

Abstract
IL-23, an IL-12 family member, has been implicated in the development of TH17 cells and the progression of autoimmune diseases. However, due to the lack of availability of sensitive antibody reagents specific for the IL-23 receptor (IL-23R), it has been difficult to characterize the cell types that express the IL-23R and are responsive to IL-23 in vivo. To address the role of IL-23 in vivo, we have generated a novel “knock-in” mouse, in which we have replaced the intracellular domain of the IL-23R by the green fluorescent protein (GFP). We show that in addition to TH17 cells, a subset of myeloid cells express IL-23R and respond to IL-23 by producing IL-17 and IL-22. Our studies further demonstrate that IL-23R expression is crucial for generation of encephalitogenic Th17 cells, but its expression on the innate immune system is dispensible in the development of experimental autoimmune encephalomyelitis (EAE).
Introduction
IL-23, a heterodimeric cytokine composed of p19 and p40 chain, plays a critical role in the development of autoimmune diseases in that the genetic loss of either p19 or p40 chain makes mice resistant to a number of autoimmune diseases (1). Similarly, polymorphisms in IL-23R have been linked to genetic susceptibility to a number of human autoimmune diseases including Psoriasis and Crohn's disease (2, 3). These data suggest an important role of IL-23:IL-23R pathway in the development of many autoimmune diseases but the mechanism by which IL-23/IL-23R pathway operates in vivo is not clear. Progress in this area is further hampered because of low expression and the lack of appropriate antibody reagents to identify and track IL-23R bearing cells in vivo. Recent studies have suggested that besides its role as growth/stabilization factor for TH17 cells, IL-23 might have an important role in innate immunity; however, innate immune cells that are responsive to IL-23 have not been characterized (4). To identify the cell types (innate or adaptive) responding to IL-23 and their effector functions under various inflammatory conditions (infection or autoimmunity), we generated an IL-23R reporter mouse. In this mouse, IL-23R-expressing cells can be followed by their expression of GFP, and their responsiveness to IL-23 can be eliminated when this mouse is bred as homozygote.
Material and Methods
Mice
C57BL/6 wild type (WT) from Jackson laboratories. WT and heterozygous IL-23R-KI mice were housed in a conventional, pathogen-free facility at 65, Landsdowne Street (Cambridge, MA) and at Harvard Institutes of Medicine. All experiments were performed in accordance with guidelines prescribed by the standing committee of animals at Harvard Medical School.
Generation of the IL-23R-KI mice
A BAC clone (RP23-204M15 containing IL-23R gene was used as a template for PCR amplification to generate 5.1 kb and 1.8 kb arms that were subcloned into EGFP containing TKPbs-LoxP-Neo cassette. The targeting construct was electroporated into Bruce4 ES cells. Targeted ES cells were injected into BALB/c blastocysts and male chimeras were bred with female C57BL/6.
Preparation of CNS mononuclear cells
CNS tissue was cut into pieces and digested with collagenase D (2.5 mg/ml, Roche Diagnostics, Indianapolis, IN) and DNAseI (1 mg/ml, Sigma, Saint Louis, MO) at 37 °C for 45 min. Mononuclear cells were isolated by a percoll gradient (70%/37%) centrifugation. Mononuclear cells were removed from the interphase, washed and resuspended in culture medium for further analysis.
Recall response with MOG35-55 peptide
WT and IL-23R-KI mice were immunized with 100ug MOG35-55 peptide emulsified in CFA. On day 8 after immunization, spleen and lymph nodes cells were prepared and cultured with 20 ug/ml of MOG35-55 and 25 ng/ml of rIL-23. After 4 days of in vitro stimulation, cells were analyzed for IL-23R/GFP expression.
Intracellular cytokine staining
Cells were re-stimulated with phorbol 12-myristate 13-acetate (PMA, 50 ng/ml, Sigma), ionomycin (1ug/ml, Sigma), and GolgiStop (1μl/ml, BD Bioscience) at 37°C/10% CO2 for 4 h followed by surface and intracellular staining according to the manufactures' (BD Bioscience) instruction. Cells were analyzed by FACS (BD Biosciences).
Cytokine analysis and real time PCR analysis
Cytokines from culture supernatants were determined by either ELISA or cytometric bead array (BD Bioscience). RNA was extracted after 48h of in vitro stimulation using RNAeasy columns (Qiagen, Valencia, CA) and subjected to quantitative RT-PCR according to the manufactures' instruction (Applied Biosystems). Primer/probe mixtures of mouse IL-17A, IL-23R, IFN-γ, T-bet, and ROR-γt were obtained from Applied Biosystems.
