

Abstract
Purpose
We previously reported that BRCA1 interacts with aryl hydrocarbon receptor nuclear translocator (ARNT) and that this interaction affects TCDD-induced CYP1A1 gene expression (Kang et al., J Biol Chem 281:14654–14662, 2006). In this study we continue this investigation and begin to define the significance of this interaction for the regulation of stress-induced transcription.
Methods
Immunoprecipitations (IPs), western blot (WB) analysis, GST pull-down assays and promoter reporter assays were used to investigate whether the aryl hydrocarbon receptor (AhR) can regulate transcription that is dependent on the activation domain 1 (AD1) domain of BRCA1.
Results
We show that AhR, a transcription factor, can bind specifically to AD1 in the C-terminal region of BRCA1 and affect BRCA1's ability to regulate transcription activity. We found that xenobiotics that positively and negatively affect AhR's activity as a transcription factor (e.g., dioxin and α-naphthoflavone, respectively), have similar effects on AhR's ability to affect AD1-domain-dependent transcription. These physical and functional AhR–AD1 interactions may require the coiled-coil motif in AD1 because point-mutations in this motif reduce these interactions.
Conclusion
Xenobiotic-activated AhR can function in two ways, as a component of the AhR/ARNT transcription factor and a regulator of AD1-dependent transcription. Consequently, BRCA1 has two distinct mechanisms for sensing xenobiotics and regulating AhR-dependent stress responses to these xenobiotics. We speculate that the normal functioning of this interaction could play a role in BRCA1's tumor suppressing ability.
Keywords: BRCA1, AD1, AhR, JunB, Xenobiotic stress, Transcription regulation
Introduction
Several mechanisms have been proposed for how BRCA1 mutations could increase breast and ovarian cancer risk. Most of these models involve universal BRCA1 functions, such as its role in the regulation of DNA repair, cell cycle progression, apoptosis and transcription. We recently reported that BRCA1 enhances xenobiotic stress-induced gene expression by stabilizing aryl hydrocarbon receptor nuclear translocator (ARNT), a subunit of the AhR/ARNT heterodimeric transcription factor [9]. Here we identify a second interaction between BRCA1 and the AhR/ARNT transcription factor, an interaction involving AhR, the other component of this heterodimeric transcription factor.
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor belonging to the basic helix-loop-helix/Per-ARNT1-Sim family of proteins [4]. AhR mediates the toxic effects of many environmental chemicals, some of which are carcinogenic. Agents that bind to and activate AhR include polycyclic and halogenated aromatic hydrocarbons such as benzo(a)pyrene [B(a)P], dioxin (e.g., TCDD), dietary compounds [e.g., Indole-3-carbinol (I3C)], natural and synthetic flavonoids, and pharmaceutical drugs [3]. Prior to ligand binding AhR is inactive, sequestered in the cytoplasm in a complex with chaperone proteins including heat shock protein 90 [18], the co-chaperone p23 [12], and immunophilin homolog XAP2 [2]. Following ligand binding, activated AhR moves to the nucleus, dissociates from the chaperone complex, forms a heterodimer with ARNT, which then binds to xenobiotic response elements (XREs) in the promoter and enhancer regions of target genes, regulating their transcription. One of these genes, cytochrome p4501A1 (CYP1A1), is often used as a model for investigating AhR-driven gene expression [20]. The cytochrome p450 genes encode enzymes involved in the metabolism of drugs, foreign chemicals, steroids and some physiologically important lipids. The cytochrome p450 enzyme system is responsible for much of the Phase I metabolism and/or detoxification of carcinogenic agents [17, 20].
BRCA1 contains at least two transcription activation domains, AD1 and AD2 that are C-terminal to where most BRCA1 risk-causing mutations are located. While multiple transcription factors are known to interact with AD2 (which is C-terminal to AD1) only a few, including JunB, are known to interact with AD1, increasing BRCA1's ability to activate transcription [5, 6]. Here, we show for the first time that AhR can also interact with the AD1 domain, also increasing BRCA1's capacity to up-regulate transcription, and that this effect is further enhanced in the presence of activating AhR ligands.
Materials and methods
Cell culture
MCF-7, T47D and HEK293 cells were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). The cells were cultured in Dulbecco's modified eagles' medium (DMEM) supplemented with 10% (HEK293) or 5% (MCF-7 or T47D) fetal bovine serum (FBS), 100 unit per ml penicillin and 100 μg per ml streptomycin. All reagents for cell cultures were purchased from BioWhittaker, Inc. (Walkersville, MD, USA).
Chemicals
[2-(4-amino-3-methylphenyl)-5-flurobenxothiazole] (5F 203, NSC 703786) and its non-fluorinated parent compounds [2-(4-amino-3-methylphenyl) benzothiazole] (DF 203, NSC 674495) were obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute. These compounds were dissolved in DMSO to make a 100 mM stock concentration and further diluted to the working concentration (1 μM) just before being used. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) was prepared as a 31 μM stock in DMSO and diluted to the working concentration (10 nM) immediately prior to use. 3-Methylcholanthrene (3-MC) and α-naphthoflavone (α-NF) were prepared as a 1 mM stock in DMSO and diluted to 1 μM for the working solution. I3C was prepared as a 100 mM stock in ethanol solution and diluted to 50 μM for the working solution. All reagents were purchased from Sigma-Aldrich (St Louis, MO, USA).
