

Summary
Late infantile neuronal ceroid lipofuscinosis (LINCL), a fatal autosomal recessive neurodegenerative lysosomal storage disorder of childhood, is caused by mutations in the CLN2 gene, resulting in deficiency of the protein tripeptidyl peptidase I (TPP-I). We have previously shown that direct CNS administration of AAVrh.10hCLN2 to adult CLN2 knockout mice, a serotype rh.10 adeno-associated virus expressing the wild type CLN2 cDNA, will partially improve neurological function and survival. In this study, we explore the hypothesis that administration of AAVrh.10hCLN2 to the neonatal brain will significantly improve the results of AAVrh.10hCLN2 therapy. To assess this concept, AAVrh.10hCLN2 vector was administered directly to the CNS of CLN2 knockout mice at 2 days, 3 wk and 7 wk of age. While all treatment groups show a marked increase in total TPP-I activity over wild-type mice, neonatally treated mice displayed high levels of TPP-I activity in the CNS 1 yr after administration which was spread throughout the brain. Using behavioral markers, 2 day treated mice demonstrate marked improvement over 3 wk, 7 wk or untreated mice. Finally, neonatal administration of AAVrh.10hCLN2 was associated with markedly enhanced survival, with a median time of death 376 days for neonatal treated mice, 277 days for 3 wk treated mice, 168 days for 7 wk treated mice, and 121 days for untreated mice. These data suggest that neonatal treatment offers many unique advantages, and that early detection and treatment may be essential for maximal gene therapy for childhood lysosomal storage disorders affecting the CNS.
Introduction
There are over 40 identifiable lysosomal storage disorders, which are rare monogenic autosomal recessive diseases characterized by accumulation of unmetabolized substrates in lysosomes (Neufeld, 1991; Vellodi, 2005). Among the lysosomal storage disorders, approximately 50% affect the central nervous system (CNS) (Cardone, 2007; Hoffmann and Mayatepek2005; Jeyakumar et al., 2005). While many of the systemic manifestations of several of the lysosomal storage disorders can be effectively treated by intravenous enzyme therapy (Brady, 2006), the blood brain barrier prevents enzymes administered systemically to reach the brain, and thus the CNS manifestations of these disorders are, for now, untreatable (Beck, 2007; Sondhi et al., 2001).
Gene therapy represents one approach to deliver the deficient enzymes to the CNS, using gene transfer vectors expressing the wild type cDNA to correct the relevant cell population in the CNS (Beck, 2007; Cardone, 2007; Mandel et al., 2006; Sondhi et al., 2001). Studies of experimental animal models and humans have shown that a significant challenge to providing effective gene therapy for the CNS manifestations of the lysosomal storage disorders is that most of these disorders diffusely affect the CNS, and thus effective therapy requires delivery of the product of the therapeutic vector throughout the brain (Beck, 2007; Sondhi et al., 2001). This challenge is further compounded by the physical limitations of administration of gene therapy vectors to the human CNS, including the number of burr holes that can be placed in the skull, the volume that can be administered per min, and the total time of anesthesia that can be used safely (Crystal et al., 2004; Hackett et al., 2005). Finally, corrective gene therapies for neurodegenerative diseases are not restorative but rather halt disease progression, making delivery prior to disease onset an important objective. With these limitations in mind, three basic strategies have been employed to achieve therapeutic levels of gene expression throughout the brain: identifying gene transfer vectors that will diffuse through the CNS more effectively (Cearley and Wolfe2006; Deglon and Hantraye2005; Shevtsova et al., 2005; Sondhi et al., 2007), optimizing the method of delivery and volume and rate of infusion (Chen et al., 1999; Hackett et al., 2005; Hsich et al., 2002), and administration at an earlier time point, when the brain is smaller and immature (Bostick et al., 2007; Broekman et al., 2007; Daly et al., 1999; Griffey et al., 2006; Li and Daly2002; Passini et al., 2003; Passini and Wolfe2001; Rafi et al., 2005; Shen et al., 2001; Waddington et al., 2004). Studies from our laboratory and others have identified several serotypes of adeno-associated virus (AAV) vectors that are more effective than the first generation vectors used for CNS gene transfer (Broekman et al., 2006; Burger et al., 2005; Cearley and Wolfe2006; Harding et al., 2006; Sondhi et al., 2007; Taymans et al., 2007), the parameters for optimal administration of vectors to the CNS have been established (Chen et al., 1999; Hackett et al., 2005; Hsich et al., 2002) and several studies have shown that administration earlier in life results in an advantage over therapy in older animals (Bostick et al., 2007; Broekman et al., 2007; Daly et al., 1999; Griffey et al., 2006; Li and Daly2002; Passini et al., 2003; Passini and Wolfe2001; Rafi et al., 2005; Shen et al., 2001; Waddington et al., 2004).
In the present study, we asked whether CNS administration to newborns of an AAV serotype rh10 vector, one of the AAV vectors known to be among the best to transfer genes to the CNS (Cearley and Wolfe2006; Sondhi et al., 2007), will have a better therapeutic advantage than the same vector administered later in life. To test this hypothesis, we have used late infantile neuronal ceroid lipofuscinosis (LINCL) as the target disorder. LINCL, a fatal, autosomal recessive neurodegenerative disease of middle childhood, is a typical lysosomal storage disorder with diffuse CNS manifestations, caused by mutations in the CLN2 gene, resulting in a deficiency of the lysosomal tripeptidyl peptidase (TPP-I) (Sleat et al., 1997; Williams et al., 1999; Worgall et al., 2007). In prior studies, we have shown that CNS administration of an AAVrh.10 vector coding for the wild type human CLN2 cDNA to 7 wk old CLN2 knockout mice increased the median age of survival from 128 days to 162 days compared to the first generation AAV serotype 2 or 5 vector which had no impact on survival (Passini et al., 2006; Sondhi et al., 2007), and Cabrera-Salazar et al (Cabrera-Salazar et al., 2007) have shown that an AAV1 vector coding for the CLN2 cDNA administered to 4 wk old CLN2-/- mice had a significant survival advantage over administration at 11 wk.
Based on these observations, and studies of several groups showing that administration of gene transfer vectors to the newborn CNS provides significant advantage to administration later in life (Bostick et al., 2007; Broekman et al., 2007; Daly et al., 1999; Griffey et al., 2006; Li and Daly2002; Passini et al., 2003; Passini and Wolfe2001; Rafi et al., 2005; Shen et al., 2001; Waddington et al., 2004), we hypothesized that if an AAVrh.10 vector coding for the wild type CLN2 cDNA were administered to the CNS of newborn CLN2-/- mice, we would achieve significant improvement in survival over administration to older animals. Compared to parallel administration in 3 or 7 wk old mice, administration at postnatal day 2 demonstrated remarkable persistent, widespread expression of the CLN2 gene product, with concomitant improvement of a number of tests of CNS function as well as significant survival advantage.
Methods
AAVrh.10hCLN2 Vector
The AAVrh.10hCLN2 vector contains an expression cassette consisting of the human CLN2 cDNA driven by a CMV/β-actin hybrid promoter consisting of the enhancer from the cytomegalovirus immediate early gene, the promoter, splice donor and intron from the chicken β-actin gene, the splice acceptor from the rabbit β-globin gene followed by a CLN2 cDNA with an optimized Kozak translation initiation sequence (Sondhi et al., 2007). The cDNA is followed by the polyadenylation sequence from rabbit β-globin. The expression cassette is surrounded by the inverted terminal repeats of AAV2. To produce AAVrh.10hCLN2, 293 cells were cotransfected using Polyfect with the vector plasmid described above and two helper plasmids: pΔF6, which expresses the required adenoviral helper functions for replication and the other with the AAV2 rep gene, and the AAVrh.10 cap gene (Sondhi et al., 2007). After 72 hr, the cells were harvested and AAV purified on iodixinol gradients, followed by QHP ion exchange chromatography. Vector preparations were assessed by TaqMan real-time polymerase chain reaction to determine genomic copies and by in vitro gene transfer in 293 ORF6 cells.