Results and Discussion
Generation of IL-23R-GFP.KI reporter mice
To identify the cell types responding to IL-23 in vivo, we generated a knock-in “reporter” mouse, in which an IRES-GFP cassette was introduced after exon 8 in the endogenous IL-23R gene (Fig.1A). We obtained three independent ES cell clones that were appropriately targeted as shown by southern blot, and with two of the targeted clones germ line transmission was achieved (Fig. 1B).
Figure 1.
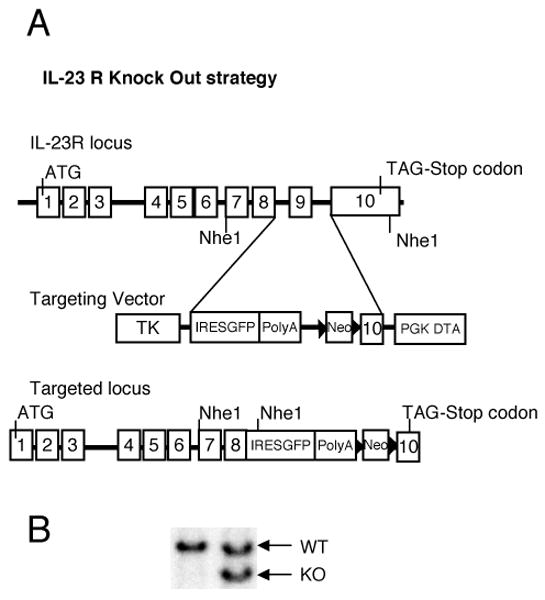
Generation of IL-23R reporter mice. A, IL-23 genomic locus and targeting strategy. An IRES-EGFP cassette was introduced right after the EXON8 of the endogenous IL-23R gene. B, Southern blot was used to identify the properly targeted ES cells. DNA purified from ES cells was digested by NheI and probed with a 1.1 kb specific probe generated by PCR. The hybridized 15.7 kb fragment corresponds to the wild type allele, and the hybridized 6.8 kb fragment corresponds to the targeted allele.
In heterozygote mice, IL-23R-expressing cells can be followed by their expression of GFP, and when bred as homozygotes, the deletion of the IL-23R abrogates their responsiveness to IL-23. IL-23R-GFP.KI and IL-23R-/- mice were healthy and fertile and had similar numbers of CD4, CD8, B cells, γδ T cells, CD11c+ and CD11b+ cells as WT control mice (data not shown).
IL-23R-expressing cells are present in the lymph node and are enriched in the lamina propria (LP)
To determine the expression of IL-23R at the single cell level, we examined the expression of GFP in the lymphoid tissues of naïve IL-23R-KI and WT mice. We did not find any IL-23R(GFP) expression on CD8+ T cells, B cells or NK cells (data not shown). However, a small percentage of IL-23R expressing cells (∼1.4%) were CD3+ T cells and were detected mainly in the lymph nodes but not in the spleen (Fig. 2A). Only a minority of the GFP expressing T cells were CD4+ T cells (∼0.2%, data not shown) while the majority of them were γδ-T cells. In fact ∼40% of the γδ-T cells present in the LN were positive for IL-23R(GFP) expression and thus constitute the major IL-23R expressing T cell subset in the lymphoid organs of naïve animals. The other subsets of cells expressing IL-23R were CD11b+ macrophages (∼6%) and CD11c+ DCs (∼4%) (Fig. 2A). Notably, the IL-23R positive subsets are mainly located in the LN but not in the spleen of naïve animals and are mainly composed of αβ–T cells, macrophages and γδ-T cells.
Figure 2.
