

Abstract
The specificity and function of T helper (Th) immune responses underlying the induction, progression, and resolution of experimental autoimmune myocarditis (EAM) in A/J mice are unclear. Published data suggest involvement of both Th1 and Th2 responses in EAM; however, the previous inability to assess antigen-specific in vivo and in vitro T cell responses in cardiac myosin immunized animals has confounded our understanding of this important model of autoimmune myocarditis. The goal of our study was to develop an alternative model of EAM based on a recombinant fragment of cardiac myosin, in hopes that the recombinant protein will permit measurement of functional T cell responses that is not possible with purified native protein. A/J mice immunized with a recombinant fragment of cardiac myosin spanning amino acids 1074–1646, termed Myo4, developed severe myocarditis characterized by cardiac hypertrophy, massive mononuclear cell infiltration and fibrosis, three weeks post-immunization. The mice also developed an IgG1 dominant humoral immune response specific for both Myo4 and purified cardiac myosin. The in vitro stimulation of splenocytes harvested from Myo4-immunized animals with Myo4 resulted in cellular proliferation with preferential production of the Th1- and Th17-associated cytokines, IFN-γ, IL-17 and IL-6, respectively. Production of IL-4 was negligible by comparison. This study describes a new model of EAM, inducible by immunization with a specific fragment of cardiac myosin, from which antigen-specific analyses reveal an importance for both Th1 and Th17 immunity.
Keywords: autoimmunity, myocarditis, Th immunity, epitope spreading
Introduction
A number of infectious and non-infectious agents, including bacteria, viruses, parasites, drugs, and toxins (1–4) may cause or contribute to the development of myocarditis. These agents can induce myocarditis via direct induction of myocyte damage and death, or by disruption of the cardiac extracellular matrix. Under these conditions, the immune system participates in the identification and elimination of infected cells and in the repair of damaged cardiac tissue. During this repair process, immune responses targeting self-antigens may develop in some individuals. Autoantibodies specific for cardiac antigens are produced in people having a number of inflammatory heart diseases, such as idiopathic dilated cardiomyopathy (2, 5–7), tuberculous pericarditis (8), Lyme carditis (9), rheumatic heart disease (10), Chagas heart disease (11) and inflammatory states of unknown etiology (12). In addition to the production of cardiac-specific autoantibodies, cardiac antigen-specific T cell clones have been isolated from the hearts of humans with rheumatic heart disease (13) and Chagas heart disease (11, 14) as well.
Animal models of human disease are powerful tools used to identify and analyze the immune mediators responsible for disease progression or remission. To explore mechanisms of autoimmune myocarditis, immunization with cardiac myosin alpha heavy chain has been utilized to induce cardiac inflammation in animals (15). Experimental autoimmune myocarditis (EAM) is histologically similar to human myocarditis, with myocyte swelling, necrosis, and fibrosis accompanied by mononuclear cell infiltration consisting of 80% monocytes and macrophages, 16% T cells (with a 4 to 1 ratio of CD4 to CD8 phenotype), and 2% B cells (16). Although plasma cells have been identified in the heart, cardiac myosin-specific autoantibodies have been shown incapable of inducing disease in mice lacking cell surface expression or extracellular matrix deposition of cardiac myosin (17).
The T helper (Th) immunity responsible for disease pathogenesis induced by cardiac myosin immunization is variable depending on the model system utilized (18). In the rat model, cardiac myosin leads to a Th1 mediated disease (19). In BALB/c mice, Stat4 and IL-12, critical components of Th1 activation, are essential for disease development (20). In the A/J mouse model, a large number of eosinophils and giant cells can be identified in the heart infiltrate (21). Cardiac myosin-specific IgG1 antibody levels correlate with disease severity, whereas cardiac myosin-specific IgG2a antibody levels do not (21). Anti-IL-4 administration decreases the severity of EAM whereas anti-IFN-γ treatment exacerbates disease (21). Therefore, EAM in the A/J mouse appears to have a Th2 phenotype.
To definitively assess the Th phenotype associated with disease pathogenesis, antigen specific cytokine production needs to be investigated. The determination of antigen specific immunity induced by cardiac myosin has been hindered in mouse models due to the toxic nature of cardiac myosin in culture (22). To circumvent myosin toxicity and to define pathogenetic epitopes of myosin, peptide models of mouse EAM were developed. A multitude of peptides have been identified in BALB/c mice and it is clear that disease is mediated by the activation of both Th1 and Th17 cells (23). Only one peptide, CM 334–352 (Allen peptide), has been shown to induce myocarditis in A/J mice and the immunity suggest that the disease is mediated by Th2 responses (21).
To further investigate the involvement of various regions of whole cardiac myosin in the induction and propagation of EAM, we produced five non-overlapping recombinant fragments spanning the entire molecule. Here, we report a new model of EAM in A/J mice based on a recombinant myosin fragment. This model facilitates the investigation of antigen-specific immunity from which we have observed cytokine profiles corresponding to both Th1 and Th17 responses.
Materials and methods
Mice
5–7 week old male A/J mice (Jackson Laboratories, Bar Harbor, ME, USA) were housed under specific pathogen-free conditions. Mice were anesthetized by a single intraperitoneal injection of 240 mg/kg avertin or each experimental manipulation. The use and care of mice were conducted in accordance with the guidelines of the Center for Comparative Medicine of Northwestern University.
