

Abstract
Human erythrocytes infected with the malaria parasite P. falciparum have increased permeabilities to many solutes. The plasmodial surface anion channel (PSAC) may mediate these changes. Despite good understanding of the biochemical and biophysical properties, the genetic basis of PSAC activity remains unknown. Functional polymorphisms in laboratory isolates and two mutants generated by in vitro selection implicate a parasite-encoded channel, though parasite-induced modifications of endogenous channels have not been formally excluded. Here, we identified stable differences in furosemide efficacy against PSAC activity induced by HB3 and 3D7A parasites. This difference was apparent in both single PSAC patch-clamp recordings and in sorbitol-mediated osmotic lysis measurements, confirming that Cl− and sorbitol are transported by a single channel type. Examination of 19 progeny from a genetic cross between HB3 and 3D7A revealed complex inheritance with some cloned progeny exhibiting furosemide affinities outside the range of parental values. Isolates generated by selfing of the 3D7A clone also exhibited altered furosemide affinities, implicating changes in one or more alleles during meiosis or passage through a primate host. PSAC may be encoded by multiple parasite genes (e.g. a multi-gene family or multiple genes that encode distinct channel subunits) or a single polymorphic gene under strong selective pressure.
Introduction
P. falciparum malaria is a leading cause of morbidity and mortality worldwide, reflecting the extraordinary success of this parasite in immune evasion and adaptation to intracellular survival. Intracellular growth, especially in the relatively inert human erythrocyte, imposes unique constraints on the eukaryotic malaria parasite. To overcome these constraints, the parasite remodels the erythrocyte by exporting many proteins to the host cytosol and membrane, proliferating membranous structures and electron dense protrusions (“knobs”) under the host membrane, and increasing host membrane permeability to various solutes including anions, organic cations, amino acids, purines, and sugars (Ginsburg et al., 1985; Upston and Gero, 1995; Kirk et al., 1994; Staines et al., 2000; Desai et al., 2000).
Patch-clamp studies have established that the increased host membrane permeability results from one or more ion channels. One candidate channel, the plasmodial surface anion channel (PSAC), has been extensively studied with both single-channel and whole-cell recordings. Single PSAC recordings in the presence of both specific and non-specific antagonists has identified pharmacological effects that correlate with both organic solute tracer flux and osmotic lysis measurements (Desai et al., 2000; Cohn et al., 2003; Alkhalil et al., 2004; Desai et al., 2005; Kang et al., 2005). Furthermore, a blasticidin S resistant mutant exhibited changes in PSAC gating and conductance that were linked to reduced organic solute uptake (Hill et al., 2007). Each of these studies suggests that PSAC accounts for the increased uptake of organic and inorganic small solutes after infection. However, a number of other ion channels have been observed in some electrophysiological studies (Staines et al., 2007), raising questions about relative contributions to overall infected erythrocyte permeability and the biological role of each putative channel.
A major stumbling block to addressing these and other questions has been the identification of the gene(s) encoding these channels. Although the other proposed channels are thought to result from activation of quiescent human channels, PSAC appears to be a parasite-encoded channel based on in vitro selection of mutants (Hill et al., 2007; Lisk et al., 2008) and functional polymorphisms when geographically divergent parasite isolates are cultured in erythrocytes from a single human donor (Alkhalil et al., 2004). In either case, cloning of the responsible genes will permit heterologous expression and definitive tallying of permeant solutes after isolation from other erythrocyte-associated transport mechanisms. If the responsible gene(s) are encoded by the parasite, their identification should also help uncover the channel's biological role, its trafficking to the host membrane, and regulation of its expression.
A number of studies based on biochemical or informatic approaches have been undertaken to identify the responsible gene(s) (Breuer et al., 1987; Desai et al., 2005; Martin et al., 2005; Baumeister et al., 2006), but a compelling candidate gene remains elusive.
Linkage analysis in P. falciparum genetic crosses represents a powerful alternative approach because it does not require assumptions about the origin or number of ion channels present on the host membrane. This approach has not been previously used to study the increased erythrocyte permeability after infection, presumably because reproducible phenotypic differences between the parents of available genetic crosses were unknown. We have identified a stable and quantitative difference in PSAC activity between the HB3 and 3D7A isolates, which are the parents of the first available P. falciparum genetic cross(Walliker et al., 1987). Quantitative trait locus analysis of 19 progeny from this cross indicates that inheritance of furosemide affinity is complex. While specific genomic loci with significant probabilities were not identified, these findings further implicate parasite genetic elements in induction of PSAC activity and may point to an essential role for this channel in intracellular parasite survival.
Results
Difference in furosemide effectiveness against PSAC in HB3- and 3D7A-infected erythrocytes
Functional polymorphisms in PSAC gating between two laboratory isolates represented the first published evidence supporting a role of parasite genetic elements in the increased permeability of infected erythrocytes (Alkhalil et al., 2004). Identification of similar differences in channel behavior between parental isolates from a P. falciparum genetic cross would allow the inheritance in recombinant progeny to be examined and may lead to mapping of PSAC's gene(s). We therefore began the present study with patch-clamp surveys of erythrocytes infected with each of the 5 parental isolates (HB3, 3D7, Dd2, 7G8, and GB4) from the 3 available P. falciparum genetic crosses (Walliker et al., 1987; Wellems et al., 1990; Hayton et al., 2008).