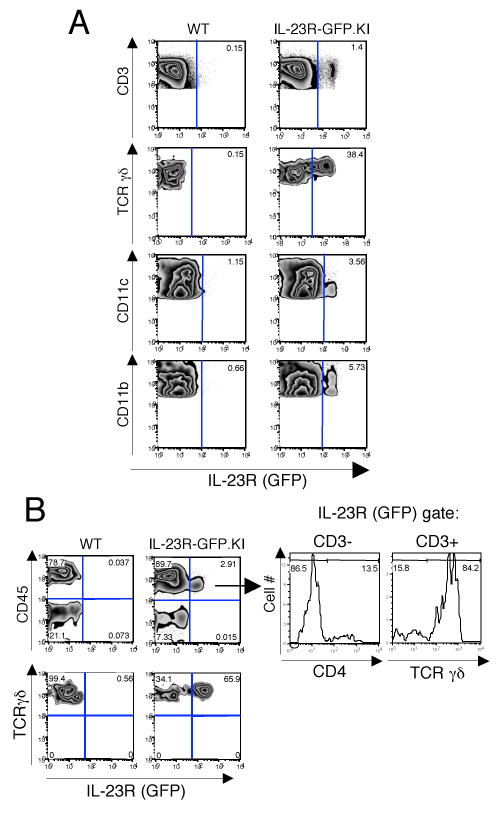
IL-23R expressing cells are enriched in the lamina propria (LP) of naive mice. Cells from lymph nodes, spleen and LP were prepared from naive WT and IL-23R-GFP.KI mice, stained and analyzed by flow cytometry. A, Lymph node cells were stained for CD3, TCRγδ, CD11c, and CD11b. B, IL-23R(GFP) expression was also analyzed in LP cells. LP cells were stained for CD45 and TCRγδ (B left panel). The expression of CD3, CD4, and TCRγδ was analyzed in the CD45+IL-23R/GFP+ compartment of LP cells (B right panel).
In contrast to the small frequency of IL-23R(GFP) expressing cells in secondary lymphoid organs, IL-23R(GFP) positive cells were prominently found in the LP. Here, about 3% of all CD45+ cells expressed IL-23R(GFP) while the majority of γδ-T cells (∼65%) expressed IL-23R(GFP) (Fig. 2B). Interestingly, LP DCs did not show any expression of IL-23R(GFP). Similarly to the LN, only a small fraction of the macrophage subsets expressed IL-23R/GFP (data not shown). When further defining the IL-23R(GFP) positive cells in the intestinal LP, we identified fraction of ∼13% CD4+ IL-23R(GFP) expressing cells that did not express CD3 (Fig. 2B right panel). These cells were derived from hematopoietic precursors since they expressed CD45 and were reminiscent of previously described Lymphoid tissue inducer (LTi) cells, which also express the transcription factor ROR-γt and produce IL-17(5,Takatori, 2009 #693).
IL-23R-/- mice have a defective TH17 response and are resistant to EAE
Since IL-23 plays an important role in EAE, we first assessed whether IL-23R-/- mice, like p19 deficient mice (6), are resistant to EAE by immunizing WT and IL-23R-/- mice with MOG35-55 emulsified in CFA. Whereas WT mice developed signs of EAE on day 14 reaching the peak of disease on day 20 after immunization, IL-23R-/- mice were completely resistant to disease (Fig. 3A). Since heterozygous IL-23R-GFP.KI mice were as susceptible to EAE as WT mice (data not shown), we took advantage of the GFP reporter to track IL-23R bearing cells during active EAE. Approximately 25% of the CD4+ T cells infiltrating the inflamed CNS were positive for IL-23R(GFP) expression and the vast majority (68%) of CD4+GFP+ cells produced IL-17. In contrast, GFP- CD4+ T cells predominantly produced IFN-γ but not IL-17 (Fig. 3B). These data establish that IL-23R is expressed on T cells at the site of inflammation and the expression correlates with IL-17 production suggesting that IL-23R defines a population of infiltrating proinflammatory TH17 cells in the target tissue. The dynamics of IL-23R expression and its in vivo functions in activated/memory T cells are not clear. To understand its functional consequences, WT and IL-23R-/- mice were immunized with MOG35-55/CFA LN cells and splenocytes were re-stimulated in vitro with MOG35-55 peptide alone or in the presence of IL-23. The relative lack of antigen specific TH17 cells in the spleen but not in the draining LN of IL-23R-/- animals (Fig. 3C) suggests TH17 cells might be induced in draining LN in an IL-23R-independent manner but may require IL-23R signalling to stably develop into TH17 cells or live long enough to leave the LN to populate the spleen and infiltrate tissues. Alternatively, IL-23 might also endow developing TH17 cells with trafficking properties essential for their egress from LN, migration into the spleen and infiltration into the target tissues. IL-17 producing CD4+ T cells were