | 50.6 ± 15.7 (n = 94) | 27.8 ± 19.1* (n = 90) | 54.5 ± 16.3 (n = 70) | 35.4 ± 19.0* (n = 204) |
| Regular | 60.8 ± 14.8 (n = 16) | 70.4 ± 13.8 (n = 4) | 61.6 ± 23.8 (n = 7) | 62.0 ± 14.9 (n = 3) |
| Bursting | 47.4 ± 16.7 (n = 18) | 22.1 ± 12.4 (n = 50) | 44.0 ± 8.5 (n = 9) | 27.7 ± 14.0 (n = 88) |
| Irregular nonbursting | 48.9 ± 14.9 (n = 60) | 30.8 ± 21.7 (n = 36) | 55.4 ± 15.7 (n = 54) | 40.7 ± 20.1 (n = 113) |
| With pauses | 52.2 ± 11.5 (n = 5) | 17.9 ± 6.9 (n = 25) | 48.0 ± 15.6 (n = 12) | 29.2 ± 16.8 (n = 83) |
| Without pauses | 50.5 ± 16.0 (n = 89) | 31.6 ± 21.4 (n = 65) | 55.9 ± 16.3 (n = 58) | 39.7 ± 19.3 (n = 121) |
Values are means ± SD expressed in hertz.
*p < 0.001, significantly different from normal (one-tailed t test).
Table 2.
Burst characteristics in normal and transgenic mice
| GPi |
GPe |
|||
|---|---|---|---|---|
| Normal(n = 18) | Transgenic(n = 51) | Normal(n = 9) | Transgenic(n = 88) | |
| No. of bursts during 30 s | 6.1 ± 6.2 | 8.7 ± 8.3 | 4.1 ± 2.3 | 16.2 ± 18.5* |
| Duration of each burst (ms) | 33.8 ± 30.8 | 34.8 ± 27.6 | 101.8 ± 86.2 | 42.4 ± 49.1** |
| No. of spikes during each burst | 8.0 ± 5.0 | 6.3 ± 4.2 | 15.2 ± 10.4 | 7.5 ± 4.8** |
| Intraburst firing rate (Hz) | 274.3 ± 80.3 | 188.7 ± 61.1** | 181.6 ± 47.3 | 200.9 ± 63.5 |
| Total length of bursts during 30 s (% of spikes) | 3.2 ± 3.2 | 8.4 ± 7.3** | 5.1 ± 4.7 | 14.1 ± 16.0* |
| Total length of bursts during 30 s (% of times) | 0.7 ± 0.7 | 0.9 ± 0.7 | 1.5 ± 1.5 | 1.7 ± 1.8 |
| Poisson surprise value for each burst | 3.7 ± 0.7 | 4.0 ± 0.5* | 4.4 ± 1.6 | 4.1 ± 0.8 |
Values are means ± SD.
*p < 0.05 and
**p < 0.01, significantly different from normal (one-tailed t test).
Table 3.
Pause characteristics in normal and transgenic mice
| GPi |
GPe |
|||
|---|---|---|---|---|
| Normal(n = 5) | Transgenic(n = 25) | Normal(n = 11) | Transgenic(n = 83) | |
| No. of pauses during 30 s | 1.2 ± 0.5 | 2.7 ± 2.0 | 2.0 ± 1.3 | 3.5 ± 3.2 |
| Duration of each pause (ms) | 410.3 ± 102.0 | 1074.8 ± 614.8** | 418.5 ± 71.4 | 871.7 ± 689.7* |
| Total length of pauses during 30 s (% of time) | 1.6 ± 0.6 | 10.0 ± 8.6* | 2.8 ± 1.9 | 10.3 ± 12.7* |
| Poisson surprise value for each pause | 6.2 ± 3.2 | 4.4 ± 1.9 | 5.7 ± 2.9 | 6.1 ± 4.5 |
Values are means ± SD.
*p < 0.05 and
**p < 0.01, significantly different from normal (one-tailed t test).
Relationships between firing patterns and firing rates were further analyzed in Table 1. Regular GPi and GPe neurons showed similar firing rates both in the normal and transgenic mice. In contrast, firing rates of bursting and irregular nonbursting neurons decreased in the transgenic mice. These results suggest that reduction of mean firing rates of GPi and GPe neurons in the transgenic mice are caused by both (1) decreased number of regular neurons and (2) reduction of firing rates of bursting and irregular nonbursting neurons. Firing rates were also compared between GPi and GPe neurons with pauses and those without pauses (Table 1). Reduction of firing rates occurred in neurons without pauses as well as in neurons with pauses.
Responses of GPi and GPe neurons to cortical stimulation
Stimulation of the forelimb and/or orofacial region of the motor cortex was applied to 91 GPi and 68 GPe neurons in six normal mice, and induced responses in 59 GPi and 40 GPe neurons (∼60%). The typical response pattern of GPi (36%) and GPe (56%) neurons in the normal mice was a triphasic response composed of early excitation, followed by inhibition, and late excitation (Figs. 3A, 4A). Other patterns such as excitation followed by inhibition, inhibition followed by excitation, monophasic or biphasic excitation, and monophasic inhibition were also observed (Figs. 3C, 4C). In most cases, these events ended within 50 ms after cortical stimulation and were not followed by late responses (Figs. 3A, 4A; Table 4).