Preparation of cardiac myosin, heart homogenate, and Allen peptide
Cardiac myosin heavy chains were purified according to the method of Shiverick et al. (24). Briefly, mouse hearts were minced and homogenized in 10 vol of ice cold KCl buffer (0.3 M KCl, 0.15 M K2HPO4, 10 mM Na4P2O7, 1 mM MgCl2, pH 6.80). Myosin was extracted from the muscle homogenate by stirring at 4°C for 90 min. The suspension was centrifuged at 140,000 × g for 1 hr at 4°C and the decanted supernatant was diluted with 20 vol of water and incubated at 4°C overnight to precipitate the myosin. The precipitate was collected by centrifugation at 12,000 × g for 30 min at 4°C and suspended in ice cold imidazole buffer (0.5 M KCl, 10 mM imidazole, 5 mM MgCl2, 5 mM Na2ATP, 2 mM DTT, pH 6.80). The solution was then centrifuged at 43,000 × g for 30 min at 4°C to remove actin. The myosin was precipitatedin 8 vol of ice cold water at 4°C overnight. The precipitate was collected by centrifugation at 12,000 × g for 30 min at 4°C and the pellet was suspended in the imidazole buffer and centrifuged at 43,000 × g for 30 min at 4°C to remove residual actin. The supernatant was again precipitated overnight at 4°C in 6.5 vol of ice cold water. The precipitate was collected by centrifugation at 12,000 × g for 30 min at 4°C and suspended in 50 mM Na4P2O7, pH 6.8. Protein concentration was determined by comparing dilutions of the purified myosin solution with known concentrations of purified rabbit myosin heavy chain standards (Sigma, Atlanta, GA, USA) by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Total heart homogenate was prepared by washing A/J hearts with Dulbecco’s phosphate buffered saline (PBS, GibcoBRL, Grand Island, NY, USA), mincing hearts with a razor blade, and performing homogenization of hearts in SDS buffer. The Allen peptide, spanning cardiac myosin amino acids 334–352 (NH2-DSAF DVLS FTAE EKAG VYK-COOH), was purchased from Global Peptide Services (Fort Collins, CO, USA). The peptide was dissolved in PBS at 5 mg/ml.
Engineering, production, and concentration of hexahistidine-tagged Myo4 protein
Total RNA was extracted from an A/J mouse tail using the Trizol extraction method and RT-PCR was used to generate cDNA. The following oligonucleotides were used for PCR amplification of the Myo4 coding region: 5′ primer GATCAGTACATATGTATGACAAGCTTCAGCTGGAA and 3′ primer CAGTCTCGAGTGAATTCTTCAGGTGTTTCTGTG. The Myo4 PCR product was directionally cloned into the pET23b vector using HindIII and EcoRI, permitting expression of a hexahistidine (His) tagged Myo4 protein and transformed into E. coli BL21 DE3 pLysS (Novagen, Gibbstown, NJ, USA). Induction of recombinant protein expression using isopropyl-β-D-thiogalactopyranoside (IPTG) and purification and detoxification of Myo4 on an Aktaprime machine by nickel chromatography (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) were conducted using standard methods. Following protein concentration with a 200 ml Stircell apparatus (Millipore, Temecula, CA, USA) using a 10 kDa membrane filter, Myo4 was dialyzed into PBS for use. The final product is a 68 kDa recombinant protein spanning amino acids 1074–1646 of cardiac myosin.
Immunization
Mice were immunized and boosted 7 days later with either purified cardiac myosin (300μg/immunization/animal), Myo4 (250 μg/immunization/animal), Allen peptide (50–150 nmols/immunization/animal ), or saline emulsified in complete Freund’s adjuvant. A total volume of 0.1 ml of emulsion was subcutaneously injected into three sites in the dorsal flank. Myo4- and Allen peptide-immunized mice were injected with 200 ng of pertussis toxin (List Biological Laboratories, Campbell, CA, USA) on days 0 and 2 post primary immunization.
Histopathology
Hearts were removed, rinsed with PBS, and fixed for at least 24 hrs in 10% buffered formalin. Fixed hearts were embedded in paraffin, sectioned, stained with hematoxylin-eosin or Masson’s trichrome, and examined by light microscopy. Each section was examined for evidence of mononuclear and polymorphonuclear cellular inflammation and fibrosis and was assigned a histologic score between 0 (no involvement noted) to 4 (100% involvement), with 1, 2, 3 representing 25, 50, and 75% involvement of the histologic section as previously described (25–29).
Delayed-type hypersensitivity
Antigen-specific delayed-type hypersensitivity (DTH) was quantitated using a standard ear swelling assay as previously described (30) Pre-challenge ear thickness in anesthetized animals was measured with a Mitutoyo model 7326 engineer’s micrometer (Mitutoyo MTI Corporation, Aurora, IL, USA). Antigen (10 μg in 0.15 M K2PO4, 0.01 Na4P2O7, 0.3 M KCl, pH 6.8) was injected intradermally into the dorsal surface of the ear using a 100 μl Hamilton syringe fitted with a 30 gauge needle. Bovine serum albumin was injected in the opposite ear as a baseline control. After 24 hrs, the net swelling of baseline injection was subtracted from that of the antigen-injection and expressed in units of 10−4 inches. Antigen-induced ear swelling is representative of mononuclear cell infiltration and exhibits typical DTH kinetics (i.e., minimal swelling at 4 hr, maximal swelling at 24–48 hr).
Serologic analysis
Levels of cardiac myosin-specific and Myo4-specific total IgG, IgG1, and IgG2a were determined by ELISA. Briefly, Maxisorp plates (Fisher Scientific, Pittsburgh, PA, USA) were coated overnight at 4°C with 100 μl of cardiac myosin or Myo4 at 2.5 μg/ml in PBS. The plates were washed with PBS containing 0.05% v/v Tween-20, blocked with 2% BSA and 5% normal goat serum, and incubated with twofold serial serum dilutions for 2 hr at 37°C. After washing, peroxidase conjugated, anti-mouse IgG, IgG1, or IgG2a was added for 1 hr at 37°C. The conjugate was detected with 3, 3′, 5, 5′-tetramethylbenzidine (TMB) substrate (BioFX Laboratories, Owings Mills, MD, USA) and quantitated by measurement of the OD450 in a Kinetic MicroPlate Reader (Molecular Devices, Sunnyvale, CA, USA)
Western blot analysis
Heart homogenate (40 μg), myosin (1 μg), Myo4 (8 μg), and BSA (8 μg) protein preparations were resolved by SDS-PAGE, stained with GelCode Blue (Fisher Scientific, Pittsburgh, PA, USA) or transferred to nitrocellulose and incubated with 1:200 dilutions of sera from immunized animals. Secondary antibody incubations were performed using a 1:2000 dilution of alkaline phosphatase-conjugated goat anti-mouse IgG. Immunoblots were developed using 5-bromo-4-chloro-3-indolyl phosphate and p-nitroblue tetrazolium phosphate (United States Biochemical, Cleveland, OH, USA) in 100 mM Tris, 1 mM MgCl2 (pH 9.5).