markedly decreased in the spleens of IL-23R-/- mice as compared to MOG-recall cultures of WT splenocytes. Activation of T cells in the presence of IL-23 further up-regulated the frequency of TH17 cells both in LN (∼58%) and spleen (∼48%) in WT but not in the IL-23R-/- CD4+ T cells from LN and spleen (Fig. 3C). Thus, IL-23R expression on recently activated T cells is essential for expanding/maintaining the TH17 population. If IL-23 were essential for the stable development of long-lasting TH17 cells, one would expect that IL-23R-/- mice would have a defect in memory TH17 responses. To address this, memory T cells from WT and IL-23R-/- mice were sorted based on their CD4+CD62L- profile and were activated in the presence of IL-23. The percentage of memory TH17 cells in WT mice was close to 4% whereas IL-23R-/- mice had about half that fraction (Fig. 3D). Memory TH17 cells from WT mice responded vigorously to IL-23 with the expansion of IL-17 producing cells (from 4 to 18%) whereas IL-23-/- mice did not expand TH17 cells above the baseline suggesting that IL-23/IL-23R signalling is absolutely required for the expansion/maintenance of TH17 memory T cells. However, since IL-23R is expressed on both effector T cells and innate immune cells, resistance to EAE in IL-23R-/- mice could be due to a defect in IL-17 production in either of these two compartments. Therefore, we immunized WT and IL-23R-/- mice with MOG35-55/CFA and 7 days after immunization harvested draining LN cells that were stimulated in vitro with MOG35-55 in the presence of IL-23. Equal numbers of activated T cell blast were adoptively transferred into IL-23R-competent naïve syngenic WT hosts. Whereas recipients of WT T cells developed EAE, MOG35-55 specific LN cells lacking IL-23R failed to transfer disease (Fig. 3E). We conclude that MOG-specific CD4+, TCR α/β+, IL-23R+ cells are crucial for inducing EAE since lack of IL-23R on MOG-specific T cells abrogates their encephalitogenicity. Our results are similar to the data recently published by Dan Cua and colleagues that have has also generated an IL-23R deficient and showed that IL-23R expression on TH17 cells is crucial for their effector function (7). However in this study the role of IL-23R on innate cells was not addressed.
Figure 3.
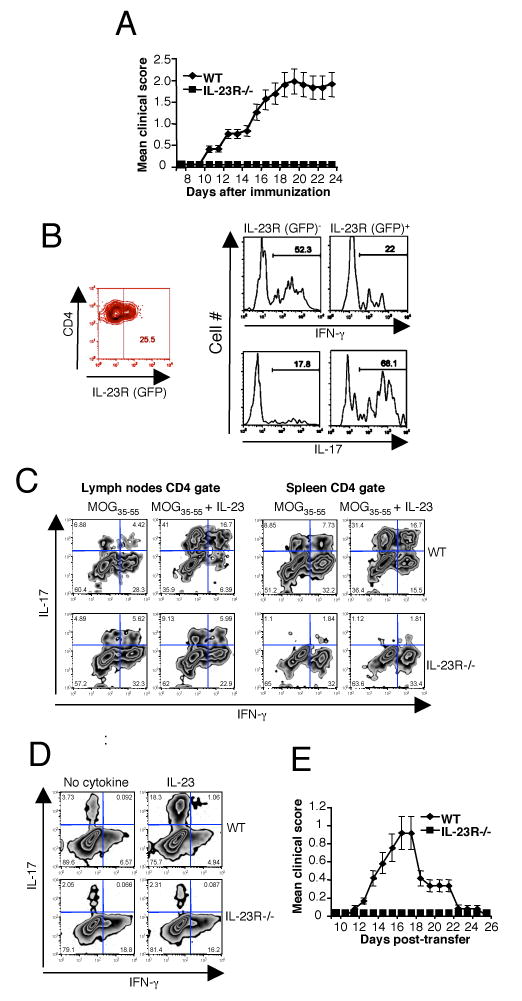
IL-23R-/- mice have a defective TH17 response and are resistant to EAE. A, EAE was induced in WT and IL-23R-/- mice by immunization with MOG35-55 emulsified in CFA. The course of EAE in these mice were monitored and shown as mean clinical score. B, IL-23R-GFP.KI mice were immunized with MOG35-55/CFA. On day 16 at the peak of disease, mononuclear cells were isolated from the CNS and stimulated with PMA/ionomycin. Intracellular cytokine staining for IFN-γ and IL-17 was performed. The histograms represent the percentages of cytokine positive cells in the CD4+IL-23R/GFP-and CD4+IL-23R/GFP+ gates, respectively. C, WT and IL-23R-/- mice were immunized with MOG35-55/CFA. On day 8, spleens (right panel) and LNs (left panel) were collected and cultured with MOG35-55 alone or in the presence of rIL-23 for 6-8 days. Cells were re-stimulated with PMA/Ionomycin before intracellular cytokine staining for IL-17 and IFN-γ was performed. Quadrants represent intracellular cytokine cytokine staining in the CD4+ T cell gate. D, CD4+CD44+ memory T cells from WT or IL-23R-/- mice were activated with anti-CD3 in the presence rIL-23 (30 ng/ml) with irradiated