We used single-channel and whole-cell recordings of infected cells to examine PSAC gating properties, voltage-dependence, single channel conductance, copy number/cell, and pharmacology. As identified previously in comparisons of currents induced by Indo 1 and 7G8 isolates (Alkhalil et al., 2004), differences in voltage-dependence were apparent in pairwise comparisons of whole-cell recordings from some of these isolates; however, we did not attempt to evaluate their statistical significance because these differences had modest signal-to-noise ratios, making them difficult to track in cross progeny (data not shown).
In contrast, examination of PSAC pharmacology revealed an unambiguous difference between channels induced by the HB3 and 3D7A isolates. Figure 1 demonstrates this difference with single channel recordings in the presence of furosemide, a relatively well characterized inhibitor of PSAC activity (Alkhalil et al., 2004; Desai, 2005; Desai et al., 2005). In the absence of inhibitors, gating (channel opening and closing behavior) and single channel Cl− conductance (net ion flux/unit time) did not differ between these isolates (Fig. 1A, typical of n = 10-12 patches for each isolate). However, addition of furosemide at a nonsaturating concentration revealed significantly greater inhibition of HB3-induced channels (Fig. 1B). This difference was reproducible in single channel patches and was even more obvious in patches containing two functional PSAC molecules (Fig. 1C). The accentuated difference in two-channel patches indicates that each channel molecule opens and closes independently and that both exhibit the same isolate-specific furosemide affinity.
Fig. 1.

Furosemide is more effective against PSAC on HB3- than on 3D7A-infected cells (left and right columns, respectively). Graphic shows single channel recordings in the absence of inhibitors (A) or with 25 μM furosemide present in both bath and pipette compartments (B). 25 μM furosemide was also symmetrically present in recordings from two-channel patches (C). Closed channel levels are indicated with dashes to either side of each trace. Currents shown reflect Cl− flux. For all traces, the membrane potential was clamped at −100 mV and the scale bars represent 200 ms (horizontal)/ 3 pA (vertical). The difference in furosemide efficacy shown is typical of > 10 channel molecules recorded from each isolate.
Previous studies suggest furosemide interacts with PSAC at a single site on the extracellular face of the channel to produce allosteric inhibition of solute transport (Desai et al., 2005; Desai, 2005). Greater inhibitory efficacy against HB3-induced channels may result either from improved furosemide access to this site or prolonged residence there. We distinguished these possibilities by tallying the durations of openings and closings in single channel recordings obtained with 25 μM furosemide in bath and pipette compartments. All points histograms of opening burst durations tallied from recordings on HB3 and 3D7A yielded identical profiles (Fig. 2A). Moreover, these histograms were well fitted by a single exponentially decaying probability density function (smooth curve). These observations can be conservatively explained by a single on rate constant that governs furosemide access, binding, and inhibition of the channel; this rate constant does not appear to vary between these two isolates' channels. In contrast, examination of closed channel durations revealed a marked difference between the two isolates, with long closings (> 10 ms duration) significantly more frequent in HB3-induced channels (Fig. 2B). These longer closing durations reflect slower furosemide unbinding from PSAC in HB3-infected cells and can account for the greater inhibitory efficacy against this isolate's channels.
Fig. 2.

Different furosemide block durations account for the difference in efficacy. (A) Histogram of open burst durations in the presence of 25 μM furosemide for HB3 and 3D7A channels (black and red lines, respectively). Bursts were defined as described in the Experimental procedures. The histogram is presented on a square root logarithmic plot to reveal populations of events having single exponential distributions (Sigworth and Sine, 1987). The smooth line represents the best fit of the data to Equation 1, consistent with a single on rate constant for furosemide block. Notice that channels from the two parasites have identical distributions. (B) Histogram of closed durations with 25 μM furosemide for HB3 and 3D7A channels (black and red lines, respectively), plotted on a square root-linear plot to accentuate long closings, which usually reflect furosemide block. Notice that block events of durations > 10 ms are more abundant in recordings from HB3 parasites.
As individual closed and blocked channel events cannot be distinguished experimentally, the histograms in Figure 2B include both furosemide-mediated block events and intrinsic channel closings. Intrinsic closings result from conformational changes within the channel protein and reflect molecular random kinetic energy as they occur without external energy. Gating analyses indicate that PSAC has at least three closed states when inhibitors are absent and that furosemide induces a single population of significantly longer blocked events (Desai, 2005). As utilized in this previous study, we fitted the closed histograms for HB3 and 3D7A to a probability density function that incorporates the three intrinsic closed states and a single population of furosemide block durations. This approach yielded estimates for mean furosemide block durations of 5.1 ms for 3D7A and 14.8 ms for HB3, suggestive of a 3-fold higher affinity for PSAC activity in HB3-infected cells.
Correlated effect on sorbitol uptake and development of a quantitative trait for linkage studies
Because single channel recordings from infected erythrocytes are arduous and provide only low throughput, we examined PSAC activity from these two isolates in a technically straightforward assay based on osmotic swelling and selective lysis of infected erythrocytes in sorbitol solutions (Wagner et al., 2003). Furosemide affinity estimates obtained by this method match those from both single channel and whole-cell recordings on infected erythrocytes (Alkhalil et al., 2004). In these experiments, HB3 and 3D7A-infected cells underwent lysis with indistinguishable kinetics when inhibitors were not added (left most trace in each panel, Fig. 3A), suggesting similar channel copy numbers and unitary sorbitol permeabilities in these isolates. When furosemide was added, however, osmotic lysis was more effectively slowed for HB3- than 3D7A-infected cells at each concentration tested; for each dose response experiment, K0.5, the furosemide concentration that doubled the lysis halftime, was determined by fitting to Equation 2. Figure 3B shows the tallied dose responses for both isolates. To evaluate reproducibility and stability of these estimates in continuous cultures with different blood donors, we performed identical osmotic lysis experiments with furosemide over a 5 month period and found stable differences between cultures of HB3 and 3D7A isolates with K0.5 values of 5.1 ± 0.3 and 15.8 ± 0.7 μM respectively (P < 10−10, Student's t test; Fig. 3C). Thus, both single channel recordings and osmotic lysis experiments implicate a 3-fold higher furosemide affinity for inhibition of PSAC activity from HB3- than 3D7A-infected cells.