Figure 3.

Responses of GPi neurons to cortical stimulation. A, Raster and PSTH for the normal mice. Cortical stimuli were delivered at time 0 (arrows). B, Raster and PSTH for the transgenic mice. Abnormal responses with long-lasting inhibition (left) and apparently normal triphasic responses (right) were observed. C, Proportions of neurons classified based on early response patterns in the normal (top) and transgenic (bottom) mice. ex, Excitation; inh, inhibition. D, Proportions of neurons with and without long-lasting inhibition in the normal (top) and transgenic (bottom) mice.
Figure 4.

Responses of GPe neurons to cortical stimulation. A, Raster and PSTH for the normal mice. B, Raster and PSTH for the transgenic mice. C, Proportions of neurons classified based on early response patterns in the normal (top) and transgenic (bottom) mice. D, Proportions of neurons with and without long-lasting inhibition in the normal (top) and transgenic (bottom) mice.
Table 4.
Latency and duration of triphasic responses evoked by cortical stimulation
| GPi |
GPe |
||||
|---|---|---|---|---|---|
| Normal(n = 21) | Transgenic(n = 7) | Normal(n = 26) | Transgenic(n = 33) | ||
| Latency (ms) | Early excitation | 3.3 ± 0.9 | 3.5 ± 0.5 | 4.3 ± 0.8 | 4.4 ± 1.1 |
| Inhibition | 11.6 ± 2.3 | 11.1 ± 1.7 | 11.1 ± 1.5 | 12.1 ± 2.5 | |
| Late excitation | 20.9 ± 2.1 | 18.6 ± 1.8** | 21.1 ± 2.2 | 21.2 ± 5.4 | |
| Duration (ms) | Early excitation | 5.2 ± 2.9 | 5.7 ± 1.3 | 4.9 ± 1.8 | 5.3 ± 1.7 |
| Inhibition | 6.1 ± 2.9 | 6.1 ± 2.0 | 8.7 ± 3.0 | 6.8 ± 3.2* | |
| Late excitation | 9.9 ± 5.0 | 34.6 ± 45.0** | 16.8 ± 18.7 | 26.3 ± 28.5 | |
Values are means ± SD expressed in milliseconds.
*p < 0.05 and
**p < 0.01, significantly different from normal (one-tailed t test).
Cortical stimulation induced responses in similar percentages of GPi and GPe neurons (GPi, 52 of 84; GPe, 111 of 178) in six transgenic mice, although response patterns differed. The most common response pattern of GPi and GPe neurons in the transgenic mice was short-latency monophasic or biphasic excitation followed by long-lasting inhibition (Figs. 3B, left; 4B, left), a pattern never observed in the normal mice. The latency and duration of the early excitation were 4.4 ± 2.6 and 15.3 ± 5.5 ms in the GPi and 4.0 ± 1.6 and 12.4 ± 7.1 ms in the GPe, respectively, whereas the duration of the long-lasting inhibition was 73.7 ± 29.4 ms in the GPi and 66.7 ± 31.3 ms in the GPe. The most common pattern of early response (within 50 ms after cortical stimulation) was monophasic or biphasic excitation in GPi (62%) and GPe (49%) neurons (Figs. 3C, 4C), and early responses were frequently followed by long-lasting inhibition (GPi, 56%; GPe, 41%) (Figs. 3D, 4D) (p < 0.001, χ2 test). In other GPi (14%) and GPe (30%) neurons in the transgenic mice, cortical stimulation induced a triphasic response similar to that in the normal mice (Figs. 3B, right; 4B, right), although the number of such neurons was decreased (Figs. 3C, 4C) (p < 0.01, χ2 test). The latency and duration of each component were also similar, with the exception of the latency and duration of the late excitation in the GPi and duration of the inhibition in the GPe, which differed significantly (Table 4) (t test).
Relationships between cortically evoked responses and spontaneous firing patterns were further analyzed (supplemental Table 1, available at www.jneurosci.org as supplemental material). The majority of neurons that showed excitation (GPi, 81%; GPe, 70%) to the cortical stimulation in the transgenic mice belonged to bursting neurons, whereas those in the normal mice belonged to irregular nonbursting neurons (GPi, 75%; GPe, 100%). In contrast, neurons with a cortically evoked triphasic response had a tendency to fire in irregular nonbursting manner both in the normal (GPi, 56%; GPe, 92%) and transgenic (GPi, 50%; GPe, 82%) mice. A large number of neurons that showed cortically evoked long-lasting inhibition in the transgenic mice belonged to bursting neurons (GPi, 73%; GPe, 91%) and neurons with pauses (GPi, 42%; GPe, 73%), whereas those neurons were rarely observed in the normal mice.