In vitro ELISPOT assay
Spleens were forced through 100μm mesh stainless steel screens to yield single-cell suspensions. Red blood cells in the spleen preparations were lysed by hypotonic shock in Tris-NH4Cl (pH 7.3), and the cells were washed and resuspended in Dulbecco’s modified Eagle medium (DMEM) containing 50μM β-mercaptoethanol, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1 M nonessential amino acids, and 10% fetal calf serum (Gemini Bioproducts, West Sacramento, CA, USA) (proliferation media). Cells were cultured in 96-well plates (Millipore, Temecula, CA, USA) that were pre-coated with 4 μg/ml anti-IFN-γ, anti-IL-6, anti-IL-17 (BD Biosciences, San Diego, CA, USA) or anti-IL-4 (Ebioscience, San Diego, CA, USA). Splenocytes were plated at 5 × 105 cells per well in proliferation media in either the presence of 10 μM Myo4, medium alone as a negative control, or anti-CD3 (500 ng/ml, 2C11 clone, a gift from Dr. William J. Karpus, Northwestern University) as a positive control for cell activation. Cells were incubated at 37°C in 5% CO2 for 24 hrs for IFN-γ, IL-6, and IL-17 detection and 48 hrs for IL-4 detection. Plates were washed to remove cells and incubated with 2 μg/ml biotin-conjugated anti-IFN-γ (BD Biosciences, San Diego, CA, USA), anti-IL-4 (Ebioscience, San Diego, CA, USA), anti-IL-6 (BD Biosciences, San Diego, CA, USA), and anti-IL-17 (BD Biosciences, San Diego, CA, USA). Plates were washed and incubated with alkaline phosphatase-conjugated anti-biotin antibody for 2 hrs and developed using 5-bromo-4-chloro-3-indolyl phosphate and p-nitroblue tetrazolium phosphate (United States Biochemical, Cleveland, OH, USA) in 100 mM Tris, 1 mM MgCl2 (pH 9.5). Cytokine expression was quantified using an ImmunoSpot apparatus (Cellular Technology Ltd., Shaker Heights, OH, USA).
Intracellular cytokine staining and flow cytometric analysis
Splenocytes and lymphocytes were cultured in 96-well microtiter plates (Corning-Costar) at 2 × 106 or 1 × 106 cells per well, respectively. To provide adequate antigen presentation 1 × 106 irradiated splenocytes were added to lymphocytes cultures. Cells were stimulated with either 10μM Myo4, medium alone, or anti-CD3 (500 ng/ml, 2C11 clone) as a positive control. Cells were incubated at 37°C, 5% CO2 for 72 hrs, washed, and re-stimulated for 3–5 hrs with 5 ng/ml phorbol 12-myristate 13-acetate (PMA), 500 ng/ml ionomycin, and a 1:1500 dilution of Golgi stop (BD Biosciences, San Diego, CA, USA). Cells were then washed, stained with a viability dye (Invitrogen, Chicago, IL, USA), and blocked for 10 min with a 1:100 anti-CD16/32 solution (Ebioscience, San Diego, CA, USA) with 2% rat serum. For preferential analysis of a T cell population, cells were stained with PE-Cy7-CD90.2 (Ebioscience, San Diego, CA, USA) for 30 min. After fixation with 2% Paraformaldehyde, intracellular cytokine staining was performed using anti-IFN-γ-FITC, anti-IL-4-APC, and anti-IL-17-PE (BD Biosciences, San Diego, CA, USA) in a 0.1% saponin solution. Flow cytometric analysis was performed using the DakoCytomation CyAn instrument and analyzed using Dako software. Only viable CD90.2 cells were analyzed.
Statistical analysis
All values with error bars are expressed as mean ± SEM. The statistical significance of all assays was analyzed by one-way ANOVA followed by a 2-tailed t-test and post hoc Bonferroni analysis. Values of p < 0.05 were considered significant.
Results
Myo4 induces cardiac hypertrophy, inflammation, and fibrosis
To investigate the dominant fragment of cardiac myosin responsible for the development of myocarditis in A/J mice, five non-overlapping cardiac fragments were generated to span the entire A/J cardiac myosin coding region (data not shown). These fragments spanned amino acids M1-Y349 (Myo1), G350-R774 (Myo2), I775-K1073 (Myo3), K1074-Q1646 (Myo4) and L1647-E1940 (Myo5). Of these five fragments, the 68 kDa Myo4 fragment was the first to be produced, purified, and concentrated (Figure 1). We sought to compare the myocarditis-inducing potential of Myo4 with purified cardiac myosin and a previously reported carditogenic peptide (31). A/J mice were immunized with each of these antigens and hearts were evaluated by a number of parameters. Hearts from myosin- and Myo4-immunized animals displayed significant cardiac hypertrophy while hypertrophy in mice immunized with the Allen peptide approached, but did not quite reach, statistical significance (Figure 2A). All animals showed significant cardiac inflammation and fibrosis compared to control animals (Figure 2B). Histologic scoring (25–29) revealed that inflammation and fibrosis were significant in myosin-, Myo4-, and peptide-immunized animals (Figure 2C). There were no significant differences in inflammation and fibrosis among the groups having disease.
Figure 1. Myo4 expression, purification, and concentration.