Fig. 3.

Transmittance assay for sorbitol uptake via PSAC confirms and quantifies difference in furosemide affinity. (A) Osmotic lysis kinetics in sorbitol with 0, 5, 10, 20, and 200 μM furosemide (left to right traces in each panel). Notice that while HB3 and 3D7A exhibit similar inhibitor-free lysis kinetics, each furosemide concentration slows lysis of HB3-infected cells to a greater extent, indicating more effective inhibition of sorbitol uptake. (B) Dose responses for inhibition of sorbitol-induced osmotic lysis by furosemide for HB3 and 3D7 (circles and triangles, respectively). Each symbol represents the mean ± S.E.M. of up to 20 measurements. Solid lines represent the best fit for each isolate to Equation 2. (C) Stable and reproducible difference in furosemide K0.5 for HB3 and 3D7A cultures (circles and triangles, respectively). Each symbol represents an experiment identical to that in panel A, performed on a separate day. Trials are shown in chronological order from left to right over a 146 day period; because 3D7A and HB3 were run independently of each other, the abscissa does not have numeric labels (n = 16 and 19, respectively).
Complex inheritance in a genetic cross
We then utilized the osmotic lysis assay to examine inheritance of furosemide affinity in 19 independent progeny from the HB3 × 3D7A cross. For each progeny clone, multiple furosemide dose responses were performed and used to determine mean K0.5 values as displayed in Figure 4A. These studies revealed a continuum of furosemide affinities in the progeny of this cross, suggesting complex inheritance of PSAC phenotypes. Some progeny clones exhibited furosemide affinities significantly outside the range of parental values. For example, clone X33 had a furosemide K0.5 of 2.3 ± 0.1 μM (n = 9), a value significantly lower than that of HB3 (P = 10−6). Clones with intermediate affinities were also identified: the K0.5 estimate of XP9 was also shown to be significantly different from those of both parents (11.3 ± 0.4, n = 12, P < 10−4 in both pairwise comparisons).
Fig. 4.
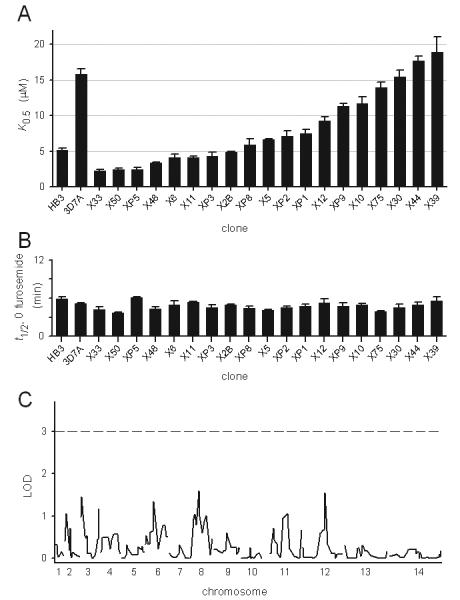
Complex inheritance of furosemide affinity in the progeny of the HB3-3D7A genetic cross. (A) Mean ± S.E.M. furosemide K0.5 for each progeny clone, determined from up to 12 independent experiments and displayed according to decreasing PSAC affinity. The parental values are included on the left to facilitate comparison. (B) Mean ± S.E.M. halftimes for lysis in sorbitol solution in the absence of PSAC inhibitors for each isolate, ordered as in (A). This parameter is inversely related to functional channel copy number/cell. (C) LOD scores for each chromosomal locus determined using QTL analysis. A conservative threshold for significance (LOD = 3) is marked with a dashed horizontal line.
We also tallied the inhibitor-free osmotic lysis halftimes for each of these isolates (Fig. 4B). The lysis halftime is inversely proportional to both the average rate of sorbitol uptake for a population of cells and the mean channel copy number/cell (Wagner et al., 2003). Lysis halftimes varied to a negligible extent amongst the progeny, suggesting conserved levels of PSAC activity induced by each isolate. There was also no correlation with furosemide K0.5, excluding systematic errors in our estimated K0.5 values.
This pattern of inheritance may be explained by the involvement of multiple genes in determination of furosemide affinity. In such cases, it is possible to identify some or all of the contributing genetic elements with quantitative trait locus (QTL) mapping (Lander and Botstein, 1989; Sen and Churchill, 2001). To this end, we tallied each progeny clone's measured K0.5 value and chromosome-wide inheritance pattern for 274 microsatellite markers that differ between the parental isolates. Linkage analysis between each genetic locus and the K0.5 values was then performed to identify loci that influence furosemide affinity. This statistical analysis of measured K0.5 values did not identify significant loci (Fig. 4C). While our analysis shows that we had an adequate number of genetic markers for uniform coverage of the parasite genome, a major limitation with the HB3-3D7A cross is that only 19 independent progeny were available for this study. It is possible that one or more significant genetic loci governing furosemide affinity may be identified if additional independent progeny become available for phenotyping and incorporation into the QTL algorithm.