Somatotopic organization in GPi and GPe
Stimulation of both the forelimb and orofacial regions of the motor cortex was successfully performed for 91 GPi and 68 GPe neurons in six normal mice and 43 GPi and 77 GPe neurons in six transgenic mice. Neurons could be classified into four groups, those with forelimb inputs, those with orofacial inputs, those with convergent inputs from both forelimb and orofacial regions, and those with no responses, based on the responses evoked by cortical stimulation. In the normal mice, small number of GPi (7%) and GPe (10%) neurons responded to stimulation of both forelimb and orofacial regions (Figs. 5A, 6A). In the transgenic mice, the number of neurons with convergent inputs from two regions was significantly increased (GPi, 28%; GPe, 35%; p < 0.001, χ2 test).
Figure 5.

Somatotopic organization in the GPi. A, Proportions of neurons classified based on cortical inputs in the normal (top) and transgenic (bottom) mice. B, Distribution of recorded GPi neurons indicated by symbols based on cortical inputs. Data from two normal (top) and two transgenic (bottom) mice are shown in frontal sections. The figures in the left top corner represent distance from bregma. Rt, Reticular thalamic nucleus; Ot, optic tract.
Figure 6.

Somatotopic organization in the GPe. A, Proportions of neurons classified based on cortical inputs in the normal (top) and transgenic (bottom) mice. B, Distribution of recorded GPe neurons. Data from two normal (top) and two transgenic (bottom) mice are shown in frontal sections. CPu, Caudate–putamen; ec, external capsule; ic, internal capsule.
The locations of recorded GPi and GPe neurons are plotted using symbols based on cortical inputs in Figures 5B and 6B. In the GPi of the normal mice, the neurons with forelimb inputs were distributed over a wide area of the GPi, although not in the most medial portion of it (Fig. 5B, top). A few neurons with orofacial inputs and with convergent inputs were found in the lateral portion of the GPi. In the transgenic mice, this segregation was not observed (Fig. 5B, bottom). The number of GPi neurons with orofacial inputs and those with convergent inputs increased (Fig. 5A), and they intruded into the central portion of the GPi, although the most medial portion remained unresponsive. Cortical inputs to GPe neurons were mainly examined in the central to lateral portion of the GPe (Fig. 6B). In the normal mice, neurons with orofacial inputs were located in the ventral portion, whereas those with forelimb inputs were located in the dorsal portion (Fig. 6B, top). Neurons with convergent inputs were found in the intermediate portion. In the transgenic mice, this type of segregation was not found. Neurons with orofacial inputs and those with convergent inputs intruded into the dorsal portion, and the three groups of neurons were intermingled (Fig. 6B, bottom). Neurons in the most dorsal portion of the GPe were unresponsive in both normal and transgenic mice.
Discussion
The present study characterized the electrophysiological properties of transgenic mice developed to express human ΔE-torsinA. These mice exhibited (1) coactivation of agonist and antagonist muscles and sustained muscle activation, (2) decreased GPi and GPe activity with bursts and pauses, (3) cortically evoked long-lasting inhibition in the GPi and GPe, and (4) somatotopic disorganization in the GPi and GPe. These neuronal abnormalities may be responsible for the behavioral abnormalities exhibited by these mice.
Coactivation of agonist and antagonist muscles and sustained muscle activation in transgenic mice
The triceps and biceps muscles were sometimes coactivated during forelimb movements in the transgenic mice (Fig. 1C). Coactivation of agonist and antagonist muscles is common to various types of dystonia (Obeso et al., 1983; Marsden and Rothwell, 1987; Cohen and Hallett, 1988; Berardelli et al., 1998; Farmer et al., 1998; Liu et al., 2004). Sustained muscle activity lasting >10 s was frequently observed in the transgenic mice (Fig. 1D). Sustained muscle activity is another important sign of dystonia (Herz, 1944; Yanagisawa and Goto, 1971; Marsden and Rothwell, 1987; Jedynak et al., 1991; Berardelli et al., 1998). It has been suggested that dystonic postures are produced by long periods of continuous EMG activity lasting several seconds (Yanagisawa and Goto, 1971; Berardelli et al., 1998). These observations suggest that the transgenic mice we examined exhibit EMG activity similar to that observed in human patients with dystonia.
Decreased GPi and GPe activity in transgenic mice
Marked reduction of the spontaneous firing rates of GPi and GPe neurons was observed in the transgenic mice (Table 1, Fig. 2). Alteration of firing patterns was also observed in them, including bursting discharges and pauses. Decreased discharge rates and irregularly grouped discharges with intermittent pauses in GPi and GPe neurons have also been observed in patients with generalized dystonia (Vitek et al., 1999; Zhuang et al., 2004; Starr et al., 2005). These findings suggest that the transgenic mice we examined may share neurological abnormalities with dystonia patients. Dystonic hamsters with paroxysmal generalized dystonia also exhibited reduced and bursting GPi activity (Gernert et al., 2002). Firing rates of both bursting and irregular nonbursting neurons decreased, suggesting that reduction of firing rate universally occurred in GPi and GPe neurons. The mechanisms responsible for decreased firing rates may include (1) alteration of membrane properties of GPi and GPe neurons; (2) increased inhibitory inputs to the GPi and GPe, such as GABAergic inputs from the striatum; and/or (3) decreased excitatory inputs to the GPi and GPe, such as glutamatergic inputs from the subthalamic nucleus (STN). Inhibitory inputs from the striatum to the GPi and GPe increase in the transgenic mice as discussed in the next section.