Administration of AAVrh.10 to the CLN2-/- Mouse Brain
All procedures were performed in accordance with NIH guidelines and under protocols approved by the Institutional Animal Care and Use Committee. Original heterozygous breeding pairs of CLN2+/- mice back crossed into C57Bl/6 were used to produce all of the CLN2-/- mice used in this study (Sleat et al., 2004). At postnatal day 20, mice were weaned and genotyped by polymerase chain reaction. The AAVrh.10hCLN2 vector was administered to the CLN2-/- mice at 7 wk, 3 wk, or 2 days of age. Untreated CLN2-/- mice, PBS injected CLN2 -/- mice, and AAVrh.10GFP-treated CLN2 -/- mice [the AAVrh.10GFP vector consists of a humanized GFP gene with expression driven by CMV promoter pseudotyped with rh.10 capsid (Zolotukhin et al., 1996)] all served as negative controls and CLN2+/+ mice served as positive controls. Pilot injections of methylene blue were performed to confirm injection locations for 3 wk and 2 day injected mice.
The 7 and 3 wk mice received bilateral intracranial injections stereotaxically in four locations per hemisphere using a 10 μl Hamilton syringe (Hamilton Company; Reno, NV) fitted with a 26 g needle. At 7 wk of age, mice were injected bilaterally in the lower striatum (A/P +0.60 mm, M/L ±1.75 mm, D/V -4.0 mm), upper striatum (A/P +0.60 mm, M/L ±1.75 mm, D/V -2.0 mm), thalamus (A/P –2.0 mm, M/L ±1.0 mm, D/V -3.0 mm), and cerebellum (A/P -6.0 mm, M/L ±0.5 mm, D/V –1.5 mm) with AAVrh.10hCLN2 (2×1010 genome copies in 3 μl per site) at a rate of 0.5 μl/min. The needle was left in position for 2 min before and 2 min following vector administration, at which point it was withdrawn slightly and left for 1 min, and then was finally withdrawn over the course of an additional minute. At 3 wk of age, mice were injected in the lower striatum (A/P +0.60 mm, M/L ±1.70 mm, D/V -3.5 mm), upper striatum (A/P +0.60 mm, M/L ±1.70 mm, D/V -2.5 mm), thalamus (A/P -2.3 mm, M/L ±1.0 mm, D/V -2.5 mm), and cerebellum (A/P -6.0 mm, M/L ±1.5 mm, D/V -1.5 mm) with AAVrh.10hCLN2 (2×1010 genome copies in 3 μl per site) at a rate of 0.5 μl/min, similarly to that for the 7 wk old mice. The total dose administered in the 7 wk and 3 wk animals was 1.6 × 1011 genome copies delivered in a total volume of 24 μl distributed equally over 8 sites. Postnatal day 2 mice were injected bilaterally in three locations per hemisphere using the 10 μl Hamilton syringe fitted with a 30 g needle. Pups were anesthetized with ice (Li and Daly, 2002), immobilized and injected at 3 separate locations per hemisphere distributed along the rostral-caudal axis (1.85 ×1010 genome copies in 0.5 μl per site). The total dose administered in the neonatal animals was 1.1×1011 genome copies delivered in a total volume of 6 μl distributed equally over 6 sites. Pups were returned to the litter and genotype determined at 20 days.
TPP-I Activity in the CNS
When observed to be close to death (severe shaking, weight loss), mice were anesthetized and transcardially perfused with PBS. Brains were excised and hemisected. The right hemisphere was submerged in 4% paraformaldehyde for morphological evaluation (see below) and the left hemisphere was cut into six, 2 mm coronal sections. All samples were flash frozen. The samples were later thawed and homogenized in NaCl (0.15 M) and Triton X-100 (1 g/L). Supernatants of homogenized samples were diluted 100 fold in phosphate buffered saline and assayed for TPP-I activity as previously described (Sondhi et al., 2007). Final TPP-I activity was adjusted to total protein concentration using a bicinchoninic acid (BCA) Protein Assay (Pierce; Rockford IL) and calculated as fluorescent units (FU)/min-mg.
Morphological Evaluation of TPP-I Expression
Efficiency of CLN2 cDNA transfer was assessed in the right hemisphere by immunohistochemical staining for the gene product TPP-I. At 72 hr post-perfusion, the brains were equilibrated in 30% sucrose at 4°C. Serial 50 μm thick sagittal sections were produced by freezing sliding microtomy. Immunohistochemical detection of TPP-I was accomplished by incubation with mouse anti-human TPP-I antibody (1:1000; 72 hr at 4°C; gift from K. Wisniewski and A. Golabek, NYS Institute for Basic Research), followed by biotin-streptavidin amplification (Vectastain kit, Vector Labs; Burlingame, CA) that was then visualized by incubation in nickel chloride enhanced diaminobenzidine (DAB kit, Vector Labs; Burlingame, CA). Images were obtained by brightfield microscopy with digital image acquisition using an Olympus BX50 and Microfire Camera (Olympus America Inc; Center Valley, PA). For multiple immunofluorescence staining, sections were incubated for 72 hr at 4°C with rabbit anti-TPP-I (1:500; gift from P. Lobel, University of Medicine and Dentistry of New Jersey) and mouse anti-NeuN (1:2500; Chemicon, Temecula, CA) or mouse anti-TPP-I (1:2000), rabbit anti-Iba-1 (1:4000; Wako Chemicals USA, Richmond, VA), and guinea pig anti-GFAP (1:2000; Advanced Immunologicals, Long Beach, CA). Secondary antisera raised in donkey were used as appropriate for species detection and were conjugated to the fluorophores Alexa 488 (Invitrogen, Carlsbad, CA) or Cy3 or Cy5 (Jackson ImmunoResearch, West Grove, PA). Fluorescence imaging was performed using an Olympus Fluoview confocal microscope (Olympus America, Center Valley, PA).
Behavioral Phenotyping
AAVrh.10hCLN2 was administered to the CNS of CLN2-/- mice at 7 wk (n=17), 3 wk (n=15), or 2 days (n=8) of age. A series of phenotypic observations were made at various intervals to assess the impact of age of AAVrh.10hCLN2 administration. The negative controls were untreated CLN2-/- (n=14) mice, PBS treated (n=14, injected at 7 wk) and AAVrh.10 GFP treated (n =6, injected at 7 wk). The positive controls were CLN2 wild-type mice (n=16). The assessments used and the testing intervals included: (1) gait (this qualitative test was performed in a subset of n = 3 mice/group at bimonthly intervals); (2) tremors (weekly; on all animals in every group); (3) appearance (weekly; on all animals in every group); (4) performance of balance beam (weekly; on all animals in every group); and (5) performance on grip strength test (weekly; on all animals in every group). All tests, except gait, were performed at the same time each week and under similar conditions on all of the surviving mice in each of the experimental groups. Apart from gait, data from each of these tests was statistically analyzed from 15 to18 wk, a period chosen based on the knowledge that CLN2 -/- mice perform normally until about 14 wk and are moribund by 18 wk (Sleat et al., 2004). There is typically a sharp decline in abilities in this time frame.
Gait analysis
Gait analysis was performed on a subset (n=3) of mice in each experimental groups at bimonthly intervals. A blind tunnel (100 cm long, 20 cm wide, 10 cm high) with one end in the dark and the other end in the light was constructed over a sheet of white paper. The hindpaws of the mouse were dipped in blue ink and the frontpaws were dipped in red ink (Crayola washable paints, Binney and Smith, Easton, PA). The mouse was placed at the light end of the tunnel and as it ran to the dark end, the footprints were recorded on the paper and saved for qualitative analysis.
Tremors
Based on previous empirical observations, a tremor scale was created and applied to all mice. Tremors were assessed while the mouse was on the balance beam (described below) for a period of at least 30 sec. The mice were rated on a scale of 1 to 5 as follows: score 1, occasional violent and severe seizures coupled with repeated tremors; score 2, tremors and shaking most of the time; score 3, tremors and shaking occasionally; score 4, a shuffling movement with splayed hind feet but no tremors or shaking; and score 5, an active and agile mouse with no visible tremors or shuffling.