Changes in furosemide affinity upon selfing
In light of the observed complex inheritance, we next used sorbitol uptake experiments to examine furosemide K0.5 for PSAC induced by the 3D7B parasite, which was generated in a self-mating experiment performed with the 3D7A isolate at the time of the original cross (Walliker et al., 1987). The 3D7B parasite should be genetically identical to 3D7A except for changes that may be specifically acquired during meiosis or passage through the chimpanzee. Interestingly, we found that 3D7B had a furosemide K0.5 significantly lower than that of the parental 3D7A (Fig. 5A; K0.5 = 6.5 ± 0.6 μM, n = 9, P < 10−7). This observation contrasts with the stability of parasite drug sensitivities after selfing and in vivo passage. Similar, but more modest, differences were observed between HB3 and its selfed progeny (not shown).
Fig. 5.

Changes observed upon selfing. (A) Furosemide K0.5 values for the parental 3D7A isolate and for 3D7B, the uncloned parasite generated by selfing of 3D7A. (B) Furosemide K0.5 values for limiting dilution clones obtained from 3D7A or 3D7B cultures. Note that each 3D7A clone has the same K0.5 as the original 3D7A isolate in panel A, but that cloning of 3D7B reveals a mixture of PSAC phenotypes. (C) DNA fingerprint profiles of indicated parasites. The positions of major peaks and their relative amplitudes corresponds to sizes and PCR amplification efficiency for individual rif genes. Note that the profile for HB3 differs from that for 3D7A and 3D7B parasites. The similar fingerprints for 3D7A and each 3D7B clone excludes significant contamination of their cultures by unrelated parasites and reveals a lack of global genomic changes upon selfing.
Because the 3D7B parasite is expected to be genetically identical to the cloned 3D7A isolate, the original study did not subject this parasite to limiting dilution cloning (Walliker et al., 1987). Moreover, we recognized that an uninteresting explanation for altered phenotype after selfing is that the 3D7A parental isolate might not be clonal for PSAC-furosemide affinity, as has been observed for another phenotype (Eksi et al., 2005). We therefore used limiting dilution of both 3D7A and 3D7B cultures to generate a collection of cloned parental and selfed isolates. PSAC-furosemide affinity was then determined for several clones (Fig. 5B). Each clone derived from 3D7A had a K0.5 essentially identical to that of the parental isolate, confirming that 3D7A is clonal for furosemide affinity and that this phenotype is stable in long-term in vitro culture. In contrast, clones generated from the selfed 3D7B culture had distinct K0.5 values differing from that of the 3D7B pool. One clone, 3D7B-34G2, resembled the 3D7A parental isolate; the other two clones, 3D7B-35B10 and 3D7B-35C9, had values similar to one another but different from both 3D7A and the selfed 3D7B pool. This collection of measurements suggests that selfing yielded a limited repertoire of changes in the responsible gene(s). These changes may arise during meiosis or may reflect selection of rare variants by immune pressure during passage through a chimpanzee. As a further test of the extent of changes occurring after selfing, we performed DNA fingerprinting analysis of each clone. The amplification profile for rif genes for each of the 3D7B clones was very similar to that of 3D7A, but was distinct from the unrelated HB3 parental isolate (Fig. 5C). This analysis excluded contamination by unrelated parasite isolates maintained in our laboratory and confirmed that there are not global changes in the 3D7A genome after selfing.
Discussion
Although the increased permeability of erythrocytes infected with malaria parasites has been known and universally accepted for decades (Overman, 1948; Homewood and Neame, 1974; Ginsburg et al., 1985; Kirk et al., 1994), the precise molecular mechanism(s) remain debated (Staines et al., 2007). Either PSAC or upregulated human channels identified in some electrophysiological surveys may mediate the uptake of each organic or inorganic solute with increased permeability. Our study represents the first use of genetic linkage analysis to characterize this transport and provides new insights into possible mechanisms.
One line of evidence that supports transport via PSAC has been the quantitatively concordant pharmacology for each solute's transport: furosemide, phloridzin, and dantrolene each inhibit Cl− flux in single PSAC patch-clamp at concentrations matching those required to inhibit organic solute uptake using osmotic lysis and tracer flux (Alkhalil et al., 2004; Desai, 2005; Lisk et al., 2006). Although quantitatively compelling, these correlations have not been universally accepted because some workers consider these small molecule antagonists to be highly nonspecific. The present findings directly address this concern because isolate-specific differences in inhibitory K0.5 implicate polymorphisms in a defined pocket on the channel that interacts with furosemide. They are not consistent with amorphous inhibition by furosemide, such as might occur with hydrophobic small molecules that partition into lipid bilayers.