Cortically evoked long-lasting inhibition in GPi and GPe neurons of transgenic mice
To investigate the mechanism of abnormal firing of GPi and GPe neurons in the transgenic mice further, responses evoked by cortical stimulation were observed. In the normal mice, cortical stimulation typically induced triphasic responses composed of early excitation, inhibition, and late excitation in GPi and GPe neurons (Figs. 3A, 4A). Similar triphasic responses were also observed in the GPi, GPe, and substantia nigra pars reticulata (SNr), another output station of the basal ganglia, in rats and monkeys. The origin of each component has been intensively studied (Ryan and Sanders, 1994; Maurice et al., 1998, 1999; Nambu et al., 2000; Kita et al., 2004; Tachibana et al., 2008). Early excitation is mediated by the cortico-STN-GPe/GPi/SNr pathway, whereas inhibition and late excitation are mediated by the cortico-striato-GPe/GPi/SNr and cortico-striato-GPe-STN-GPe/GPi/SNr pathways, respectively. However, in the transgenic mice we examined, cortical stimulation induced early excitation followed by late long-lasting inhibition in GPi and GPe neurons (Figs. 3B, 4B). Because the excitability of motor cortex was unchanged in the transgenic mice, these abnormal patterns of response may be generated through the cortico-basal ganglia pathways. The early excitation may, at least its early phase, be mediated by the cortico-STN-GPe/GPi pathway, as in the normal mice, because the latency of the early excitation in the transgenic mice was short and similar to that in the normal mice. These observations also suggest that the activity of STN neurons is unchanged in the transgenic mice. The origin of the late long-lasting inhibition may be (1) increased inhibitory input via the striato-GPe/GPi pathway or (2) decreased excitatory input via the STN-GPe/GPi pathway. The latter explanation seems less likely to be correct, because activity along the cortico-STN-GPi/GPe pathway appeared to be unchanged, as discussed above. The early excitation sometimes included two excitatory peaks (biphasic excitation) (Figs. 3B, 4B). Strong inhibition in the GPe evoked by the cortico-striato-GPe pathway might disinhibit STN neurons and produce excitation in the GPi and GPe, corresponding to the second excitation of the early response in the transgenic mice. The above observations, together with positive relationships between cortically evoked responses and spontaneous firing patterns, also suggest that spontaneous excitation in the cortex is transmitted to the GPi and GPe through the cortico-basal ganglia pathways, and induces short-latency excitation and long-lasting inhibition, which might be the origins of bursts and pauses, respectively.
Somatotopic disorganization of GPi and GPe in transgenic mice
Motor territories in the GPi and GPe of monkeys are somatotopically organized, as indicated by their somatosensory inputs, activity during voluntary movements, and cortically evoked responses (DeLong et al., 1985; Yoshida et al., 1993). The present study confirmed the somatotopic organization in the normal mice. In the transgenic mice, however, somatotopic disorganization was observed, and many GPi and GPe neurons received convergent inputs from both forelimb and orofacial regions (Figs. 5, 6). Widened somatosensory receptive fields in pallidal neurons have been reported in patients with generalized (Vitek et al., 1999) and focal (Lenz et al., 1998; Sanger et al., 2001) dystonia. Because the somatotopic arrangement and excitability of the motor cortex in the transgenic mice appeared normal in the present study, information-crossing may have occurred through the cortico-basal ganglia pathways. One explanation for this is that single GPi or GPe neurons receive inputs from more striatal neurons in the transgenic mice than in the normal mice (Fig. 7). This explanation agrees well with the assumption of increased inhibitory input via the striato-GPe/GPi pathway noted in the previous section.
Figure 7.

Schematic diagrams showing information processing through the basal ganglia in normal condition (A) and dystonia (B). In dystonia, cortical activation induces strong inhibition over wide areas of the GPi, as well as strong excitation in the thalamus and cortex as a result of disinhibition, resulting in the motor hyperactivity and involuntary muscle contractions.