syngenic APCs. Four days later cells were re-stimulated with PMA/Ionomycin and intracellular cytokine staining was performed for IL-17 and IFN-γ. E, For passive transfer of EAE, MOG35-55 specific T cells from immunized WT or IL-23R-/- mice were cultured in the presence of MOG35-55 with rIL-23 (30 ng/ml). On day 8 CD4+ T cells were reactivated with plate bound anti-CD3 and anti-CD28 for 48 hrs. Equal number of MOG35-55 specific CD4+ T cells either from WT or IL-23-/- mice were transferred into WT recipient. The course of the EAE was monitored and is shown as mean clinical score.
IL-23R expression on innate immune system is dispensable for the development of EAE
To address which subsets of innate cells express IL-23R(GFP), we analyzed IL-23R-GFP.KI mice crossed onto a RAG2-deficient background (Fig 4A). In the LN of these mice we could detect a subset of IL-23R(GFP) expressing cells that also expressed the myeloid markers CD11b and CD11c (data not shown). We also immunized IL-23R-GFP.KI mice with MOG35-55/CFAand found that in the draining LN a similar subset of CD11b+CD11c+ DCs that expressed MHC class II (Fig. 4B). This subset of IL-23R(GFP)+ myeloid cells, in contrast to its IL-23R/GFP- counterparts, responded to IL-23 by expression of ROR-γt, and IL-17 (Fig. 4B right panel). Furthermore, in response to IL-23, IL-23R(GFP)+ CD11c+ LN cells also produced large amounts of IL-6 (Fig.4C). Because these cells expressed TGF-β as well, our data suggest that a subset of LN-derived myeloid DCs that express IL-23R might be particularly well equipped to induce TH17 response. Hence, in addition to T cells, macrophages/monocytes also express IL-23R and these cells may not only induce tissue inflammation by producing IL-17, but may also be involved in de novo differentiation of TH17 cells via their production of IL-6 and TGF-β(8, 9). To assess whether innate cells that express IL-23R also contribute to the development of EAE, we first transferred MOG specific TH17 cells into RAG2-/- or IL23R-/-RAG2-/- recipients. Both genotypes developed EAE with similar clinical course (Fig 4D), indicating that TH17 cells do not require IL-23R expression on innate immunity cells to promote EAE. However, these innate cells that express IL-23R may be involved in inducing pathogenic T cells. To address this issue, we transferred naïve T cells derived from MOG specific TCR transgenic mice into RAG or RAG IL-23R-/- recipients and immunized these mice with MOG/CFA for development of EAE. Both genotypes developed severe EAE with a similar kinectics and disease severity (Fig. 4E) suggesting that the major effect of IL-23 on EAE is to expand and stabilize proinflammatory Th17 cells while its effect on innate cells is dispensable for the development of EAE. This raises the issue of what is the role of IL-23R in innate immune system in EAE. One can speculate that responsiveness to IL-23 in innate immune cells in the CNS may induce the initial wave of IL-17 and make the target tissues responsive to tissue inflammation. Furthermore, IL-23R expression may define a subset of APCs that are particularly equipped to mediate epitope spreading by inducing pathogenic TH responses within the target tissue that thus further propagate autoimmunity and tissue inflammation in the CNS. Therefore it will be interesting to study the role of IL-23R in the model of PLP induced EAE in which epitope spreading is crucial for the relapsing remitting course of the disease.
Figure 4.