The 3-fold greater efficacy of furosemide for HB3-induced channels than for 3D7A channels was quantitatively reproduced both in single channel studies that measure Cl− flux and in sorbitol-mediated osmotic lysis experiments. This observation strongly supports uptake of Cl− and sorbitol primarily via PSAC and is consistent with previous studies that have examined furosemide's effects on the channel (Alkhalil et al., 2004; Desai, 2005; Desai et al., 2005). Analyses of single PSAC recordings in the presence of multiple furosemide concentrations have determined that this inhibitor interacts with an extracellular site on the channel protein and that inhibition of solute transport does not result from occlusion of the channel pore by furosemide. Additional evidence for PSAC's role in the uptake of diverse solutes has come from in vitro selection of two distinct PSAC mutants with altered selectivity and pharmacology (Hill et al., 2007; Lisk et al., 2008). The concordant differences in furosemide effectiveness between the two parental isolates also put to rest concerns about use of hypertonic solutions required for single PSAC recordings (Bouyer et al., 2007): isolate-specific differences in channel behavior can be quantitatively reproduced in both molar salt solutions used in patch-clamp studies and in isotonic sorbitol, a solution with lower than physiological ionic strength.
Heritable differences in a defined extracellular pocket on this channel provide new evidence favoring a parasite-encoded protein over human ion channels activated by the parasite. In the scenario of a parasite-encoded channel, the observed difference in furosemide affinity may reflect one or more nonsynonymous single nucleotide polymorphisms (SNPs) in the channel gene; given that the inhibitor-free activity of HB3 and 3D7A channels are indistinguishable, this polymorphism is likely to be neutral to in vivo selective pressure. In contrast, up-regulated human channels are unlikely to exhibit parasite isolate-specific differences in furosemide binding. One of these alternative models proposes that up-regulation of human channels is mediated by parasite-encoded protein kinases (Bouyer et al., 2007). While it is formally possible that heritable polymorphisms in a parasite protein kinase may differentially phosphorylate and activate a putative human channel, it is hard to envision how such differences, occurring at the channel's cytosolic face, could yield a difference in the extracellular furosemide binding pocket without producing observable changes in inhibitor-free channel behavior. Nonspecific mechanisms such as parasite-mediated oxidative stress (Huber et al., 2002) have even greater difficulty in accounting for our findings because differences in isolate-specific generation of oxidative radicals are unknown. The observed conservation of inhibitor-free sorbitol permeability (a measure of channel copy number) amongst the progeny clones is also inconsistent with simple induction via oxidation or other non-specific modulators: variations in modulator concentrations amongst these clones sufficient to yield differing furosemide affinities would be expected to produce differences in functional channel copy number as well.
If our data indeed reflect heritable polymorphisms in one or more parasite genes, linkage analysis represents a novel approach to identification of the channel's gene or genes. An important advantage of this approach for the study of PSAC is that it does not require informatic assumptions such as orthology with known ion channels in other organisms or the number of transmembrane domains encoded. To successfully identify genetic loci, linkage analysis of inheritance in a cross requires a robust and reproducible phenotypic difference between the parents, a sufficient number of progeny carrying independent recombination events, and an adequate collection of genome-wide markers for tracking the inheritance pattern of each chromosomal locus in independent progeny. By comparison to other quantitative traits tracked in previous studies (Wellems et al., 1990; Rohrbach et al., 2006), the magnitude and reproducibility of the difference in PSAC-furosemide affinity between the parental isolates appear to be adequate for genetic mapping. We were, however, limited by the number of independent progeny cloned to date from the HB3-3D7A cross.
In spite of this limitation, transport studies of progeny from HB3-3D7A mating and from selfing of 3D7A demonstrate that inheritance of PSAC phenotypes is not simply Mendelian. This finding is surprising in light of single channel patch-clamp studies that implicate a single functional unit responsible for increased permeability after infection. Moreover, because blood stage parasites are haploid, the progeny in our experiments are presumed to carry only the allele from one parent or the other at each genomic locus. Explanations for inheritance of PSAC phenotypes will need to account for 1) the stability of furosemide affinity under in vitro parasite culture (Fig. 3C), 2) the detection of at least two distinct non-parental phenotypes in the progeny clones (e.g. the low furosemide K0.5 of X33 and the intermediate value of XP9), and 3) the altered phenotype after selfing of the 3D7A isolate (Fig. 5).
One possible explanation is that the furosemide binding site on PSAC is encoded by two or more genes encoding distinct subunits of a functional ion channel complex. This possibility could also account for changes after selfing of 3D7A if the subunits are encoded by paralogous genes capable of meiotic gene conversion (Szostak et al., 1983). Indeed, gene conversion is an established mechanism of non-Mendelian inheritance in microbial genetic crosses (Bowring and Catcheside, 1996; Freitas-Junior et al., 2000). This explanation for PSAC inheritance is particularly appealing in light of parallels to a study of individual alleles of the var gene family of P. falciparum (Freitas-Junior et al., 2000), which also identified stability of parental alleles in asexual stage parasite cultures, non-parental alleles in genetic cross progeny, and changes after selfing. As recognized by those workers, the gene conversion events that drive this diversity may occur significantly more frequently during meiosis than during asexual replication, accounting for stability of phenotypes in long term asexual cultures.
Another possible explanation is that PSAC activity is encoded by individual members of a multigene family that exhibits mono-allelic expression and switching, also documented for var genes and other gene families in this parasite (Scherf et al., 1998; Lavazec et al., 2007). To account for our findings, some copies of this gene would need to carry polymorphisms that alter the structure of the furosemide binding site. Although switching between alleles might be expected to yield gradually drifting HB3 and 3D7A furosemide affinities in continuous culture (∼ 70 generations in Fig. 3C), two observations suggest that the observed stability can be reconciled with a multigene family model for PSAC. First, similar inhibitor-free transport rates for all isolates suggest that domains critical for solute translocation are highly conserved between members of this putative gene family. Then, in vitro culture conditions are unlikely to favor one PSAC allele over others, rendering detection of cells that have undergone gene switching difficult in population osmotic lysis experiments. Second, gene switching rates appear to vary over a broad range and may be affected by a number of variables (Lavazec et al., 2007). In light of these considerations, it is quite possible that various progeny clones acquired and maintained unique alleles through low level gene switching and immune selective pressure during in vivo passage. They may then have become apparent through limiting dilution cloning at the time of the original HB3-3D7A genetic cross. A gene switching model is also attractive in light of relatively rapid changes in phenotype observed upon removal and reapplication of selective pressure to a blasticidin S resistant PSAC mutant (Hill et al., 2007).