Neural mechanisms of motor hyperactivity in transgenic mice
The GPi, an output nucleus of the basal ganglia, is composed of GABAergic inhibitory neurons and fires at high frequency in normal states. Its target structures, such as the thalamus and frontal cortex, are thus continuously inhibited. Striatal inputs reduce GPi activity in temporal manner, excite thalamic and cortical neurons via disinhibition, and finally release appropriate movements with appropriate timing (Fig. 7A) (Nambu et al., 2000, 2002). However, in the transgenic mice, cortical excitation induced long-lasting inhibition in the GPi, suggesting that even tiny amounts of neuronal activity originating in the cortex are transmitted through the cortico-basal ganglia pathways and finally induce strong and long-lasting inhibition in the GPi (Fig. 7B). Moreover, somatotopic disorganization was noted in the GPi and cortical activation induced inhibition over a wide area of this region. Wide areas of the thalamus and cortex are thus activated in uncontrollable manner, resulting in the motor hyperactivity and involuntary muscle contractions observed in the transgenic mice. A similar mechanism may underlie the symptoms of human dystonia. This may also explain the motor overflow in dystonia, which results in unintentional muscle contraction during voluntary movements. Activation of the forelimb region in motor cortex, for example, may inhibit large areas of the GPi and finally induce involuntary movements of multiple body parts. Indeed, reduction of the discharge rate of GPi neurons plays a role in induction of hyperkinesia or involuntary movements. Inactivation of the GPi by muscimol injection induced increase in thalamic activity with tonic activation of the triceps muscle (Inase et al., 1996). Deactivation of the GPi by kynurenate injection elicited dyskinesia (Robertson et al., 1989), and inactivation of the GPi resulted in tonic and phasic coactivation of the flexor and extensor muscles (Mink and Thach, 1991). STN blockade by muscimol injection or lesions induced decrease in spontaneous firing rate and cortically evoked long-lasting inhibition in GPi neurons as well as severe hemiballism (Hamada and DeLong, 1992a,b; Nambu et al., 2000; Tachibana et al., 2008).
Relationships with other transgenic models
The transgenic mice investigated in the present study exhibited hyperkinesia and dystonic-like movements. Recently, other mouse models have been developed via overexpression of ΔE-torsinA (Sharma et al., 2005; Grundmann et al., 2007; Zhao et al., 2008) or GAG deletion in the DYT1 gene (Dang et al., 2005, 2006). However, because these models have failed to replicate such severe symptoms, the phenotype of the present transgenic mice may not be attributable entirely to overexpression of ΔE-torsinA. Electrophysiological analyses of other models of transgenic mice may improve our understanding of the pathophysiology of dystonia and enable the development of more effective treatments of it.
Footnotes
This work was supported by Grants-in-Aid for Exploratory Research 18650089 and Grants-in-Aid for Scientific Research (B) 18300135 from Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT), The Uehara Memorial Foundation, Takeda Science Foundation, and United States–Japan Brain Research Cooperative Program (A.N.); Grants-in-Aid for Scientific Research (C) (19500354) from MEXT (S.C.); and National Institute of Neurological Disorders and Stroke–National Institutes of Health Grant NS 043038 (P.S.). We thank K. Kobayashi and Y. Yasoshima for crucial technical advice and A. Ito, K. Miyamoto, and M. Ohara for technical assistance.
References
- Abeles M. Quantification, smoothing, and confidence limits for single-units' histograms. J Neurosci Methods. 1982;5:317–325. doi: 10.1016/0165-0270(82)90002-4. [DOI] [PubMed] [Google Scholar]
- Aldridge JW, Gilman S. The temporal structure of spike trains in the primate basal ganglia: afferent regulation of bursting demonstrated with precentral cerebral cortical ablation. Brain Res. 1991;543:123–138. doi: 10.1016/0006-8993(91)91055-6. [DOI] [PubMed] [Google Scholar]
- Berardelli A, Rothwell JC, Hallett M, Thompson PD, Manfredi M, Marsden CD. The pathophysiology of primary dystonia. Brain. 1998;121:1195–1212. doi: 10.1093/brain/121.7.1195. [DOI] [PubMed] [Google Scholar]
- Bergstrom DA, Bromley SD, Walters JR. Dopamine agonists increase pallidal unit activity: attenuation by agonist pretreatment and anesthesia. Eur J Pharmacol. 1984;100:3–12. doi: 10.1016/0014-2999(84)90309-1. [DOI] [PubMed] [Google Scholar]