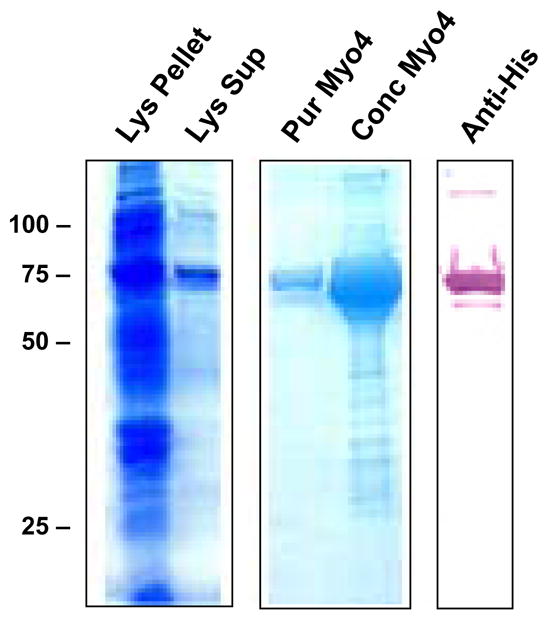
Myo4 is expressed as an abundant 68 kDa protein in E. coli found in both the pellet (Lys Pellet) and supernatant (Lys Sup) of a bacterial lysate. Myo4 is easily purified by nickel chromatography (Pur Myo4) and can be concentrated (Conc Myo4) to approximately 5 mg/ml for use in experiments. Myo4 can be detected by western blotting with anti-his antibodies that detect the C-terminal hexahistidine tag used for protein purification (Anti-His).
Figure 2. Immunization of A/J mice with cardiac myosin, purified Myo4 or Allen peptide induces similar cardiac hypertrophy, inflammation, and fibrosis 21 days post immunization.



A/J mice were immunized with 300 μg of cardiac myosin/CFA, 250 μg of Myo4/CFA, 50–150 nmols of Allen peptide/CFA, or PBS/CFA and sacrificed 21 days post immunization. (A) Cardiac hypertrophy was assessed by determination of the heart weight to body weight ratio. (B) Cardiac fibrosis and inflammation were assessed by staining with Masson’s trichrome or hematoxylin and eosin. Bars = 100 μm. (C) Quantitation of inflammation and fibrosis. Error bars represent standard error of the mean (SEM). n = 10 mice per group. * p ≤ 0.05 vs PBS control; ¥ p = 0.057 vs PBS control.
Serologic responses of Myo4-immunized animals
An isotype-specific ELISA was used to investigate the serologic response in Myo4-immunized animals (Figure 3). Sera obtained from mice at days 7, 14, and 21 post immunization were assessed for myosin-specific and Myo4-specific IgG, IgG1, and IgG2a. IgG1 responses directed against both Myo4 and cardiac myosin appeared to dominate IgG2a production at day 14 but, by day 21, Myo4-specific antibodies of all isotypes were present at extremely high titers. To further characterize the total IgG responses, western blot analysis of whole heart homogenate, purified cardiac myosin, Myo4, and BSA was performed using pools of sera from each group. Analysis of heart homogenate was performed in part to detect potential T cell-dependent antibody production indicative of “epitope spreading” to other cardiac antigens resulting from development of immunity to antigens released as a result of the initial myosin-specific autoimmune response (32–34). Myosin-immunized animals displayed strong IgG responses against a number of cardiac proteins ranging from 200 kDa to 37 kDa, as well as strong myosin and Myo4 responses (Figure 4). Many of the higher molecular mass proteins are likely myosin degradation products since they also appear in the lane containing purified cardiac myosin. Interestingly, Myo4-and Allen peptide-immunized animals displayed IgG responses against similar cardiac proteins, although the concentration of antigen-specific antibodies from the Allen peptide mice appears lower.
Figure 3. Myo4-immunization induces dominant anti-Myo4 and anti-myosin IgG1 antibody production.

Serum obtained from Myo4-immunized animals at days 7, 14, and 21 post immunization was analyzed by endpoint antibody titration for anti-myosin and anti-Myo4 antibodies by ELISA. In addition to total IgG production, serum was specifically analyzed for both IgG1 and IgG2a isoforms. Error bars represent SEM from at least 5 mice per group. Values without error bars were at the maximum detection capacity for the assay.
Figure 4. Antibodies from Myo4-immunized mice react with multiple cardiac proteins.

Sera from mice immunized with cardiac myosin, Myo4, Allen peptide, or PBS were obtained 21 days post-immunization and was pooled for western blot analysis. Heart homogenate (H), cardiac myosin (C), Myo4 (M), and BSA (B) were resolved by SDS-PAGE and either stained with GelCode Blue or transferred to nitrocellulose and incubated with 1:200 dilutions of sera. Secondary antibody incubations were performed using a 1:2000 dilution of alkaline phosphatase-conjugated goat anti-mouse IgG and blots were developed using 5-bromo-4-chloro-3-indolyl phosphate and p-nitroblue tetrazolium phosphate.
Myo4-immunized animals display delayed-type hypersensitivity to both Myo4 and cardiac myosin
We examined the delayed-type hypersensitivity (DTH) reponses in immunized mice to evaluate T cell immunity in Myo4-induced myocarditis (Figure 5). Not surprisingly, myosin-immunized mice displayed significant DTH responses to myosin and Myo4. However, a significant response was not generated to Allen peptide. As expected, Myo4-immunized mice displayed significant DTH responses to Myo4 and also developed strong DTH specific for myosin and the Allen peptide. Since the Allen peptide is not contained within the Myo4 fragment, this result indicates that intramolecular epitope spreading occurred within the myosin molecule. Finally, Allen peptide-immunized animals displayed significant DTH responses against peptide alone, although responses to Myo4 and myosin were elevated compared to PBS control.
Figure 5. Myo4-immunization induces strong delayed-type hypersensitivity responses specific for Myo4, purified cardiac myosin, and Allen peptide.

Cellular immune responses to Myo4, myosin, and Allen peptide were measured by a standard 24 hr ear swelling, delayed-type hypersensitivity (DTH) assay, in mice immunized with myosin, Myo4, Allen peptide or PBS. Right ears were injected with 10 μg of test antigen and left ears were injected with 10 μg BSA and net ear swelling (right minus left) was determined. Error bars, when shown, represent SEM. The absence of an error bar indicates that the SEM could not be calculated due to an insufficient number of measurements. * p ≤ 0.05.