Coat appearance
Based on previous empirical observations, a scale of coat appearance was created and applied to all mice. Coat appearance was assessed while the mouse was on the balance beam (described below) for a period of at least 30 sec. Mice were rated on a 3 point scale of 1 to 5 as follows: score 1, greasy, clumped, and unkempt fur; score 3, greasy but mostly kempt fur; and score 5, clean, smooth and shiny fur.
Balance beam
A horizontal wooden beam, 3 cm × 125 cm was secured 100 cm from the ground over a tub filled with pads to protect mice that fell off the beam. The mouse was placed in the middle of the beam and the time until fall was recorded. Mice that did not fall after 3 min were returned to their cage and the time was recorded as 3 min.
Grip strength
A string was stretched between two wooden dowels. The mouse was placed in the middle of the suspended string and the time until fall was recorded. Mice that did not fall after 3 min were returned to their cage and the time was recorded as 3 min.
Survival
Mice were observed 3 times a week for signs of deterioration in health. If deemed moribund (severe shaking, weight loss), mice were sacrificed and age of death was recorded.
Statistical Analysis
Data is provided as mean values ± SEM. Analysis of phenotype ratings and time were performed by ANCOVA with the post-hoc Fischer test with each measurement contributing to the value used for means testing with treatment group as factor and time as covariant. Since some parameters such as tremor and appearance are not normally distributed, significance was confirmed by non-parametric analysis at selected timepoints. Survival analysis was assessed using Kaplan–Meier test.
Results
CNS TPP-I Levels
All CLN2-/- mice treated with AAVrh.10hCLN2 exhibited higher levels of TPP-I activity (FU/min-mg) in the brain, compared to uninjected CLN2-/- mice and uninjected CLN2+/+ mice (Figure 1). Mice treated at postnatal day 2, 3 wk and 7 wk all showed a nearly 100-fold increase in activity above wild-type levels. In addition, the 2 day treated mice possessed a wider rostral-caudal distribution compared to 7 and 3 wk treated mice, with significantly higher levels of TPP-I activity in the frontal lobe (p<0.01).
Figure 1.

TPP-I Activity in the CNS following direct CNS administration of AAVrh.10hCLN2. TPP-I activity in the brain following administration of AAVrh.10hCLN2 (2 × 1010 genome copies/site) bilaterally at multiple locations along the rostral-caudal axis at 7 wk, 3 wk, or 2 days of age. Mice were sacrificed when moribund. Samples included untreated controls (n=14, CLN2-/-); wild-type (n=16, CLN2+/+); 7 wk AAVrh.10 treated (n=17, CLN2 -/-); 3 wk AAVrh.10 treated (n=11, CLN2 -/-); and 2 day AAVrh.10 treated (n=5, CLN2 -/-). Mean specific activity was measured in brain homogenates across six, 2 mm coronal sections. The y-axis is represented as a log scale.
Distribution of TPP-I Expression
In agreement with the measure of enzymatic activity, no TPP-I-positive staining was detected in the brains of untreated CLN2 -/- mice (not shown) or those that received the PBS vehicle injection (Figure 2A, E). In all groups receiving the AAVrh.10hCLN2 administration, regions of injection could be identified by robust cellular expression of TPP-I (Figure 2 B-D and F-H). Multiple immunofluorescence staining showed that TPP-I expression was detected within cells colocalized with expression of NeuN, confirming their neuronal identity (not shown). Outside of regions of injection, the extent and staining patterns of TPP-I-positive cells varied as a function of the age of administration of the AAVrh.10hCLN2 (Figure 2). Assessment of mice treated at 7 wk resulted in sparse punctate staining within scattered neurons in most regions examined that were adjacent to the site of delivery. Assessment of mice treated at 3 wk resulted in stronger labeling within neurons, so that enough of the cell soma was filled to produce a recognizable cellular profile, and TPP-I-positive neurons were observed at greater frequency. Strikingly, evaluation following administration at 2 days postnatal showed strong intracellular TPP-I expression. Most neurons were TPP-I-positive in all regions examined, including regions such as the midbrain.
Figure 2.
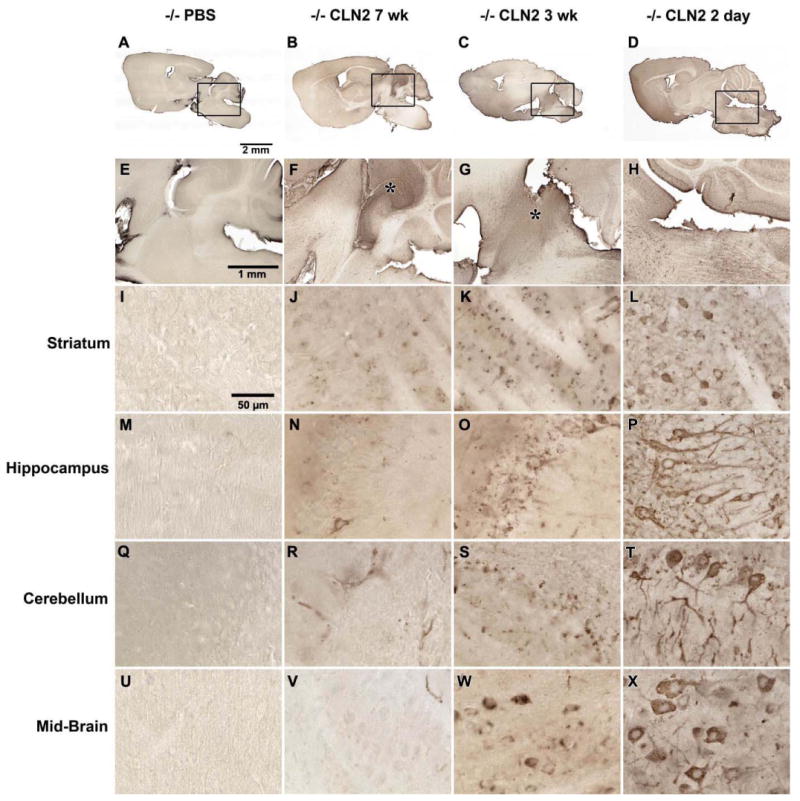
Immunohistological assessment of the expression of TPP-I in the CNS following gene transfer. A-D. Sagittal sections of whole brain after: A. delivery of PBS; B. delivery of AAVrh.10hCLN2 at 7 wk; C. delivery of AAVrh.10hCLN2 at 3 wk; and D. delivery of AAVrh.10hCLN2 at 2 days. E - H represent higher magnification views of panels A - D respectively. E. Close up of PBS injected mice showing no detectable TPP-I staining; F,G. Close up of AAVrh.10hCLN2 mice injected at 7 wk and 3 wk respectively showing a distribution gradient in detection of TPP-I between strong staining in the region of injection (asterisk) and reduced staining further from the region of injection. H. Close up of mice injected at 2 days showing uniform distribution of TPP-I staining across the equivalent region. Specific structures outside the region of injection were examined at higher magnification including: I-L. striatum; M-P. hippocampus; Q-T. cerebellum and U-X. mid-brain. I. Striatum of mice receiving PBS in which no TPP-I positive cells were seen; J,K. striatum of mice injected with AAVrh.10hCLN2 at 7 wk or 3 wk producing punctate cellular staining in some cells; L. striatum of mice injected with AAVrh.10hCLN2 at 2 days with no obvious region of injection and equivalent regions of the striatum containing numerous cells with strong cytoplasmic staining extending into cell processes. M. Hippocampus of PBS injected mice containing no detectable TPP-I staining;. N,O. hippocampus of mice injected with AAVrh.10hCLN2 at 7 or 3 wk, showing few TPP-I-positive cells with punctate staining; P. hippocampus in mice injected at 2 days containing robust TPP-I staining filling cell bodies and processes in all parts. Q. Cerebellum of PBS injected mice containing no detectable TPP-I staining; R,S. cerebellum outside of the region of injection in mice injected with AAVrh.10hCLN2 at 7 wk or 3 wk with TPP-I staining only in blood vessels or few TPP-I-positive cells with punctate staining; T. cerebellum of mice injected at 2 days with AAVrh.10hCLN2 showing all parts of the cerebellum with robust TPP-I staining filling cell bodies and processes. U. Midbrain of PBS-injected mice containing no TPP-I staining; V. midbrain of mice injected with AAVrh.10hCLN2 at 7 wk with only a few TPP-I positive blood vessels; W. midbrain of mice injected with AAVrh.10hCLN2 at 3 wk showing some TPP-I positive cells with uneven filling of their cytoplasm; X. Midbrain of mice injected with AAVrh.10hCLN2 at 2 days with frequent, strongly-stained cells distributed across the midbrain. Calibration bars are: A-D, 2 mm; E-H, 1 mm; and I-X, 50 μm.