- Bressman SB. Dystonia genotypes, phenotypes, and classification. Adv Neurol. 2004;94:101–107. [PubMed] [Google Scholar]
- Chiken S, Tokuno H. Ablation of striatal interneurons influences activities of entopeduncular neurons. Neuroreport. 2003;14:675–678. doi: 10.1097/00001756-200304150-00003. [DOI] [PubMed] [Google Scholar]
- Cohen LG, Hallett M. Hand cramps: clinical features and electromyographic patterns in a focal dystonia. Neurology. 1988;38:1005–1012. doi: 10.1212/wnl.38.7.1005. [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, McNaught KS, Jengelley TA, Jackson T, Li J, Li Y. Generation and characterization of Dyt1 ΔGAG knock-in mouse as a model for early-onset dystonia. Exp Neurol. 2005;196:452–463. doi: 10.1016/j.expneurol.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Pence MA, Li Y. Motor deficits and hyperactivity in Dyt1 knockdown mice. Neurosci Res. 2006;56:470–474. doi: 10.1016/j.neures.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong MR, Crutcher MD, Georgopoulos AP. Primate globus pallidus and subthalamic nucleus: functional organization. J Neurophysiol. 1985;53:530–543. doi: 10.1152/jn.1985.53.2.530. [DOI] [PubMed] [Google Scholar]
- Donoghue JP, Wise SP. The motor cortex of the rat: cytoarchitecture and microstimulation mapping. J Comp Neurol. 1982;212:76–88. doi: 10.1002/cne.902120106. [DOI] [PubMed] [Google Scholar]
- Elias S, Joshua M, Goldberg JA, Heimer G, Arkadir D, Morris G, Bergman H. Statistical properties of pauses of the high-frequency discharge neurons in the external segment of the globus pallidus. J Neurosci. 2007;27:2525–2538. doi: 10.1523/JNEUROSCI.4156-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S. Concept and classification of dystonia. Adv Neurol. 1988;50:1–8. [PubMed] [Google Scholar]
- Fahn S, Bressman SB, Marsden CD. Classification of dystonia. Adv Neurol. 1998;78:1–10. [PubMed] [Google Scholar]
- Farmer SF, Sheean GL, Mayston MJ, Rothwell JC, Marsden CD, Conway BA, Halliday DM, Rosenberg JR, Stephens JA. Abnormal motor unit synchronization of antagonist muscles underlies pathological co-contraction in upper limb dystonia. Brain. 1998;121:801–814. doi: 10.1093/brain/121.5.801. [DOI] [PubMed] [Google Scholar]
- Gernert M, Bennay M, Fedrowitz M, Rehders JH, Richter A. Altered discharge pattern of basal ganglia output neurons in an animal model of idiopathic dystonia. J Neurosci. 2002;22:7244–7253. doi: 10.1523/JNEUROSCI.22-16-07244.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann K, Reischmann B, Vanhoutte G, Hübener J, Teismann P, Hauser TK, Bonin M, Wilbertz J, Horn S, Nguyen HP, Kuhn M, Chanarat S, Wolburg H, Van der Linden A, Riess O. Overexpression of human wildtype torsinA and human ΔGAG torsinA in a transgenic mouse model causes phenotypic abnormalities. Neurobiol Dis. 2007;27:190–206. doi: 10.1016/j.nbd.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Hamada I, DeLong MR. Excitotoxic acid lesions of the primate subthalamic nucleus result in transient dyskinesias of the contralateral limbs. J Neurophysiol. 1992a;68:1850–1858. doi: 10.1152/jn.1992.68.5.1850. [DOI] [PubMed] [Google Scholar]
- Hamada I, DeLong MR. Excitotoxic acid lesions of the primate subthalamic nucleus result in reduced pallidal neuronal activity during active holding. J Neurophysiol. 1992b;68:1859–1866. doi: 10.1152/jn.1992.68.5.1859. [DOI] [PubMed] [Google Scholar]
- Herz Dystonia. 1. Historical review: analysis of dystonic symptoms and physiologic mechanisms involved. Arch Neurol Psychiatry. 1944;51:305–318. [Google Scholar]
- Inase M, Buford JA, Anderson ME. Changes in the control of arm position, movement, and thalamic discharge during local inactivation in the globus pallidus of the monkey. J Neurophysiol. 1996;75:1087–1104. doi: 10.1152/jn.1996.75.3.1087. [DOI] [PubMed] [Google Scholar]
- Jedynak CP, Bonnet AM, Agid Y. Tremor and idiopathic dystonia. Mov Disord. 1991;6:230–236. doi: 10.1002/mds.870060307. [DOI] [PubMed] [Google Scholar]
- Karmon B, Bergman H. Detection of neuronal periodic oscillations in the basal ganglia of normal and parkinsonian monkeys. Isr J Med Sci. 1993;29:570–579. [PubMed] [Google Scholar]
- Keane PE, Biziere K. The effects of general anaesthetics on GABAergic synaptic transmission. Life Sci. 1987;41:1437–1448. doi: 10.1016/0024-3205(87)90708-9. [DOI] [PubMed] [Google Scholar]
- Kita H, Nambu A, Kaneda K, Tachibana Y, Takada M. Role of ionotropic glutamatergic and GABAergic inputs on the firing activity of neurons in the external pallidum in awake monkeys. J Neurophysiol. 2004;92:3069–3084. doi: 10.1152/jn.00346.2004. [DOI] [PubMed] [Google Scholar]