Myo4-induced acute myocarditis is mediated by Th1 and Th17 immunity
To investigate the cytokine profile associated with acute myocarditis induced by immunization with Myo4, an ELISPOT assay was performed on splenocytes isolated at days 7, 10, 14, 17, and 21 post immunization (Figure 6A). Specific focus was placed on the secretion of IFN-γ, IL-4, IL-6 and IL-17 in order to characterize Th1, Th2, and Th17 immune responses, respectively. At all time points analyzed, the frequency of IFN-γ-producing cells was higher than that of IL-4 producing cells. This clearly illustrates a preponderance of Th1-type immunity over Th2 during the onset of EAM in the Myo4 model. Strong IL-6 and increasing IL-17 responses were observed over the time course. Although IL-4 production was slightly increased at day 17, the number of cells secreting this cytokine are low compared to either Th1- or Th17-associated cytokines throughout the developmental phase of disease, strongly suggesting these proinflammatory responses are critical to pathogenesis. To support this speculation, intracellular cytokine staining of splenocytes and draining lymph node cells from Myo4- and PBS-immunized animals was performed (Figure 6B). Following in vitro stimulation with Myo4, T cells were analyzed for the synthesis of IFN-γ, IL-4, and IL-17. As early as 7 days post immunization, Myo4-specific T cells, from both the spleen and lymph nodes, displayed IFN-γ production that became elevated in the lymph nodes at day 14 and dropped substantially by day 21. The opposite was observed in the case of splenocytes, where a slight dip in IFN-γ production was observed at day 14 followed by a significant increase by day 21. Similar results were observed with IL-17, where early production from both Myo4-specific splenocytes and lymphocytes was followed by an increase and subsequent decrease in production from lymphocytes and the direct opposite in splenocytes. IL-4 producing T cells were only detected in the lymph nodes at day 7 and decreased to undetectable levels at day 21. Taken together, ELISPOT and intracellular flow cytometric analysis clearly suggest important roles for both Th1 and Th17 types of immunity in the induction of Myo4-induced autoimmune myocarditis, with a negligible impact of Th2 immunity during the acute phase of disease.
Figure 6. Myo4-induced myocarditis is mediated by Th1 and Th17 immunity in the acute phase of disease.


(A) Myo4-specific production of IFN-γ (Th1), IL-4 (Th2), IL-6 and IL-17 (Th17) was analyzed using a standard ELISPOT assay. Following the isolation of splenocytes from Myo4-immunized animals at days 7, 10, 14, 17 and 21 days post immunization, cells were stimulated with purified Myo4 (10 μM) for 24 hrs for IFN-γ, IL-6, and IL-17 or 48 hrs for IL-4. (B) For a more detailed investigation of cytokine production, intracellular staining with antibodies specific for IFN-γ, IL-4 and IL-17 was performed. CD90.2+ T cells were analyzed from splenocyte (SP) and lymphocyte (LN) preparations from either Myo4- or PBS-immunized mice. All experiments were repeated at least three times. For ELISPOT analysis, error bars represent SEM.
Discussion
Myosin-induced EAM is a biphasic mouse model of inflammatory heart disease with an inflammatory phase mediated by CD4+ T cells (22) followed by a phase of repair and fibrosis (15, 19). Although much of the histopathology and serology of disease pathogenesis of myosin EAM has been characterized in A/J mice, the antigen-specific Th polarity of T cell responses has not been conclusively determined (21). The investigation of this important aspect of disease has been largely hindered due to the toxic nature of cardiac myosin to T cells in culture at concentrations higher than 10 μg/ml. Concentrations at or below this provides suboptimal T cell activation ex vivo, preventing in depth analysis of the critical role of myosin-specific immunity during EAM (22).
We found that a recombinant fragment of cardiac myosin, Myo4, is capable of inducing severe myocarditis and permits antigen-specific T cell analysis ex vivo. Myo4 spans amino acids K1074-Q1646 (Myo4) of cardiac myosin and can be readily purified as a histidine-tagged, 68 kDa recombinant protein. Myo4 is highly soluble, easily detoxified, and can be concentrated to 5 mg/ml for use in both immunizations and ex vivo assays. This recombinant antigen has other advantages over purified cardiac myosin and Allen peptide (21, 31). It takes large numbers of hearts and more than a week to purify enough cardiac myosin for a typical study (24), and the purified protein possesses small amounts of contaminants that potentially confound interpretation of antigen specificity. In contrast, Myo4 can be expressed, purified and concentrated within a matter of days. The Allen peptide is insoluble, precluding the accurate determination of its concentration, while Myo4 is soluble and is easy to quantitate. Although the present report is focused primarily on the acute phase of EAM, we have observed persistent inflammation and fibrosis at 60 days post immunization (data not shown), indicating that the Myo4 model closely mimics the original myosin-induced EAM model.
Myo4 induced development of cardiac hypertrophy, inflammation and fibrosis, all of which are associated with other EAM models (Figure 2). The myosin-specific humoral immunity showed a dominance of IgG1 over IgG2a (Figure 3), in agreement with published findings of IgG production in cardiac myosin-immunized A/J mice (21, 35) and BALB/c mice (36). Examination of the antibody responses to myosin, Myo4 and Allen peptide immunization showed the responses to be similar to heart homogenate, purified myosin and purified Myo4 (Figure 4). The only notable difference was that the IgG-specific antibody responses from Myo4-immunized animals displayed the strongest reactivity against the heart lysate, myosin, and Myo4. However, several interesting features of the humoral immune response in these models of disease were revealed in the analysis. First, myosin-immunized mice showed clear reactivity against multiple bands in the cardiac myosin preparation other than cardiac myosin. While some of these bands are likely degradation products of cardiac myosin, it is also possible that some are contaminating heart proteins that are isolated along with cardiac myosin during the purification process. If these lower molecular weight bands are, in fact, impurities, then the antibody recognition of non-myosin proteins in the heart lysate are likely not due to antigenic spreading, but rather due to specific immunity induced during immunization. However, the array of seroreactivity against heart lysate observed in animals immunized with Myo4 and Allen peptide, provides strong evidence for antigenic spreading in EAM. The contribution of immunity induced against non-myosin proteins to disease pathogenesis has not been assessed in murine-EAM but immunity to non-myosin cardiac proteins has been shown to induce myocarditis in other models (37). The importance of antigenic spreading to the propagation of cardiac autoimmunity and overall disease pathogenesis still need to be addressed. Other models of disease, of both infectious and autoimmune origins, have shown epitope spreading to be essential to disease pathogenesis (38).