Another explanation, a single gene susceptible to frequent mutations, may also yield complex, heritable differences in the furosemide binding pocket. Any one of these possible explanations, if verified in future studies, may reflect strategies used to reduce immune recognition of the channel and sustain parasite survival in the bloodstream.
Consideration of these possible explanations should help guide the evaluation of candidate genes as they are identified. Based on functional transport studies, candidate genes should also be highly conserved in other plasmodial species (Lisk and Desai, 2005) and may be absent from other apicomplexa (Alkhalil et al., 2007). Our informatic analyses of the P. falciparum genome sequence have not revealed unambiguous orthologs of ClC, CFTR, Ca++-activated, or ligand-gated chloride channel genes present in other organisms. This, combined with multiple studies documenting PSAC's unusual functional properties, strongly suggest one or more novel genes that are unique to plasmodia.
Experimental procedures
Electrophysiology
Single channel patch-clamp recordings were obtained as described previously (Alkhalil et al., 2004). Briefly, pipettes were fabricated from quartz glass to tip diameters < 0.5 μm and resistances of 1-4 MΩ. They were then filled with (in mM): 1000 choline-Cl, 115 NaCl, 10 MgCl2, 5 CaCl2, 20 Na-HEPES, pH 7.4. This hypertonic recording solution facilitates detection of small conductance ion channels by providing higher concentrations of permeant ions and by reducing noise due to series resistances in the pipette and ground. Furosemide was added from concentrated DMSO stock solutions where indicated. The same solution was present in the bath compartment in all experiments. High resistance seals (> 100 GΩ) were obtained on the infected erythrocyte membrane by maintaining protein-free filtered solutions and minimizing perfusion and other sources of vibration (Lisk and Desai, 2006). Recordings were acquired with an AxoPatch 200B amplifier (Molecular Devices), filtered at 5 kHz with an 8 pole Bessel filter (Frequency Devices), and digitized at 100 kHz (Digidata 1322A, Molecular Devices). Under these conditions, we detect negligible currents on uninfected cells and only PSAC activity on infected cells.
Dwell time distributions were obtained using code that detects mid-threshold crossings, uses linear interpolation of adjacent sample values to estimate the time of threshold crossing (Sigworth and Sine, 1987), and corrects for a Gaussian filter risetime of 66.4 μs as described (Colquhoun and Sigworth, 1983). These distributions were tallied from a minimum of 90 s of single channel recording for each parasite isolate. Histogram ordinate values were normalized to the percent of the total number of events detected under each condition and are displayed on a square root-logarithmic plot to identify open duration time constants (Sigworth and Sine, 1987) or on a square root-linear plot to detect differences in furosemide-induced block durations. Details of the algorithm were described previously (Desai, 2005). We defined bursts of openings as clusters of channel activity flanked by closed events of ≥ 3 ms, a value that captures nearly all furosemide-induced blocked events. Fitting of the distribution of these open burst durations to the probability density function expected for a single reversible transition was then performed with:
| (1) |
where b and c are constants and t is the duration of individual channel events.
Osmotic lysis kinetics
The kinetics of infected RBC osmotic lysis in sorbitol solutions was followed as described previously (Wagner et al., 2003). In brief, trophozoite-infected RBCs were grown by standard culture methods, enriched to 95-99% by percoll/sorbitol separation, washed in 150 mM NaCl, 20 mM Na-phosphate, pH 7.5, and resuspended to 0.05% hematocrit in 280 mM sorbitol, 20 mM Na-HEPES, 0.1 mg/ml BSA, pH 7.4 at 37 °C. The transmittance of 700 nm light through this cell suspension was then continuously followed as a marker of osmotic swelling and lysis. Lysis kinetics with each isolate were determined with furosemide concentrations between 0 and 200 μM; these dose responses were fitted with
| (2) |
where x is the furosemide concentration, K0.5 is a measure of furosemide affinity, and α is a constant. This equation is based on a 1:1 stoichiometry for channel-inhibitor interactions (Wagner et al., 2003) and produced adequate fits for all isolates.
Quantitative trait locus (QTL) analysis
274 polymorphic microsatellite markers distributed across the 14 chromosomes of P. falciparum were identified and used to genotype progeny clones. QTL analysis was carried out using these genotype data and mean furosemide K0.5 values determined in osmotic lysis experiments. Computational analysis was carried out using R/qtl software (available at http://www.rqtl.org/) as described (Broman et al., 2003). Because erythrocytic stages of this parasite are haploid, the analysis was analogous to that for recombinant inbred crosses. Similar results were obtained with the expectation minimization, Haley-Knott regression, and multiple imputation algorithms described in the software package.