- Kita H, Tachibana Y, Nambu A, Chiken S. Balance of monosynaptic excitatory and disynaptic inhibitory responses of the globus pallidus induced after stimulation of the subthalamic nucleus in the monkey. J Neurosci. 2005;25:8611–8619. doi: 10.1523/JNEUROSCI.1719-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H, Chiken S, Tachibana Y, Nambu A. Origins of GABAA and GABAB receptor-mediated responses of globus pallidus induced after stimulation of the putamen in the monkey. J Neurosci. 2006;26:6554–6562. doi: 10.1523/JNEUROSCI.1543-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PL, Ozelius L, Brin MF, Fahn S, Kidd KK, Gusella J, Breakefield XO. Molecular genetics of an autosomal dominant form of torsion dystonia. Adv Neurol. 1988;50:57–66. [PubMed] [Google Scholar]
- Legéndy CR, Salcman M. Bursts and recurrences of bursts in the spike trains of spontaneously active striate cortex neurons. J Neurophysiol. 1985;53:926–939. doi: 10.1152/jn.1985.53.4.926. [DOI] [PubMed] [Google Scholar]
- Lenz FA, Suarez JI, Metman LV, Reich SG, Karp BI, Hallett M, Rowland LH, Dougherty PM. Pallidal activity during dystonia: somatosensory reorganisation and changes with severity. J Neurol Neurosurg Psychiatry. 1998;65:767–770. doi: 10.1136/jnnp.65.5.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Rouiller EM, Wiesendanger M. Modulation of sustained electromyographic activity by single microstimuli: comparison of two forelimb motor cortical areas of the rat. Somatosens Mot Res. 1993;10:51–61. doi: 10.3109/08990229309028823. [DOI] [PubMed] [Google Scholar]
- Liu X, Tailor J, Wang S, Yianni J, Gregory R, Stein J, Aziz T. Reversal of hypertonic co-contraction after bilateral pallidal stimulation in generalised dystonia: a clinical and electromyogram case study. Mov Disord. 2004;19:336–340. doi: 10.1002/mds.10655. [DOI] [PubMed] [Google Scholar]
- Löscher W, Rohlfs A, Rundfeldt C. Reduction in firing rate of substantia nigra pars reticulata neurons by valproate: influence of different types of anesthesia in rats. Brain Res. 1995;702:133–144. doi: 10.1016/0006-8993(95)01030-4. [DOI] [PubMed] [Google Scholar]
- Marsden CD, Rothwell JC. The physiology of idiopathic dystonia. Can J Neurol Sci. 1987;14:521–527. doi: 10.1017/s031716710003804x. [DOI] [PubMed] [Google Scholar]
- Maurice N, Deniau JM, Glowinski J, Thierry AM. Relationships between the prefrontal cortex and the basal ganglia in the rat: physiology of the corticosubthalamic circuits. J Neurosci. 1998;18:9539–9546. doi: 10.1523/JNEUROSCI.18-22-09539.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice N, Deniau JM, Glowinski J, Thierry AM. Relationships between the prefrontal cortex and the basal ganglia in the rat: physiology of the cortico-nigral circuits. J Neurosci. 1999;19:4674–4681. doi: 10.1523/JNEUROSCI.19-11-04674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mink JW, Thach WT. Basal ganglia motor control. III. Pallidal ablation: normal reaction time, muscle cocontraction, and slow movement. J Neurophysiol. 1991;65:330–351. doi: 10.1152/jn.1991.65.2.330. [DOI] [PubMed] [Google Scholar]
- Nambu A, Yoshida S, Jinnai K. Discharge patterns of pallidal neurons with input from various cortical areas during movement in the monkey. Brain Res. 1990;519:183–191. doi: 10.1016/0006-8993(90)90076-n. [DOI] [PubMed] [Google Scholar]
- Nambu A, Tokuno H, Hamada I, Kita H, Imanishi M, Akazawa T, Ikeuchi Y, Hasegawa N. Excitatory cortical inputs to pallidal neurons via the subthalamic nucleus in the monkey. J Neurophysiol. 2000;84:289–300. doi: 10.1152/jn.2000.84.1.289. [DOI] [PubMed] [Google Scholar]
- Nambu A, Tokuno H, Takada M. Functional significance of the cortico-subthalamo-pallidal “hyperdirect” pathway. Neurosci Res. 2002;43:111–117. doi: 10.1016/s0168-0102(02)00027-5. [DOI] [PubMed] [Google Scholar]
- Neafsey EJ, Sievert C. A second forelimb motor area exists in rat frontal cortex. Brain Res. 1982;232:151–156. doi: 10.1016/0006-8993(82)90617-5. [DOI] [PubMed] [Google Scholar]