To investigate cellular immunity in Myo4 EAM, a combination of DTH, ELISPOT of secreted cytokines, and intracellular cytokine staining/flow cytometry were employed. Myosin-immunized mice showed significant Myo4-specific DTH, albeit lower than that specific to whole cardiac myosin. DTH responses to the Allen peptide were not significant in these mice, suggesting that this peptide may not be the dominant epitope to which cellular immune responses are directed in myosin-induced disease. The lack of Allen peptide-specific DTH was surprising since this peptide was identified through a study of a hybridoma cells derived from myosin-immunized mice (31). While capable of inducing myocarditis, the Allen peptide may be simply initiating a cascade of autoimmune responses that diverge to other regions of cardiac myosin or other cardiac proteins during the progression of disease. The myosin-specific DTH in Myo4-immunized animals could be a result of the recognition of its own epitopes within whole cardiac myosin. However, Myo4-immunization has also produced DTH responses against other, non-overlapping fragments of cardiac myosin encompassing its head and tail regions (data not shown). These results, considered with the high but statistically insignificant (due to low n) DTH to Allen peptide, strongly suggest potential for both intra and intermolecular spreading in Myo4-induced EAM.
Our primary aim in developing the Myo4 EAM model was to be able to measure antigen-specific cytokine production. ELISPOT and intracellular cytokine staining both showed that the Th1, IFN-γ response, dominates the Th2, IL-4 response, during the acute phase of disease and that Th17 immunity, characterized by robust IL-6 and IL-17 production, also plays a potential role in the pathogenesis of Myo4 EAM (Figure 6). Only one published study focused on the role of Th1 and Th2 immune responses in EAM in A/J mice (21). In that report, cardiac myosin-specific IgG1, but not IgG2a, correlated with disease severity. Anti-IL-4 administration on days −1, 2, 6, 9, 12, 15, and 18 decreased disease severity and anti-IFN-γ treatment on days −1, 6, and 12 exacerbated disease, suggesting a critical role for Th2 immunity in EAM in A/J mice. The authors also concluded that IFN-γ plays a protective role in disease and IL-4 specifically has a disease promoting role. Our current results identify both similarities and differences between the two experimental models. Both models clearly display similar cardiac hypertrophy, inflammation, fibrosis (Figure 2), and the antibody reactivity is consistent with IgG1 dominance over IgG2a, although high titers of IgG2a antibodies are detected at day 21 in Myo4-immunized animals (Figure 4). Although the histopathology and serology are similar in both models, the disease initiating antigen is different and, coupled with the differences in the assays used to assess the contribution of Th immunity and the detection of antigen specific cytokine production, could explain the differences observed. For instance, animals immunized with cardiac myosin and stimulated ex vivo with 10 μg/ml myosin (a suboptimal concentration for stimulation (22)) produce mainly IL-4 and minimal IFN-γ (21). Here, we clearly demonstrate using ELISPOT and intracellular cytokine analysis of T cells from Myo4-immunized animals that IFN-γ responses dominate IL-4 (Figure 6). The methods used to assess the Th phenotype is consistent with several other models of EAM that were shown to be Th1-mediated and were assessed either through specific knock out animals or direct assessment of cytokine production from T cells (19, 20, 39).
Th17 immunity has been linked to a number of disease models, suggesting importance for this response in mediating autoimmune pathology (23, 40). Th17 cells are produced in Th1 and Th2 dominated immune responses and may serve to assist in promoting pathology in both cases, or solely mediate pathology in the absence of either (23). Our data show that Th1- and Th17-producing cells increase during the acute phase of EAM and could potentially both promote inflammation. Based on the cytokine profile of Th17 cells, it is reasonable to believe that they enhance the magnitude of the inflammatory infiltrate by promoting the recruitment of Th1 responder cells (monocyte, macrophages, and neutrophils), promoting their proliferation, and enhancing their survival. They can also enhance cytotoxity and upregulate iNOS expression. Interestingly, IL-17 is a potent neutrophil chemoattractant, in mice, and in the presence of an intact Th1-dominated response, macrophages are typically the dominant monocyte of the inflammatory infiltrate, as is seen in both EAM and EAE (16, 41). However, published reports and unpublished observations show that neutrophils dominate the inflammatory infiltrate in Th1-defective, Th17-driven processes (42, 43). Manipulations of the composition of an inflammatory infiltrate or the production of cytokines could very well indicate an alternative disease-inducing pathway rather than verify the natural pathway responsible for driving inflammation. Further analysis of the composition of the inflammatory infiltrate in naturally occurring EAM would have to be analyzed in order to verify this hypothesis. Technical advances that allow for the investigation of the contribution of Th1, Th17, and Th2 cells independently are necessary to clarify the pathogenic potential of each subtype in EAM.
Conclusion
We report the development of a recombinant model of experimental autoimmune myocarditis, induced by immunization with a 68 kDa fragment of cardiac myosin (Myo4), spanning amino acids K1074-Q1646 (Myo4). Myo4 is capable of inducing severe autoimmune myocarditis in A/J mice 21 days post immunization that is characterized by Th1 and Th17 immunity. Myo4 EAM has advantages over other models in ease of production of the immunogen and ability to measure antigen-specific functional immunity in ex vivo assays. The development of Myo4-EAM will allow for novel investigations into the contribution of epitope spreading to disease pathology as well as many studies that are currently underway to address prophylactic and therapeutic treatments in alleviating acute and chronic autoimmune myocarditis.
Acknowledgments
This work was supported in part by NIH grant R01 HL080692 (DME and MDD) and a Predoctoral Fellowship from the American Heart Association (KVH).