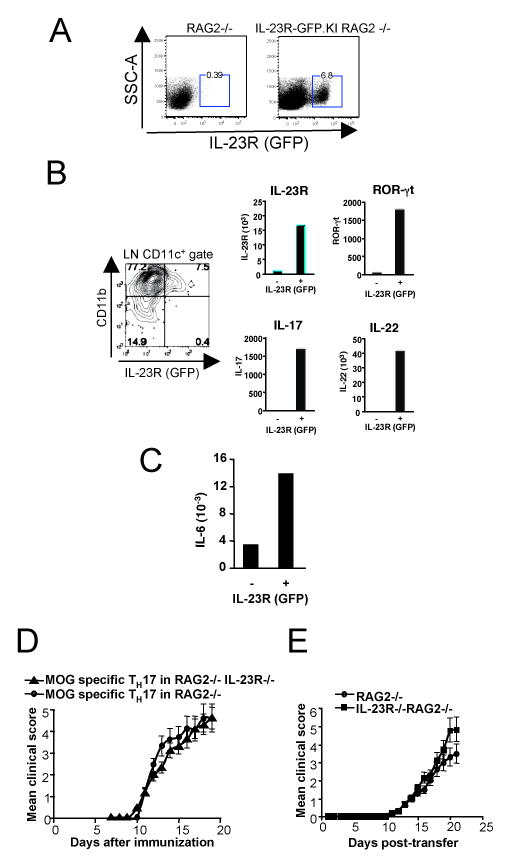
IL-23R expression on APCs is dispensable for the development of EAE. A, Single cell suspensions were prepared from Lymph nodes (LNs) from naive either RAG2 -/- or IL-23R-GFP.KI RAG2 -/- mice and IL-23R(GFP) expression was analyzed on total LNs' cells. B, IL-23R-GFP.KI mice were immunized with MOG35-55/CFA. On day 17, IL-23R(GFP) expression was analyzed on myeloid cell populations as indicated in histogram. C, MOG specific TH17 cells were i.v transferred into RAG2-/- and RAG2-/-IL-23R-/- mice. The course of EAE in these mice were monitored and shown as mean clinical score. D, Naïve T cells derived from MOG specific mice were i.v transferred into RAG2-/- or IL-23R-/- RAG2-/- mice. 24 h later, the mice were immunized with MOG35-55 emulsified in CFA. The course of EAE in these mice were monitored and shown as mean clinical score.
In summary, our studies demonstrate in vivo that IL-23R is not only expressed on T cells but it expressed on various innate immune cells. The innate immune cells respond to IL-23 by producing IL-17 and IL-22, however, the IL-23R is dispensable of the induction of acute EAE. On the other hand, loss of IL-23R is absolutely required for the myelin specific T cells to induce EAE. This is surprising since recent studies suggest that IL-17 is dispensable for induction of EAE. It is important to emphasize though that loss of tissue inflammation and resistance to autoimmunity, observed in the IL-23R mice, may not just be due to loss of IL-17 but also to the loss of other cytokines like IL-22 produced by TH17 upon activation with IL-23.
Acknowledgments
We thank D. Kozoriz for cell sorting and technical assistance.
Footnotes
This work was supported by grants from National Institutes of Health to M.O., E.B., and V.K.K. And National Multiple Sclerosis Society (RG-2571-D-9 to V.K.K. and RG-3882-A-1 to M.O. A.A. and E.B. by a transition award (TA 3014A1/1) from the National Multiple Sclerosis Society (NMSS), New York. L.R.B. is supported by a post-doctoral fellowship from EMBO. A.J. is the recipient of a PhD scholarship by the Boehringer Ingelheim Fonds. T.K. is supported by the Deutsche Forschungsgemeinschaft.
Abbreviations used in this paper: IL-23R, IL-23 receptor, EAE, experimental autoimmune encephalomyelitis, LP, lamina propria, LN, lymph nodes.
References
- 1.Kastelein RA, Hunter CA, Cua DJ. Discovery and Biology of IL-23 and IL-27: Related but Functionally Distinct Regulators of Inflammation. Annu Rev Immunol. 2007;25:221–224. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 2.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, Matsunami N, Ardlie KG, Civello D, Catanese JJ, Leong DU, Panko JM, McAllister LB, Hansen CB, Papenfuss J, Prescott SM, White TJ, Leppert MF, Krueger GG, Begovich AB. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eberl G, Littman DR. Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science. 2004;305:248–251. doi: 10.1126/science.1096472. [DOI] [PubMed] [Google Scholar]
- 6.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 7.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O'Shea JJ, Cua DJ. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 9.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]