Limiting dilution cloning and DNA fingerprinting
Parasite cultures, maintained by standard methods, were subjected to limiting dilution cloning as described previously (Rosario, 1981). Fingerprinting of clones was performed by PCR amplification of a polymorphic microsatellite within the rif multigene family as described (Su et al., 1998). The results are displayed as fluorescence intensity profiles in arbitrary units against amplicon size, as generated by analysis on an ABI 3100 automated sequencer (Applied Biosystems).
Acknowledgments
We thank Tsione Solomon for help with early experiments and Jianbing Mu for assistance with DNA fingerprinting. This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases and by the Medicines for Malaria Venture (MMV).
References
- Alkhalil A, Cohn JV, Wagner MA, Cabrera JS, Rajapandi T, Desai SA. Plasmodium falciparum likely encodes the principal anion channel on infected human erythrocytes. Blood. 2004;104:4279–4286. doi: 10.1182/blood-2004-05-2047. [DOI] [PubMed] [Google Scholar]
- Alkhalil A, Hill DA, Desai SA. Babesia and plasmodia increase host erythrocyte permeability through distinct mechanisms. Cell Microbiol. 2007;9:851–860. doi: 10.1111/j.1462-5822.2006.00834.x. [DOI] [PubMed] [Google Scholar]
- Baumeister S, Winterberg M, Duranton C, Huber SM, Lang F, Kirk K, Lingelbach K. Evidence for the involvement of Plasmodium falciparum proteins in the formation of new permeability pathways in the erythrocyte membrane. Mol Microbiol. 2006;60:493–504. doi: 10.1111/j.1365-2958.2006.05112.x. [DOI] [PubMed] [Google Scholar]
- Bouyer G, Egee S, Thomas SL. Toward a unifying model of malaria-induced channel activity. Proc Natl Acad Sci USA. 2007;104:11044–11049. doi: 10.1073/pnas.0704582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowring FJ, Catcheside DE. Gene conversion alone accounts for more than 90% of recombination events at the am locus of Neurospora crassa. Genetics. 1996;143:129–136. doi: 10.1093/genetics/143.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer WV, Kutner S, Sylphen J, Ginsburg H, Cabantchik ZI. Covalent modification of the permeability pathways induced in the human erythrocyte membrane by the malarial parasite Plasmodium falciparum. J Cell Physiol. 1987;133:55–63. doi: 10.1002/jcp.1041330107. [DOI] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- Cohn JV, Alkhalil A, Wagner MA, Rajapandi T, Desai SA. Extracellular lysines on the plasmodial surface anion channel involved in Na+ exclusion. Mol Biochem Parasitol. 2003;132:27–34. doi: 10.1016/j.molbiopara.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth FJ. Fitting and statistical analysis of single-channel records. In: Sakmann B, Neher E, editors. Single-channel recording. Plenum Press; New York: 1983. pp. 191–263. [Google Scholar]
- Desai SA. Open and closed states of the plasmodial surface anion channel. Nanomedicine. 2005;1:58–66. doi: 10.1016/j.nano.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Desai SA, Alkhalil A, Kang M, Ashfaq U, Nguyen ML. PSAC-independent phloridzin resistance in Plasmodium falciparum. J Biol Chem. 2005;280:16861–16867. doi: 10.1074/jbc.M414629200. [DOI] [PubMed] [Google Scholar]
- Desai SA, Bezrukov SM, Zimmerberg J. A voltage-dependent channel involved in nutrient uptake by red blood cells infected with the malaria parasite. Nature. 2000;406:1001–1005. doi: 10.1038/35023000. [DOI] [PubMed] [Google Scholar]
- Eksi S, Haile Y, Furuya T, Ma L, Su X, Williamson KC. Identification of a subtelomeric gene family expressed during the asexual-sexual stage transition in Plasmodium falciparum. Mol Biochem Parasitol. 2005;143:90–99. doi: 10.1016/j.molbiopara.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Freitas-Junior LH, Bottius E, Pirrit LA, Deitsch KW, Scheidig C, Guinet F, et al. Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature. 2000;407:1018–1022. doi: 10.1038/35039531. [DOI] [PubMed] [Google Scholar]
- Ginsburg H, Kutner S, Krugliak M, Cabantchik ZI. Characterization of permeation pathways appearing in the host membrane of Plasmodium falciparum infected red blood cells. Mol Biochem Parasitol. 1985;14:313–322. doi: 10.1016/0166-6851(85)90059-3. [DOI] [PubMed] [Google Scholar]
- Hayton K, Gaur D, Liu A, Takahashi J, Henschen B, Singh S, et al. Erythrocyte binding protein PfRH5 polymorphisms determine species-specific pathways of Plasmodium falciparum invasion. Cell Host Microbe. 2008;4:40–51. doi: 10.1016/j.chom.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Pillai AD, Nawaz F, Hayton K, Doan L, Lisk G, Desai SA. A blasticidin S-resistant Plasmodium falciparum mutant with a defective plasmodial surface anion channel. Proc Natl Acad Sci USA. 2007;104:1063–1068. doi: 10.1073/pnas.0610353104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homewood CA, Neame KD. Malaria and the permeability of the host erythrocyte. Nature. 1974;252:718–719. doi: 10.1038/252718a0. [DOI] [PubMed] [Google Scholar]