Consistent with the observation that administration at 2 days produced higher levels at the most rostral brain segments when measured by enzymatic assay, the frontal cortex showed extensive TPP-I expression despite being distant from the sites of injection (Figure 3). With 7 and 3 wk administration times, nearly all TPP-I-positivity was detected in tubular and circular vascular profiles indicative of endothelial cell-staining in small blood vessels and capillaries, likely as a result of mechanical disruption and seepage of vector into vascular system and transport to distant loci during surgical delivery. In contrast, TPP-I staining was well defined in neurons of the frontal cortex in the 2 day administration group. To further evaluate the phenotype of cells expressing TPP-I, immunofluorescence staining was performed to colocalize TPP-I protein (red) with microglia (green) or astrocytes (blue; Figure 3). The multiple labeling revealed that TPP-I expression was not in astrocytes or microglia regardless of the age of the mice at vector administration. There was no apparent difference in microglial density or morphology between conditions suggesting that microglia were not activated in either the untreated CLN2 -/- or the animals receiving gene delivery.
Figure 3.
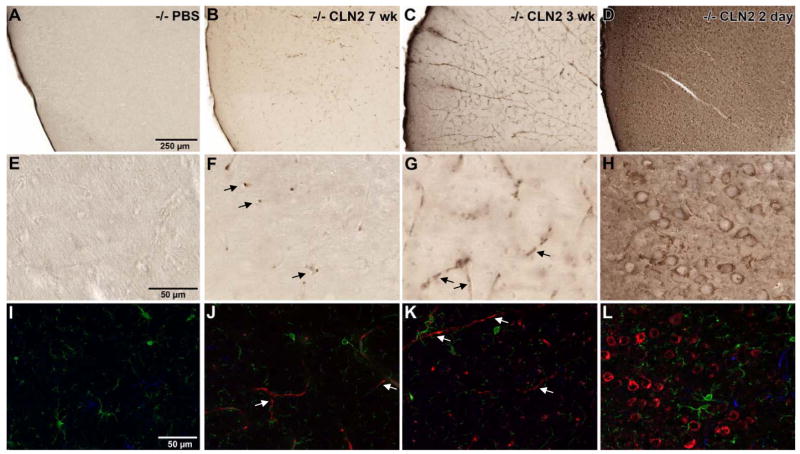
Widespread expression of TPP-I in brain regions outside the area of vector administration is achieved by early (2 day) postnatal delivery of AAVrh.10hCLN2. The frontal cortex was examined as a region distant from the sites of injection to assess the effect of age at vector administration with the extent of transgene expression. Panel A - H represent immunoperoxidase staining for TPP-I while panels I - L represent immunofluorescent staining. A, E. Low magnification and high magnification images from PBS-administered animals, where immunoperoxidase staining revealed no detectable TPP-I staining. B, F. Tubular and circular vascular endothelial staining of blood vessels (arrows) for TPP-I was observed in animals injected at 7 wk as evident from both the low and the high magnification images; and C, G. In addition to vascular staining (arrows), occasional punctate staining of TPP-I was seen within a few, dispersed cells in both the low and high magnification images of animals injected at 3 wk. D, H. Injection at 2 days produced widespread cellular staining within the frontal cortex as seen in both the low and high magnification views. To evaluate the phenotype of cells expressing TPP-I, immunofluorescence staining was performed to colocalize TPP-I protein (red) with microglia (green) or astrocytes (blue). Detection of TPP-I was the same as seen with the immunoperoxidase staining. I. No TPP-I staining in PBS-injected animals, J. TPP-I-positive blood vessels in 7 wk (arrows); and K. blood vessel staining in 3 wk injected animals (arrows). L. Widespread, robust TPP-I staining was observed in 2 day injected animals. Neither microglia nor astrocytes coexpressed TPP-I. A-D., bar = 250 μm; E-L., bar = 50 μm.
Impact on Gait
Gait analysis of mice was conducted bimonthly on a subset of n=3 mice per group. The representative data shown here is gait analysis done in mice with advanced disease that are close to the mean age of death for that group (Figure 4). In wild-type mice (assessed at age 18 wk, the typical age of death for untreated CLN2 -/- mice), front and hind paw prints were largely superimposed and there was no limb dragging evident (Figure 4A). In contrast, untreated CLN2-/-mice also assessed at 18 wk age displayed dragging of feet, poor coordination of front and hind paws, and overall disordered gait (Figure 4B). CLN2-/- mice treated with AAVrh.10hCLN2 showed marked improvement in gait associated with earlier age of administration (Figure 4C-E). The mice treated at 7 wk age assessed at 22 wk age showed some dragging and no overlap of hind and front paws. In mice treated at 3 wk assessed at 36 wk age, there was minimal dragging but still poor coordination of front and hind paws. In contrast, in the mice treated as newborns and assessed at 52 wk age, the coordination of front and hind paws was excellent, approaching that of wild-type mice. Quantitative analysis by measuring separation of foot prints was impossible due to the inability to separate the front paws from the hind paws due to excessive dragging in the untreated CLN2 -/- controls. Another approach was to assess the total ink (red or blue) deposited as a measure of foot dragging, but this was also not possible due to the inconsistencies between experimental subjects depending on how much ink was initially applied to the feet.
Figure 4.
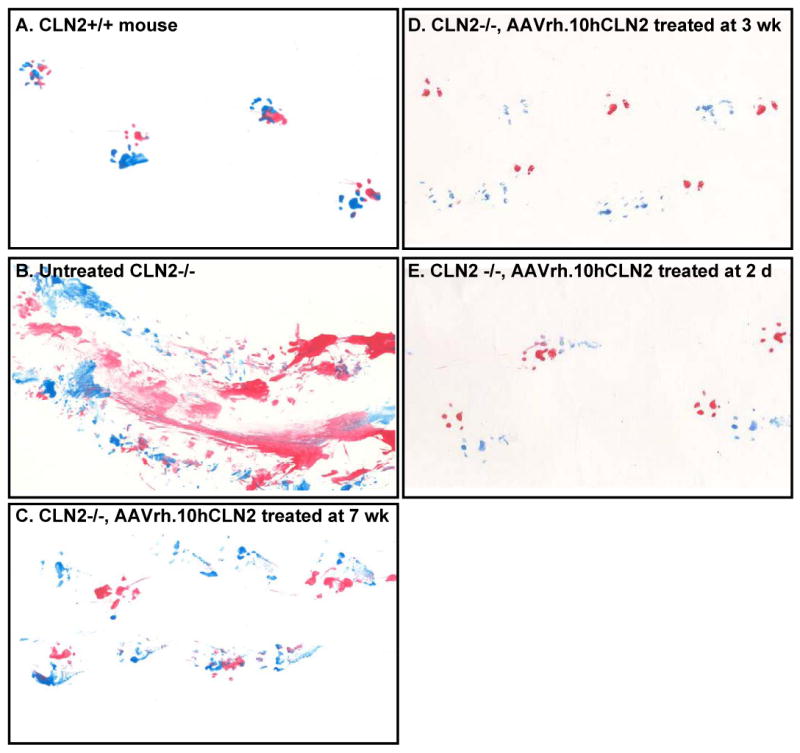
Impact of age of AAVrh.10hCLN2 administration on gait. Hindpaws were covered in blue ink and frontpaws in red ink. Deficits in gait are revealed as mouse ran along a sheet of paper. The data shown here is representative of the gait analysis carried out in mice with advanced disease close to the mean age of death for that group. A. Wild-type; B. Untreated CLN2 -/-; C. CLN2 -/- treated at 7 wk; D. CLN2 -/- treated at 3 wk; and E. CLN2 -/- treated at 2 days.