- Neafsey EJ, Bold EL, Hass G, Hurley-Gius KM, Guirk G, Sievert CF, Terreberry RR. The organization of the rat motor cortex: a microstimulation mapping study. Brain Res Rev. 1986;11:77–96. doi: 10.1016/s0006-8993(86)80191-3. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rothwell JC, Lang AE, Marsden CD. Myoclonic dystonia. Neurology. 1983;33:825–830. doi: 10.1212/wnl.33.7.825. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett J, Kramer P, Bressman SB, Shalish C, de Leon D, Rutter M, Risch N, Brin MF, Markova ED, Limborska SA, Ivanova-Smolenskaya IA, McCormick MK, Fahn S, Buckler AJ, Gusella JF, Breakefield XO. Fine localization of the torsion dystonia gene (DYT1) on human chromosome 9q34: YAC map and linkage disequilibrium. Genome Res. 1997a;7:483–494. doi: 10.1101/gr.7.5.483. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997b;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Robertson RG, Farmery SM, Sambrook MA, Crossman AR. Dyskinesia in the primate following injection of an excitatory amino acid antagonist into the medial segment of the globus pallidus. Brain Res. 1989;476:317–322. doi: 10.1016/0006-8993(89)91252-3. [DOI] [PubMed] [Google Scholar]
- Rouiller EM, Moret V, Liang F. Comparison of the connection properties of the two forelimb areas of the rat sensorimotor cortex: support for the presence of a premotor or supplementary motor cortical area. Somatosens Mot Res. 1993;10:269–289. doi: 10.3109/08990229309028837. [DOI] [PubMed] [Google Scholar]
- Ryan LJ, Sanders DJ. Subthalamic nucleus and globus pallidus lesions alter activity in nigrothalamic neurons in rats. Brain Res Bull. 1994;34:19–26. doi: 10.1016/0361-9230(94)90181-3. [DOI] [PubMed] [Google Scholar]
- Sanger TD, Tarsy D, Pascual-Leone A. Abnormalities of spatial and temporal sensory discrimination in writer's cramp. Mov Disord. 2001;16:94–99. doi: 10.1002/1531-8257(200101)16:1<94::aid-mds1020>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Sharma N, Baxter MG, Petravicz J, Bragg DC, Schienda A, Standaert DG, Breakefield XO. Impaired motor learning in mice expressing torsinA with the DYT1 dystonia mutation. J Neurosci. 2005;25:5351–5355. doi: 10.1523/JNEUROSCI.0855-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashidharan P, Sandu D, Potla U, Armata IA, Walker RH, McNaught KS, Weisz D, Sreenath T, Brin MF, Olanow CW. Transgenic mouse model of early-onset DYT1 dystonia. Hum Mol Genet. 2005;14:125–133. doi: 10.1093/hmg/ddi012. [DOI] [PubMed] [Google Scholar]
- Starr PA, Rau GM, Davis V, Marks WJ, Jr, Ostrem JL, Simmons D, Lindsey N, Turner RS. Spontaneous pallidal neuronal activity in human dystonia: comparison with Parkinson's disease and normal macaque. J Neurophysiol. 2005;93:3165–3176. doi: 10.1152/jn.00971.2004. [DOI] [PubMed] [Google Scholar]
- Tachibana Y, Kita H, Chiken S, Takada M, Nambu A. Motor cortical control of internal pallidal activity through glutamatergic and GABAergic inputs in awake monkeys. Eur J Neurosci. 2008;27:238–253. doi: 10.1111/j.1460-9568.2007.05990.x. [DOI] [PubMed] [Google Scholar]
- Tang JK, Moro E, Mahant N, Hutchison WD, Lang AE, Lozano AM, Dostrovsky JO. Neuronal firing rates and patterns in the globus pallidus internus of patients with cervical dystonia differ from those with Parkinson's disease. J Neurophysiol. 2007;98:720–729. doi: 10.1152/jn.01107.2006. [DOI] [PubMed] [Google Scholar]
- Vitek JL. Pathophysiology of dystonia: a neuronal model. Mov Disord. 2002;17(Suppl 3):S49–S62. doi: 10.1002/mds.10142. [DOI] [PubMed] [Google Scholar]
- Vitek JL, Chockkan V, Zhang JY, Kaneoke Y, Evatt M, DeLong MR, Triche S, Mewes K, Hashimoto T, Bakay RA. Neuronal activity in the basal ganglia in patients with generalized dystonia and hemiballismus. Ann Neurol. 1999;46:22–35. doi: 10.1002/1531-8249(199907)46:1<22::aid-ana6>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Soares J. Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J Neurophysiol. 2006;95:2120–2133. doi: 10.1152/jn.01013.2005. [DOI] [PubMed] [Google Scholar]
- Yanagisawa N, Goto A. Dystonia musculorum deformans. Analysis with electromyography. J Neurol Sci. 1971;13:39–65. doi: 10.1016/0022-510x(71)90206-1. [DOI] [PubMed] [Google Scholar]
- Yasoshima Y, Kai N, Yoshida S, Shiosaka S, Koyama Y, Kayama Y, Kobayashi K. Subthalamic neurons coordinate basal ganglia function through differential neural pathways. J Neurosci. 2005;25:7743–7753. doi: 10.1523/JNEUROSCI.1904-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Nambu A, Jinnai K. The distribution of the globus pallidus neurons with input from various cortical areas in the monkeys. Brain Res. 1993;611:170–174. doi: 10.1016/0006-8993(93)91791-p. [DOI] [PubMed] [Google Scholar]
- Zhao Y, DeCuypere M, LeDoux MS. Abnormal motor function and dopamine neurotransmission in DYT1 ΔGAG transgenic mice. Exp Neurol. 2008;210:719–730. doi: 10.1016/j.expneurol.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang P, Li Y, Hallett M. Neuronal activity in the basal ganglia and thalamus in patients with dystonia. Clin Neurophysiol. 2004;115:2542–2557. doi: 10.1016/j.clinph.2004.06.006. [DOI] [PubMed] [Google Scholar]