- Huber SM, Uhlemann AC, Gamper NL, Duranton C, Kremsner PG, Lang F. Plasmodium falciparum activates endogenous Cl− channels of human erythrocytes by membrane oxidation. EMBO J. 2002;21:22–30. doi: 10.1093/emboj/21.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Lisk G, Hollingworth S, Baylor SM, Desai SA. Malaria parasites are rapidly killed by dantrolene derivatives specific for the plasmodial surface anion channel. Mol Pharmacol. 2005;68:34–40. doi: 10.1124/mol.104.010553. [DOI] [PubMed] [Google Scholar]
- Kirk K, Horner HA, Elford BC, Ellory JC, Newbold CI. Transport of diverse substrates into malaria-infected erythrocytes via a pathway showing functional characteristics of a chloride channel. J Biol Chem. 1994;269:3339–3347. [PubMed] [Google Scholar]
- Lander ES, Botstein D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics. 1989;121:185–199. doi: 10.1093/genetics/121.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavazec C, Sanyal S, Templeton TJ. Expression switching in the stevor and Pfmc-2TM superfamilies in Plasmodium falciparum. Mol Microbiol. 2007;64:1621–1634. doi: 10.1111/j.1365-2958.2007.05767.x. [DOI] [PubMed] [Google Scholar]
- Lisk G, Desai SA. Improved perfusion conditions for patch-clamp recordings on human erythrocytes. Biochem Biophys Res Commun. 2006;347:158–165. doi: 10.1016/j.bbrc.2006.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisk G, Desai SA. The plasmodial surface anion channel is functionally conserved in divergent malaria parasites. Eukaryot Cell. 2005;4:2153–2159. doi: 10.1128/EC.4.12.2153-2159.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisk G, Kang M, Cohn JV, Desai SA. Specific inhibition of the plasmodial surface anion channel by dantrolene. Eukaryot Cell. 2006;5:1882–1893. doi: 10.1128/EC.00212-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisk G, Pain M, Gluzman IY, Kambhampati S, Furuya T, Su XZ, et al. Changes in the plasmodial surface anion channel reduce leupeptin uptake and can confer drug resistance in P. falciparum-infected erythrocytes. Antimicrob Agents Chemother. 2008;52:2346–2354. doi: 10.1128/AAC.00057-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RE, Henry RI, Abbey JL, Clements JD, Kirk K. The ‘permeome’ of the malaria parasite: an overview of the membrane transport proteins of Plasmodium falciparum. Genome Biol. 2005;6:R26. doi: 10.1186/gb-2005-6-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman RR. Reversible cellular permeability alterations in disease. In vivo studies on sodium, potassium and chloride concentrations in erythrocytes of the malarious monkey. Am J Physiol. 1948;152:113–121. doi: 10.1152/ajplegacy.1947.152.1.113. [DOI] [PubMed] [Google Scholar]
- Rohrbach P, Sanchez CP, Hayton K, Friedrich O, Patel J, Sidhu AB, et al. Genetic linkage of pfmdr1 with food vacuolar solute import in Plasmodium falciparum. EMBO J. 2006;25:3000–3011. doi: 10.1038/sj.emboj.7601203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario V. Cloning of naturally occurring mixed infections of malaria parasites. Science. 1981;212:1037–1038. doi: 10.1126/science.7015505. [DOI] [PubMed] [Google Scholar]
- Scherf A, Hernandez-Rivas R, Buffet P, Bottius E, Benatar C, Pouvelle B, et al. Antigenic variation in malaria: in situ switching, relaxed and mutually exclusive transcription of var genes during intra-erythrocytic development in Plasmodium falciparum. EMBO J. 1998;17:5418–5426. doi: 10.1093/emboj/17.18.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Churchill GA. A statistical framework for quantitative trait mapping. Genetics. 2001;159:371–387. doi: 10.1093/genetics/159.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staines HM, Alkhalil A, Allen RJ, De Jonge HR, Derbyshire E, Egee S, et al. Electrophysiological studies of malaria parasite-infected erythrocytes: current status. Int J Parasitol. 2007;37:475–482. doi: 10.1016/j.ijpara.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staines HM, Rae C, Kirk K. Increased permeability of the malaria-infected erythrocyte to organic cations. Biochim Biophys Acta. 2000;1463:88–98. doi: 10.1016/s0005-2736(99)00187-x. [DOI] [PubMed] [Google Scholar]
- Su XZ, Carucci DJ, Wellems TE. Plasmodium falciparum: parasite typing by using a multicopy microsatellite marker, PfRRM. Exp Parasitol. 1998;89:262–265. doi: 10.1006/expr.1998.4299. [DOI] [PubMed] [Google Scholar]
- Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- Upston JM, Gero AM. Parasite-induced permeation of nucleosides in Plasmodium falciparum malaria. Biochim Biophys Acta. 1995;1236:249–258. doi: 10.1016/0005-2736(95)00055-8. [DOI] [PubMed] [Google Scholar]
- Wagner MA, Andemariam B, Desai SA. A two-compartment model of osmotic lysis in Plasmodium falciparum-infected erythrocytes. Biophys J. 2003;84:116–123. doi: 10.1016/S0006-3495(03)74836-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walliker D, Quakyi IA, Wellems TE, McCutchan TF, Szarfman A, London WT, et al. Genetic analysis of the human malaria parasite Plasmodium falciparum. Science. 1987;236:1661–1666. doi: 10.1126/science.3299700. [DOI] [PubMed] [Google Scholar]
- Wellems TE, Panton LJ, Gluzman IY, do Rosario VE, Gwadz RW, Walker-Jonah A, Krogstad DJ. Chloroquine resistance not linked to mdr-like genes in a Plasmodium falciparum cross. Nature. 1990;345:253–255. doi: 10.1038/345253a0. [DOI] [PubMed] [Google Scholar]