Impact on Tremors and Coat Appearance
The tremors of CLN2-/- mice treated with AAVrh.10hCLN2 at 2 days, 3 wk, and 7 wk of age and controls were characterized weekly. For all groups, the data was analyzed from 15 to 18 wk, a period chosen based on the knowledge that untreated CLN2 -/- mice perform normally until about 14 wk and are moribund by 18 wk (Sleat et al., 2004). There is typically a sharp decline in abilities about this time. The therapeutic effect of gene therapy on tremors depended on the age of treatment with the severity of tremors decreasing with earlier treatment (Figure 5A). Untreated CLN2-/- mice exhibited moderate tremors and received significantly worse tremor ratings than wild-type controls that did not exhibit any signs of tremor activity (p<0.0001). The two negative control groups exhibited a tremor rating that was not significantly different from the untreated CLN2 -/- mice (PBS, p>0.4, and AAVrh.10GFP, p>0.4, not shown). In comparison all CLN2-/- mice treated with AAVrh.10hCLN2 experienced significantly milder tremor activity (p<0.0001). The 2 day and 3 wk treatment groups received significantly better tremor ratings than mice treated at 7 wk of age (p<0.0001) and the 2 day treated were better than the 3 wk treated (p<0.005), although not at the level of the wild-type mice (p<0.0001).
Figure 5.




Impact of age of AAVrh.10hCLN2 administration on phenotype and behavioral task performance. All groups were assessed weekly for rated phenotypes (0 to 5 scale, see Methods) and performance in behavioral assays. Means were calculated over a 4 wk period (15 to 18 wk) immediately preceding the death of the uninjected CLN2 -/- control group. A. Effect of the age of treatment on ratings of tremors; B. Effect of the age of treatment on ratings of coat appearance; C. Effect of the age of treatment on performance in balance beam test; and D. Effect of the age of treatment on performance in grip strength test. The numbers of mice for each group are as follows: Uninjected controls (n=14, CLN2 -/-); wild-type controls (n=16, CLN2 +/+); 7 wk AAVrh.10 injected (n=17, CLN2 -/-); 3 wk AAVrh.10 injected (n=15, CLN2 -/-); 2 day AAVrh.10 injected (n=8, CLN2 -/-).
The impact of AAVrh.10hCLN2 treatment on coat appearance of CLN2-/- mice depended on the age of treatment (Figure 5B). CLN2-/- mice treated the earliest received the best ratings for coat appearance. Untreated CLN2-/- had difficulty grooming their coats as the disease progresses and therefore received significantly lower coat appearance ratings than wild-type controls (p<0.0001). The two negative control groups also had difficulty grooming to the extent that they were not significantly different from the untreated CLN2 -/- mice (PBS, p>0.1, and AAVrh.10GFP, p>0.2, not shown). By contrast, CLN2-/- mice treated with AAVrh.10hCLN2 at all ages received significantly higher coat appearance ratings (p<0.005). The 2 day treatment group received higher ratings for coat appearances than both the 7 wk (p<0.05) and 3 wk (both p<0.0001) groups and was improved to the extent that their performance was equivalent to wild-type controls (p>0.05).
Performance on Behavioral Tasks
Assessment of CLN2-/- mice by performance on the balance beam indicated that the time the mouse could remain on the beam depended on the age of treatment (Figure 5C). Untreated CLN2-/- mice remained on the balance beam for significantly less time than wild-type controls (p<0.0001). The two negative control groups were not significantly different from the untreated CLN2 -/- mice in the ability to remain on the beam (PBS, p>0.05, and AAVrh.10GFP, p>0.1, not shown). By contrast, all CLN2-/- mice with AAVrh.10hCLN2 administered to the CNS, regardless of their age of treatment, showed significantly better performance than untreated CLN2-/- mice, but the earlier the age of treatment, the greater the improvement in balance beam test performance. The 2 day and 3 wk treatment groups, while not different from each other (p>0.05), both performed better than mice treated at 7 wks of age (p<0.001), with the performance of the 2 day and 3 wk treated animals was equivalent to wild-type controls (p>0.05).
Assessment of CLN2-/- mice by performance on grip strength also showed that grip strength depends on the age of treatment (Figure 5D). The grip strength of untreated CLN2-/-mice was significantly less than that of wild-type controls (p<0.0001). The two negative control groups were not significantly different from the untreated CLN2 -/- mice in their ability to maintain grip (PBS, p>0.05, and AAVrh.10GFP, p>0.05, data not shown). By contrast, CLN2-/- mice with AAVrh.10hCLN2 administered to the CNS at all ages showed significantly stronger grip strength than untreated CLN2-/- mice, which was greatest for mice treated at the earliest age. Mice treated at either 2 days or 3 wk of age, while not different from each other (p>0.05), both performed better than mice treated at 7 wk of age (p<0.01) and as well as wild-type controls (p>0.05).
Impact on Survival
The most striking result was the correlation between earlier age of administration and extended lifespan of CLN2-/- mice (Figure 6). The median age of survival for untreated CLN2-/-mice was 121 days. Similarly the two negative control groups had a low median age of survival (PBS 117 days and AAVrh.10 GFP 120 days, not shown). By contrast, the lifespans of 2 day- and 3 wk-treated CLN2-/- mice were more than double that of the untreated CLN2-/- mice. The median ages of survival for CLN2-/- mice treated at 7 wk, 3 wk, and 2 days of age were 168 days, 277 days, and 373 days, respectively. Kaplan-Meier analysis of the survival data revealed a significant survival advantage for the 3 wk-treated-mice over 7 wk-treated-mice (p<0.001). Importantly, the lifespan of 2 day-treated-mice was significantly longer than both the 7 wk-treated mice (p<0.0001) and 3 wk-treated mice (p<0.01).
Figure 6.

Impact of age of AAVrh.10hCLN2 administration on survival of CLN2 -/- mice. Mice were administered AAVrh.10hCLN2 (1.05 to 1.6 × 1011 genome copies) by direct CNS administration at 7 wk, 3 wk, or 2 days of age. Uninjected controls (n=14, CLN2-/-), wild-type controls (n=16, CLN2 +/+), 7 wk AAVrh.10 injected (n=17, CLN2 -/-), 3 wk AAVrh.10 injected (n=11, CLN2 -/-), and 2 day AAVrh.10 injected (n=5, CLN2 -/-) mice were assessed 3 times per wk for signs of terminal decline. Mice that appeared moribund were sacrificed and age recorded.
Discussion
The biggest challenge for gene therapy for the treatment of the CNS manifestations of lysosomal storage diseases is to provide therapy that will have an impact throughout the brain. In this study, the impact of the age of treatment by an AAV serotype rh.10 vector on ameliorating the symptoms of LINCL was studied in the LINCL knockout mouse model. The data demonstrate the benefit of neonatal treatment with a 3-fold increase in life expectancy compared to untreated controls, a higher level and wider distribution of TPP-I protein within the brain than treatment at older ages, and a corresponding improvement in behavioral performance as a function of age. Together, these data suggest that neonatal treatment offers many unique advantages, and that both early detection and treatment are essential for effective genetic therapy with this type of disease. Also, in the context that LINCL is not included in routine newborn genetic screens (Fletcher, 2006; Meikle et al., 2006; NNSGRC, 2008), together with observations from other investigators of the success of neonatal gene transfer (Bostick et al., 2007; Broekman et al., 2007; Daly et al., 1999; Griffey et al., 2006; Li and Daly2002; Passini et al., 2003; Passini and Wolfe2001; Rafi et al., 2005; Shen et al., 2001; Waddington et al., 2004), the studies presented here have social implications in that they suggest that to achieve maximal therapeutic impact of currently available gene transfer vectors, newborn screening with early identification and therapy of this disorder will achieve better results than waiting until the disease manifests clinically.