References
- 1.Pisani B, Taylor DO, Mason JW. Inflammatory myocardial diseases and cardiomyopathies. Am J Med. 1997;102:459–469. doi: 10.1016/S0002-9343(97)00331-8. [DOI] [PubMed] [Google Scholar]
- 2.Caforio AL, McKenna WJ. Recognition and optimum management of myocarditis. Drugs. 1996;52:515–525. doi: 10.2165/00003495-199652040-00005. [DOI] [PubMed] [Google Scholar]
- 3.Feldman AM, McNamara D. Myocarditis. N Engl J Med. 2000;343:1388–1398. doi: 10.1056/NEJM200011093431908. [DOI] [PubMed] [Google Scholar]
- 4.Anandasabapathy S, Frishman WH. Innovative drug treatments for viral and autoimmune myocarditis. J Clin Pharmacol. 1998;38:295–308. doi: 10.1002/j.1552-4604.1998.tb04428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Latif N, Baker CS, Dunn MJ, Rose ML, Brady P, Yacoub MH. Frequency and specificity of antiheart antibodies in patients with dilated cardiomyopathy detected using SDS-PAGE and western blotting. J Am Coll Cardiol. 1993;22:1378–1384. doi: 10.1016/0735-1097(93)90546-d. [DOI] [PubMed] [Google Scholar]
- 6.Konstadoulakis MM, Kroumbouzou H, Tsiamis E, Trikas A, Toutouzas P. Clinical significance of antibodies against tropomyosin, actin and myosin in patients with dilated cardiomyopathy. J Clin Lab Immunol. 1993;40:61–67. [PubMed] [Google Scholar]
- 7.Goldman JH, Keeling PJ, Warraich RS, Baig MK, Redwood SR, Dalla Libera L, Sanderson JE, Caforio AL, McKenna WJ. Autoimmunity to alpha myosin in a subset of patients with idiopathic dilated cardiomyopathy. Br Heart J. 1995;74:598–603. doi: 10.1136/hrt.74.6.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maisch B, Maisch S, Kochsiek K. Immune reactions in tuberculous and chronic constrictive pericarditis. Am J Cardiol. 1982;50:1007–1013. doi: 10.1016/0002-9149(82)90409-x. [DOI] [PubMed] [Google Scholar]
- 9.Aberer E, Brunner C, Suchanek G, Klade H, Barbour A, Stanek G, Lassmann H. Molecular mimicry and Lyme borreliosis: A shared antigenic determinant between Borrelia burgdorferi and human tissue. Ann Neurol. 1989;26:732–737. doi: 10.1002/ana.410260608. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan MH, Frengley JD. Autoimmunity to the heart in cardiac disease. Current concepts of the relation of autoimmunity to rheumatic fever, postcardiotomy and postinfarction syndromes and cardiomyopathies. Am J Cardiol. 1969;24:459–473. doi: 10.1016/0002-9149(69)90489-5. [DOI] [PubMed] [Google Scholar]
- 11.Cunha-Neto E, Coelho V, Guilherme L, Fiorelli A, Stolf N, Kalil J. Autoimmunity in Chagas’ disease: Identification of cardiac myosin-B13 Trypanosoma cruzi protein crossreactive T cell clones in heart lesions of a chronic Chagas’ cardiomyopathy patient. J Clin Invest. 1996;98:1709–1712. doi: 10.1172/JCI118969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neumann DA, Burek CL, Baughman KL, Rose NR, Herskowitz A. Circulating heart-reactive antibodies in patients with myocarditis or cardiomyopathy. J Am Coll Cardiol. 1990;16:839–846. doi: 10.1016/s0735-1097(10)80331-6. [DOI] [PubMed] [Google Scholar]
- 13.Guilherme L, Cunha-Neto E, Coelho V, Snitcowsky R, Pomerantzeff PM, Assis RV, Pedra F, Neumann J, Goldberg A, Patarroyo ME, Pilleggi F, Kalil J. Human heart-infiltrating t-cell clones from rheumatic heart disease patients recognize both streptococcal and cardiac proteins. Circulation. 1995;92:415–420. doi: 10.1161/01.cir.92.3.415. [DOI] [PubMed] [Google Scholar]
- 14.Cunha-Neto E, Kalil J. Heart-infiltrating and peripheral T cells in the pathogenesis of human chagas’ disease cardiomyopathy. Autoimmunity. 2001;34:187–192. doi: 10.3109/08916930109007383. [DOI] [PubMed] [Google Scholar]
- 15.Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol. 1987;139:3630–3636. [PubMed] [Google Scholar]
- 16.Pummerer C, Berger P, Fruhwirth M, Ofner C, Neu N. Cellular infiltrate, major histocompatibility antigen expression and immunopathogenic mechanisms in cardiac myosin-induced myocarditis. Lab Invest. 1991;65:538–547. [PubMed] [Google Scholar]
- 17.Neu N, Ploier B, Ofner C. Cardiac myosin-induced myocarditis: Heart autoantibodies are not involved in the induction of the disease. J Immunol. 1990;145:4094–4100. [PubMed] [Google Scholar]
- 18.Cunningham MW. Cardiac myosin and the Th1/Th2 paradigm in autoimmune myocarditis. Am J Pathol. 2001;159:5–12. doi: 10.1016/S0002-9440(10)61665-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuse K, Kodama M, Ito M, Okura Y, Kato K, Hanawa H, Aoki S, Aizawa Y. Polarity of helper T cell subsets represents disease nature and clinical course of experimental autoimmune myocarditis in rats. Clin Exp Immunol. 2003;134:403–408. doi: 10.1111/j.1365-2249.2003.02312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Afanasyeva M, Wang Y, Kaya Z, Stafford EA, Dohmen KM, Sadighi Akha AA, Rose NR. Interleukin-12 receptor/STAT4 signaling is required for the development of autoimmune myocarditis in mice by an interferon-gamma-independent pathway. Circulation. 2001;104:3145–3151. doi: 10.1161/hc5001.100629. [DOI] [PubMed] [Google Scholar]
- 21.Afanasyeva M, Wang Y, Kaya Z, Park S, Zilliox MJ, Schofield BH, Hill SL, Rose NR. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am J Pathol. 2001;159:193–203. doi: 10.1016/S0002-9440(10)61685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol. 1991;147:2141–2147. [PubMed] [Google Scholar]