Time of Therapy
The benefit of treatment as neonates may arise from a combination of factors, including, but not limited to: (1) therapeutically relevant levels of the deficient TPP-I enzyme can be achieved early in life, which may prevent the development of disease as opposed to reversing the established disease; and (2) earlier administration leads to a higher density and more widespread distribution of vector within the much smaller, and immature neonatal brain (Kohn and Parkman1997; Waddington et al., 2004).
The value of the neonatal window for treatment has been shown previously in an animal model of Krabbe's disease where animals treated at birth with an adenovirus vector coding for galactocerebrosidase showed an improvement compared to those treated 2 wk after birth (Shen et al., 2001). Passini et al (Passini et al., 2003; Passini and Wolfe2001) have shown in the mucopolysaccharidosis-VII (MPS-VII) animal model that intraparenchymal or intraventricular injection of an AAV2 vector encoding the beta-glucuronidase (GUSB) gene into neonatal mice led to widespread and prolonged GUSB activity. They hypothesized that the wider distribution of the GUSB transgene throughout the brain following intraventricular injection was due to more efficient transduction of the deeper neuronal structures (Passini et al., 2003). These reports, along with the data described in this paper, have important implications for supporting newborn screening programs and the timing of experimental therapies.
Disease Progression in Lysosomal Storage Diseases
Several lines of evidence suggest that earlier administration offers several unique advantages to treating lysosomal storage disorders (Kohn and Parkman1997; Waddington et al., 2004). Foremost among these is that, treating individuals before disease onset prevents the occurrence of irreversible damage. In the case of LINCL, there is widespread neurodegeneration (Haltia, 2003; Lane et al., 1996; Williams et al., 1999). As with other neurodegenerative diseases, the spatially limited endogenous neurogenesis in the postnatal brain is insufficient to restore function, thus it is important to halt the pathology of LINCL before irreparable damage occurs (Ming and Song, 2005). The onset of LINCL in the mouse knockout model has been estimated to be at 5 wk of age, with the earliest observed point of autofluorescent accumulation 2 wk prior to visible symptoms (Sleat et al., 2004). Studies testing the age of administration on the treatment of LINCL have operated under this assumption, testing therapy before and after at this age of onset. For example, Cabrera-Salazar et al. (Cabrera-Salazar et al., 2007) have shown that treatment of LINCL knockout mice at 4 wk of age has greatly increased therapeutic benefits as compared to 11 wk treated mice. In that study 80% survival was observed at 330 days when CLN2 knockout mice were injected with AAV1 vector expressing CLN2 gene at 4wk of age. In the present study, the enhanced survival observed for 2 day treated mice compared to 3 wk mice suggests an important pathological process may occur even further upstream of the presumed age of onset. The newborn mice survived 100% to >330 days, better than the mice treated at 4 wk of age by AAV1 vector (Cabrera-Salazar et al., 2007) as well as better than the mice treated with AAVrh.10 vector at 3 wk of age in this study.
A similar phenomenon has been observed in previous studies with the CLN1 deficient knockout mice, which models another member of the lysosomal storage disorders family (Griffey et al., 2006). In this case, morphologic abnormalities were detected in 7-month old mice, including autofluorescence-associated neuronal loss prior to the mice exhibiting the moribund phenotype (Griffey et al., 2006). Together, these studies argue strongly for the development of better detection methods in the lysosomal disorders, such as improved magnetic resonance imaging or more sensitive behavioral parameters. Earlier detection will therefore be a critical preventative measure to insure the disease does not progress into a destructive stage.
Unique Properties of the Neonatal Brain
In addition to preventing disease onset, therapeutic administration immediately following birth may also capitalize on unique features of the neonatal brain. The increased spread of the TPP-I transgene observed in 2 day treated mice, as visualized by histology and enzymatic assays, correlates with improved neuromotor function and extended life-span. Only in the 2 day administration group were most neurons throughout the brain found to express TPP-I (neuronal identity confirmed by co-expression of TPP-I with a mature neuronal marker, NeuN). In animals injected at later ages some TPP-I was detected at a distance from region of injection but only in the vasculature suggesting access through blood brain barrier disruption at the time of injection. There may be a few explanations for this wider distribution. First, earlier administration may lead to a higher density of vector particle to brain volume in the much smaller neonatal brain. Secondly, the reduced barriers to diffusion in the still developing neonatal brain may facilitate widespread distribution of delivered vector (Passini et al., 2003; Watson et al., 2005). Similar widespread distribution in neonatal brains compared to adult brain has been reported in cell grafting studies (Englund et al., 2002; Gates et al., 1998; Olsson et al., 1997; Sidman et al., 2007; Zhao et al., 2007; Zhou and Lund1993). Interestingly, even though TPP-I levels in the CLN2 -/- mice injected with AAVrh.10CLN2 as newborns were over 100 times higher than in wildtype mice, the injected mice did not survive as long as wildtype mice. Therefore, even if the distribution of TPP-I is wider when mice are injected as newborns, some critical areas of the brain may still not be receiving enough TPP-I for normal activity.
Significance for Human Studies
Given the data and the considerations described above, the implications for human studies are significant, as these data strongly support the concept that therapy will be most effective in neonates. Although the ethical issues regarding novel therapies are complex in neonates, the potential benefit is clearly much greater. For example, infantile Krabbe's disease was treated by transplanting umbilical-cord blood, rich in stem cells, in newborns and infants ranging from 12 to 352 days of age (Escolar et al., 2005). Compared to individuals treated post-symptomatically, these individuals showed improved motor and cognitive function, without the central demyelination that accompanies Krabbe's (Escolar et al., 2005).
In the case of LINCL, the diagnosis is typically not made until the age of >3 yr, at which point significant damage has already occurred (Steinfeld et al., 2002; Williams et al., 1999; Worgall et al., 2007). Diagnosis in newborns only occurs when an older sibling has already presented with the disease, leaving at risk newborns from parents who are unaware that they are carriers. The ease of newborn genetic screening argues for including the common mutations for the neurological lysosomal storage disorders in newborn genetic screening programs (Hayes et al., 2007; Kemper et al., 2007; Wilcken, 2007). For example, in 2006, New York became the first state to include Krabbe's disease in its Newborn Screening Program (2008; Therrell and Adams2007), setting the precedent for other infantile neuronal disorders. This study supports the inclusion of LINCL in similar programs, and for a broader spectrum of genetic tests for rare childhood monogenic disorders in general.
Acknowledgments
We thank P. Lobel and D. Sleat from University of Medicine and Dentistry of New Jersey for providing the CLN2 +/- breeding pairs and the rabbit anti-TPP-I antibody; K. Wisniewski and A. Golabek from NYS Institute for Basic Research for providing the anti-hCLN2 antibody; J. Ronda, M. Baad and S. Schuck for technical assistance; and N. Mohamed for help in preparing this manuscript. These studies were supported, in part, by U01 NS047458; Nathan's Battle Foundation, Greenwood, IN; and the Will Rogers Memorial Fund, Los Angeles, CA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beck M. New therapeutic options for lysosomal storage disorders: enzyme replacement, small molecules and gene therapy. Hum Genet. 2007;121:1–22. doi: 10.1007/s00439-006-0280-4. [DOI] [PubMed] [Google Scholar]
- Bostick B, Ghosh A, Yue Y, Long C, Duan D. Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Ther. 2007;14:1605–1609. doi: 10.1038/sj.gt.3303029. [DOI] [PubMed] [Google Scholar]
- Brady RO. Enzyme replacement for lysosomal diseases. Annu Rev Med. 2006;57:283–296. doi: 10.1146/annurev.med.57.110104.115650. [DOI] [PubMed] [Google Scholar]
- Broekman ML, Baek RC, Comer LA, Fernandez JL, Seyfried TN, Sena-Esteves M. Complete Correction of Enzymatic Deficiency and Neurochemistry in the GM1-gangliosidosis Mouse Brain by Neonatal Adeno-associated Virus-mediated Gene Delivery. Mol Ther. 2007;15:30–37. doi: 10.1038/sj.mt.6300004. [DOI] [PubMed] [Google Scholar]
- Broekman ML, Comer LA, Hyman BT, Sena-Esteves M. Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or -2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 2006;138:501–510. doi: 10.1016/j.neuroscience.2005.11.057. [DOI] [PubMed] [Google Scholar]
- Burger C, Nash K, Mandel RJ. Recombinant adeno-associated viral vectors in the nervous system. Hum Gene Ther. 2005;16:781–791. doi: 10.1089/hum.2005.16.781. [DOI] [PubMed] [Google Scholar]
- Cabrera-Salazar MA, Roskelley EM, Bu J, Hodges BL, Yew N, Dodge JC, Shihabuddin LS, Sohar I, Sleat DE, Scheule RK, Davidson BL, Cheng SH, Lobel P, Passini MA. Timing of therapeutic intervention determines functional and survival outcomes in a mouse model of late infantile batten disease. Mol Ther. 2007;15:1782–1788. doi: 10.1038/sj.mt.6300249. [DOI] [PubMed] [Google Scholar]
- Cardone M. Prospects for gene therapy in inherited neurodegenerative diseases. Curr Opin Neurol. 2007;20:151–158. doi: 10.1097/WCO.0b013e32809f97ff. [DOI] [PubMed] [Google Scholar]
- Cearley CN, Wolfe JH. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol Ther. 2006;13:528–537. doi: 10.1016/j.ymthe.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Chen MY, Lonser RR, Morrison PF, Governale LS, Oldfield EH. Variables affecting convection-enhanced delivery to the striatum: a systematic examination of rate of infusion, cannula size, infusate concentration, and tissue-cannula sealing time. J Neurosurg. 1999;90:315–320. doi: 10.3171/jns.1999.90.2.0315. [DOI] [PubMed] [Google Scholar]
- Crystal RG, Sondhi D, Hackett NR, Kaminsky SM, Worgall S, Stieg P, Souweidane M, Hosain S, Heier L, Ballon D, Dinner M, Wisniewski K, Kaplitt M, Greenwald BM, Howell JD, Strybing K, Dyke J, Voss H. Clinical protocol. Administration of a replication-deficient adeno-associated virus gene transfer vector expressing the human CLN2 cDNA to the brain of children with late infantile neuronal ceroid lipofuscinosis. Hum Gene Ther. 2004;15:1131–1154. doi: 10.1089/hum.2004.15.1131. [DOI] [PubMed] [Google Scholar]
- Daly TM, Vogler C, Levy B, Haskins ME, Sands MS. Neonatal gene transfer leads to widespread correction of pathology in a murine model of lysosomal storage disease. Proc Natl Acad Sci USA. 1999;96:2296–2300. doi: 10.1073/pnas.96.5.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deglon N, Hantraye P. Viral vectors as tools to model and treat neurodegenerative disorders. J Gene Med. 2005;7:530–539. doi: 10.1002/jgm.707. [DOI] [PubMed] [Google Scholar]
- Englund U, Fricker-Gates RA, Lundberg C, Bjorklund A, Wictorin K. Transplantation of human neural progenitor cells into the neonatal rat brain: extensive migration and differentiation with long-distance axonal projections. Exp Neurol. 2002;173:1–21. doi: 10.1006/exnr.2001.7750. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, Wenger DA, Pietryga D, Wall D, Champagne M, Morse R, Krivit W, Kurtzberg J. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Fletcher JM. Screening for lysosomal storage disorders--a clinical perspective. J Inherit Metab Dis. 2006;29:405–408. doi: 10.1007/s10545-006-0246-7. [DOI] [PubMed] [Google Scholar]
- Gates MA, Olsson M, Bjerregaard K, Bjorklund A. Region-specific migration of embryonic glia grafted to the neonatal brain. Neuroscience. 1998;84:1013–1023. doi: 10.1016/s0306-4522(97)00512-5. [DOI] [PubMed] [Google Scholar]
- Griffey MA, Wozniak D, Wong M, Bible E, Johnson K, Rothman SM, Wentz AE, Cooper JD, Sands MS. CNS-directed AAV2-mediated gene therapy ameliorates functional deficits in a murine model of infantile neuronal ceroid lipofuscinosis. Mol Ther. 2006;13:538–547. doi: 10.1016/j.ymthe.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Hackett NR, Redmond DE, Sondhi D, Giannaris EL, Vassallo E, Stratton J, Qiu J, Kaminsky SM, Lesser ML, Fisch GS, Rouselle SD, Crystal RG. Safety of direct administration of AAV2(CU)hCLN2, a candidate treatment for the central nervous system manifestations of late infantile neuronal ceroid lipofuscinosis, to the brain of rats and nonhuman primates. Hum Gene Ther. 2005;16:1484–1503. doi: 10.1089/hum.2005.16.1484. [DOI] [PubMed] [Google Scholar]
- Haltia M. The neuronal ceroid-lipofuscinoses. J Neuropathol Exp Neurol. 2003;62:1–13. doi: 10.1093/jnen/62.1.1. [DOI] [PubMed] [Google Scholar]
- Harding TC, Dickinson PJ, Roberts BN, Yendluri S, Gonzalez-Edick M, Lecouteur RA, Jooss KU. Enhanced gene transfer efficiency in the murine striatum and an orthotopic glioblastoma tumor model, using AAV-7- and AAV-8-pseudotyped vectors. Hum Gene Ther. 2006;17:807–820. doi: 10.1089/hum.2006.17.807. [DOI] [PubMed] [Google Scholar]
- Hayes IM, Collins V, Sahhar M, Wraith JE, Delatycki MB. Newborn screening for mucopolysaccharidoses: opinions of patients and their families. Clin Genet. 2007;71:446–450. doi: 10.1111/j.1399-0004.2007.00783.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann B, Mayatepek E. Neurological manifestations in lysosomal storage disorders -from pathology to first therapeutic possibilities. Neuropediatrics. 2005;36:285–289. doi: 10.1055/s-2005-872810. [DOI] [PubMed] [Google Scholar]
- Hsich G, Sena-Esteves M, Breakefield XO. Critical issues in gene therapy for neurologic disease. Hum Gene Ther. 2002;13:579–604. doi: 10.1089/10430340252837198. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Dwek RA, Butters TD, Platt FM. Storage solutions: treating lysosomal disorders of the brain. Nat Rev Neurosci. 2005;6:713–725. doi: 10.1038/nrn1725. [DOI] [PubMed] [Google Scholar]
- Kemper AR, Hwu WL, Lloyd-Puryear M, Kishnani PS. Newborn screening for Pompe disease: synthesis of the evidence and development of screening recommendations. Pediatrics. 2007;120:e1327–e1334. doi: 10.1542/peds.2007-0388. [DOI] [PubMed] [Google Scholar]
- Kohn DB, Parkman R. Gene therapy for newborns. FASEB J. 1997;11:635–639. doi: 10.1096/fasebj.11.8.9240965. [DOI] [PubMed] [Google Scholar]
- Lane SC, Jolly RD, Schmechel DE, Alroy J, Boustany RM. Apoptosis as the mechanism of neurodegeneration in Batten's disease. J Neurochem. 1996;67:677–683. doi: 10.1046/j.1471-4159.1996.67020677.x. [DOI] [PubMed] [Google Scholar]
- Li J, Daly TM. Adeno-associated virus-mediated gene transfer to the neonatal brain. Methods. 2002;28:203–207. doi: 10.1016/s1046-2023(02)00224-4. [DOI] [PubMed] [Google Scholar]
- Mandel RJ, Manfredsson FP, Foust KD, Rising A, Reimsnider S, Nash K, Burger C. Recombinant adeno-associated viral vectors as therapeutic agents to treat neurological disorders. Mol Ther. 2006;13:463–483. doi: 10.1016/j.ymthe.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Meikle PJ, Grasby DJ, Dean CJ, Lang DL, Bockmann M, Whittle AM, Fietz MJ, Simonsen H, Fuller M, Brooks DA, Hopwood JJ. Newborn screening for lysosomal storage disorders. Mol Genet Metab. 2006;88:307–314. doi: 10.1016/j.ymgme.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–250. doi: 10.1146/annurev.neuro.28.051804.101459. [DOI] [PubMed] [Google Scholar]
- National Newborn Screening and Genetics Resource Center (NNSGRC) [Wed April 2, 2008];National Newborn Screening Status Report. 2008 http://genes-r-us.uthscsa.edu/nbsdisorders.pdf Last.