- 23.Rangachari M, Mauermann N, Marty RR, Dirnhofer S, Kurrer MO, Komnenovic V, Penninger JM, Eriksson U. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–2019. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiverick KT, Thomas LL, Alpert NR. Purification of cardiac myosin: Application to hypertrophied myocardium. Biochim Biophys Acta. 1975;393:124–133. doi: 10.1016/0005-2795(75)90222-6. [DOI] [PubMed] [Google Scholar]
- 25.Leon JS, Daniels MD, Toriello KM, Wang K, Engman DM. A cardiac myosin-specific autoimmune response is induced by immunization with Trypanosoma cruzi proteins. Infect Immun. 2004;72:3410–3417. doi: 10.1128/IAI.72.6.3410-3417.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leon JS, Wang K, Engman DM. Captopril ameliorates myocarditis in acute experimental Chagas disease. Circulation. 2003;107:2264–2269. doi: 10.1161/01.CIR.0000062690.79456.D0. [DOI] [PubMed] [Google Scholar]
- 27.Leon JS, Wang K, Engman DM. Myosin autoimmunity is not essential for cardiac inflammation in acute chagas disease. J Immunol. 2003;171:4271–4277. doi: 10.4049/jimmunol.171.8.4271. [DOI] [PubMed] [Google Scholar]
- 28.Godsel LM, Leon JS, Wang K, Fornek JL, Molteni A, Engman DM. Captopril prevents experimental autoimmune myocarditis. J Immunol. 2003;171:346–352. doi: 10.4049/jimmunol.171.1.346. [DOI] [PubMed] [Google Scholar]
- 29.Godsel LM, Wang K, Schodin BA, Leon JS, Miller SD, Engman DM. Prevention of autoimmune myocarditis through the induction of antigen-specific peripheral immune tolerance. Circulation. 2001;103:1709–1714. doi: 10.1161/01.cir.103.12.1709. [DOI] [PubMed] [Google Scholar]
- 30.Minoprio P, el Cheikh MC, Murphy E, Hontebeyrie-Joskowicz M, Coffman R, Coutinho A, O’Garra A. Xid-associated resistance to experimental Chagas’ disease is IFN-gamma dependent. J Immunol. 1993;151:4200–4208. [PubMed] [Google Scholar]
- 31.Donermeyer DL, Beisel KW, Allen PM, Smith SC. Myocarditis-inducing epitope of myosin binds constitutively and stably to I-Ak on antigen-presenting cells in the heart. J Exp Med. 1995;182:1291–1300. doi: 10.1084/jem.182.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: Implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 33.Lawson CM. Evidence for mimicry by viral antigens in animal models of autoimmune disease including myocarditis. Cell Mol Life Sci. 2000;57:552–560. doi: 10.1007/PL00000717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fairweather D, Kaya Z, Shellam GR, Lawson CM, Rose NR. From infection to autoimmunity. J Autoimmun. 2001;16:175–186. doi: 10.1006/jaut.2000.0492. [DOI] [PubMed] [Google Scholar]
- 35.Afanasyeva M, Hill SL, Kaya Z, Rose NR, Wang Y. Characterization of murine autoimmune myocarditis induced by self and foreign cardiac myosin. J Am Coll Cardiol. 2000;36:1992–1999. doi: 10.1016/s0735-1097(00)00939-6. [DOI] [PubMed] [Google Scholar]
- 36.Liao L, Sindhwani R, Rojkind M, Factor S, Leinwand L, Diamond B. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J Exp Med. 1995;181:1123–1131. doi: 10.1084/jem.181.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto Y, Tsukada Y, Miyakoshi A, Sakuma H, Kohyama K. C protein-induced myocarditis and subsequent dilated cardiomyopathy: Rescue from death and prevention of dilated cardiomyopathy by chemokine receptor DNA therapy. J Immunol. 2004;173:3535–3541. doi: 10.4049/jimmunol.173.5.3535. [DOI] [PubMed] [Google Scholar]
- 38.Vanderlugt CL, Neville KL, Nikcevich KM, Eagar TN, Bluestone JA, Miller SD. Pathologic role and temporal appearance of newly emerging autoepitopes in relapsing experimental autoimmune encephalomyelitis. J Immunol. 2000;164:670–678. doi: 10.4049/jimmunol.164.2.670. [DOI] [PubMed] [Google Scholar]
- 39.Maier R, Miller S, Kurrer M, Krebs P, de Giuli R, Kremer M, Scandella E, Ludewig B. Quantification and characterization of myosin peptide-specific CD4+ T cells in autoimmune myocarditis. J Immunol Methods. 2005;304:117–125. doi: 10.1016/j.jim.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 40.Rohn TA, Jennings GT, Hernandez M, Grest P, Beck M, Zou Y, Kopf M, Bachmann MF. Vaccination against IL-17 suppresses autoimmune arthritis and encephalomyelitis. Eur J Immunol. 2006:2857–67. doi: 10.1002/eji.200636658. [DOI] [PubMed] [Google Scholar]
- 41.Dal Canto MC, Melvold RW, Kim BS, Miller SD. Two models of multiple sclerosis: Experimental allergic encephalomyelitis (EAE) and Theiler’s murine encephalomyelitis virus (TMEV) infection. A pathological and immunological comparison. Microsc Res Tech. 1995;32:215–229. doi: 10.1002/jemt.1070320305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ley K, Smith E, Stark MA. IL-17a-producing neutrophil-regulatory T lymphocytes. Immunol Res. 2006;34:229–242. doi: 10.1385/IR:34:3:229. [DOI] [PubMed] [Google Scholar]
- 43.Cruz A, Khader SA, Torrado E, Fraga A, Pearl JE, Pedrosa J, Cooper AM, Castro AG. Cutting edge: IFN-gamma regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. J Immunol. 2006;177:1416–1420. doi: 10.4049/jimmunol.177.3.1416. [DOI] [PubMed] [Google Scholar]