- Neufeld EF. Lysosomal storage diseases. Annu Rev Biochem. 1991;60:257–280. doi: 10.1146/annurev.bi.60.070191.001353. [DOI] [PubMed] [Google Scholar]
- Olsson M, Bentlage C, Wictorin K, Campbell K, Bjorklund A. Extensive migration and target innervation by striatal precursors after grafting into the neonatal striatum. Neuroscience. 1997;79:57–78. doi: 10.1016/s0306-4522(96)00606-9. [DOI] [PubMed] [Google Scholar]
- Passini MA, Dodge JC, Bu J, Yang W, Zhao Q, Sondhi D, Hackett NR, Kaminsky SM, Mao Q, Shihabuddin LS, Cheng SH, Sleat DE, Stewart GR, Davidson BL, Lobel P, Crystal RG. Intracranial delivery of CLN2 reduces brain pathology in a mouse model of classical late infantile neuronal ceroid lipofuscinosis. J Neurosci. 2006;26:1334–1342. doi: 10.1523/JNEUROSCI.2676-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Watson DJ, Vite CH, Landsburg DJ, Feigenbaum AL, Wolfe JH. Intraventricular brain injection of adeno-associated virus type 1 (AAV1) in neonatal mice results in complementary patterns of neuronal transduction to AAV2 and total long-term correction of storage lesions in the brains of beta-glucuronidase-deficient mice. J Virol. 2003;77:7034–7040. doi: 10.1128/JVI.77.12.7034-7040.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Wolfe JH. Widespread gene delivery and structure-specific patterns of expression in the brain after intraventricular injections of neonatal mice with an adeno-associated virus vector. J Virol. 2001;75:12382–12392. doi: 10.1128/JVI.75.24.12382-12392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafi MA, Zhi RH, Passini MA, Curtis M, Vanier MT, Zaka M, Luzi P, Wolfe JH, Wenger DA. AAV-mediated expression of galactocerebrosidase in brain results in attenuated symptoms and extended life span in murine models of globoid cell leukodystrophy. Mol Ther. 2005;11:734–744. doi: 10.1016/j.ymthe.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Shen JS, Watabe K, Ohashi T, Eto Y. Intraventricular administration of recombinant adenovirus to neonatal twitcher mouse leads to clinicopathological improvements. Gene Ther. 2001;8:1081–1087. doi: 10.1038/sj.gt.3301495. [DOI] [PubMed] [Google Scholar]
- Shevtsova Z, Malik JM, Michel U, Bahr M, Kugler S. Promoters and serotypes: targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp Physiol. 2005;90:53–59. doi: 10.1113/expphysiol.2004.028159. [DOI] [PubMed] [Google Scholar]
- Sidman RL, Li J, Stewart GR, Clarke J, Yang W, Snyder EY, Shihabuddin LS. Injection of mouse and human neural stem cells into neonatal Niemann-Pick A model mice. Brain Res. 2007;1140:195–204. doi: 10.1016/j.brainres.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Wiseman JA, El-Banna M, Kim KH, Mao Q, Price S, Macauley SL, Sidman RL, Shen MM, Zhao Q, Passini MA, Davidson BL, Stewart GR, Lobel P. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24:9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondhi D, Hackett NR, Apblett RL, Kaminsky SM, Pergolizzi RG, Crystal RG. Feasibility of gene therapy for late neuronal ceroid lipofuscinosis. Arch Neurol. 2001;58:1793–1798. doi: 10.1001/archneur.58.11.1793. [DOI] [PubMed] [Google Scholar]
- Sondhi D, Hackett NR, Peterson DA, Stratton J, Baad M, Travis KM, Wilson JM, Crystal RG. Enhanced Survival of the LINCL Mouse Following CLN2 Gene Transfer Using the rh. 10 Rhesus Macaque-derived Adeno-associated Virus Vector. Mol Ther. 2007;15:481–491. doi: 10.1038/sj.mt.6300049. [DOI] [PubMed] [Google Scholar]
- Steinfeld R, Heim P, von GH, Meyer K, Ullrich K, Goebel HH, Kohlschutter A. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am J Med Genet. 2002;112:347–354. doi: 10.1002/ajmg.10660. [DOI] [PubMed] [Google Scholar]
- Taymans JM, Vandenberghe LH, Haute CV, Thiry I, Deroose CM, Mortelmans L, Wilson JM, Debyser Z, Baekelandt V. Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Hum Gene Ther. 2007;18:195–206. doi: 10.1089/hum.2006.178. [DOI] [PubMed] [Google Scholar]
- Therrell BL, Adams J. Newborn screening in North America. J Inherit Metab Dis. 2007;30:447–465. doi: 10.1007/s10545-007-0690-z. [DOI] [PubMed] [Google Scholar]
- Vellodi A. Lysosomal storage disorders. Br J Haematol. 2005;128:413–431. doi: 10.1111/j.1365-2141.2004.05293.x. [DOI] [PubMed] [Google Scholar]
- Waddington SN, Kennea NL, Buckley SM, Gregory LG, Themis M, Coutelle C. Fetal and neonatal gene therapy: benefits and pitfalls. Gene Ther. 2004;11 Suppl 1:S92–S97. doi: 10.1038/sj.gt.3302375. [DOI] [PubMed] [Google Scholar]
- Watson DJ, Passini MA, Wolfe JH. Transduction of the choroid plexus and ependyma in neonatal mouse brain by vesicular stomatitis virus glycoprotein-pseudotyped lentivirus and adeno-associated virus type 5 vectors. Hum Gene Ther. 2005;16:49–56. doi: 10.1089/hum.2005.16.49. [DOI] [PubMed] [Google Scholar]
- Wilcken B. Recent advances in newborn screening. J Inherit Metab Dis. 2007;30:129–133. doi: 10.1007/s10545-007-0538-6. [DOI] [PubMed] [Google Scholar]
- Williams RE, Gottlob I, Lake BD, Goebel HH, Winchester BG, Wheeler RB. CLN2 Classic Late Infantile NCL. In: Goebel HH, editor. The Neuronal Ceroid Lipofuscinoses (Batten Disease) IOS Press; 1999. pp. 37–53. [Google Scholar]
- Worgall S, Kekatpure MV, Heier L, Ballon D, Dyke JP, Shungu D, Mao X, Kosofsky B, Kaplitt MG, Souweidane MM, Sondhi D, Hackett NR, Hollmann C, Crystal RG. Neurological deterioration in late infantile neuronal ceroid lipofuscinosis. Neurology. 2007;69:521–535. doi: 10.1212/01.wnl.0000267885.47092.40. [DOI] [PubMed] [Google Scholar]
- Zhao G, McCarthy NF, Sheehy PA, Taylor RM. Comparison of the behavior of neural stem cells in the brain of normal and twitcher mice after neonatal transplantation. Stem Cells Dev. 2007;16:429–438. doi: 10.1089/scd.2006.0092. [DOI] [PubMed] [Google Scholar]
- Zhou H, Lund RD. Effects of the age of donor or host tissue on astrocyte migration from intracerebral xenografts of corpus callosum. Exp Neurol. 1993;122:155–164. doi: 10.1006/exnr.1993.1116. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Potter M, Hauswirth WW, Guy J, Muzyczka N. A “humanized” green fluorescent protein cDNA adapted for high-level expression in mammalian cells. J Virol. 1996;70:4646–4654. doi: 10.1128/jvi.70.7.4646-4654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]