Protein-DNA array
Transcription factors were able to bind the AD1 domain of BRCA1 were identified by using the Protein-DNA array II blot, and was purchased from Panomics Inc. (Fremont, CA, USA). The method using this blot allows one to determine if a target protein (tagged with HA) can bind to any of the proteins in a cell extract that can themselves bind to one or more of a mixture of 96 different pre-determined biotin-labeled duplex DNAs, each of which encodes both a unique identification sequence and the pre-determined protein binding sequence. In the blot used here, all the pre-determined protein binding sequences are designed to bind transcription factors. In brief, nuclear extracts prepared from HEK293 cells transfected for 24 h with expression vectors for either the wild-type AD1 domain of BRCA1 (Gal4-HA-DBD-AD1) or a mutant AD1 domain (Gal4-HA-DBD-AD1 L1407P) were incubated with the mixture of 96 different biotin-labeled duplex DNAs encoding consensus binding sequences for transcription factors. Immunoprecipitation with anti-HA antibodies (12CA5, Roche Applied Science) pulls down the HA-AD1 and one or more different biotin-labeled duplexes, if any proteins in the extract being examined can bind both HA-AD1 and a biotinylated duplex DNA. The bridging, interacting transcription factors are then identified indirectly, by using the IP'd biotinylated-DNA to probe a membrane spotted with complementary sequences. The location of positive spots (developed with streptavidin-HRP and a substrate) identifies DNA sequences that bind transcription factors (and/or other proteins) that may interact with the AD1 domain.
Co-immunoprecipitation and western blot analysis
MCF-7, T47D or HEK293 cells (2 × 106 cells) on 100-mm dishes were transfected and treated with various reagents as described in the figure legends. All IP procedures were carried out at 4°C as previously described [9, 10]. Cells were washed twice with PBS, then lysed in [50 mM Tris (pH 8.0), 250 mM NaCl, 1% NP-40 and protease inhibitor cocktail] and immunoprecipitated (IP'd) with either anti-AhR rabbit antibody (H-211, Santa Cruz Biotechnology) or a combination of anti-human BRCA1 mouse antibodies against N- and C-terminal epitopes on BRCA1 (Ab-1 + Ab-2 + Ab-3, Oncogene Research Products) [9, 10]. IP'd proteins were detected by WB analysis using anti-AhR rabbit antibody (H-211, Santa Cruz Biotechnology) or anti-BRCA1 rabbit antibody (C-20, Santa Cruz Biotechnology) and secondary antibodies conjugated with HRP. For identifying interactions between BRCA1 or AD1 and AhR, HEK293 cells co-transfected with GFP-AhR and HA-BRCA1 (AD1 wild-type or mutant) using Lipofectamine Plus (Invitrogen). After incubation for 24 h, cells were lysed in [50 mM Tris (pH 8.0), 250 mM NaCl, 1% NP-40 and protease inhibitor cocktail] and IP'd with either anti-GFP mouse antibody (8362-1, BD Science) or anti-HA mouse antibodies (12CA5, Roche Applied Science). IP'd proteins were detected by WB analysis using anti-GFP mouse antibody (8362-1, BD Science) or anti-HA mouse antibody (12CA5, Roche Applied Science). For competition of AhR with JunB, HEK293 cells were co-transfected with HA-AD1, GFP-AhR and two different doses of pCMV-JunB expression vector. Cell lysates were IP'd with anti-HA mouse antibody and WB analysis was performed.
GST pull-down assays
The glutathione-S-transferase (GST) pull-down assays were done as previously described [9]. In brief, GST fusion proteins produced in Escherichia coli BL21 cells (and purified by sonication and centrifugation) were incubated with glutathione-Sepharose-4B beads (Amersham Bioscience, Inc.). Washed beads were then incubated with continuous mixing overnight at 4°C with a probe (in vitro translated 35S-labeled AhR) in a buffer [RNase inhibitor (40 units), 40 mM HEPES–KOH (pH 7.6), 6 mM ATP, 20% (v/v) Glycerol, 8 mM MgCl2, 0.1% (v/v) NP-40, and 6 mM creatine phosphate], centrifuged and washed. Proteins bound to the GST fusion were eluted from the beads and fractionated by SDS-PAGE. The gels were dried under vacuum and autoradiographed using X-ray film (ImageTeK-B, American X-ray Supply, Inc.).
Reporter gene assay
Cells seeded in 24-well-plates and transfected with a reporter gene (125 ng/well) and one or more expression vectors as indicated in the figure legends using Lipofect-amine Plus (Invitrogen) were incubated overnight, and then treated with various agents for 24 h when they were harvested, lysed and used for the assays described in the figure legends. The luciferase assays were performed as previously described [9, 10]. The expression vectors for the Gal4 derivatives used in the mammalian two-hybrid assay were described previously [6]. GFP-AhR and FAhRR expression vectors were obtained from Dr. Oliver Hankinson (UCLA) and Dr. Mark Hann (Woods Hole Oceanographic Institution, MA, USA), respectively. The human AhRR clone (clone fH08618 of KIAA1234) was kindly provided by Dr. Takahiro Nagase, Kazusa DNA Research Institute, Chiba, Japan.
Statistical analysis
The two-tailed Student's t test was used for all the tests for statistical significance. The symbols used to indicate statistical significance levels in the figures (*P < 0.05) and (** or ##P < 0.005).
Results
Protein-DNA array
We used a recently developed Protein-DNA array method [7, 8] to identify transcription factors that interact with the AD1 domain of BRCA1. Nuclear extracts of HEK293 cells transfected with expression vectors for HA-AD1 (Gal4-HA-DBD-AD1) or a negative control, a HA-mutant AD1, L1407P (in the coiled-coil motif in AD1) were incubated with a mixture of biotin-labeled DNA duplexes, each containing a consensus sequence for a specific transcription factor. Pull-down with anti-HA antibodies will pull-down biotin-labeled duplexes that can directly or indirectly interact with the HA-AD1 domain. The pulled-down duplexes are then indirectly identified (see “Materials and methods”). Thus, identifying the duplexes identifies the transcription factors that bind these duplexes. The duplexes preferentially pulled-down by using HA-AD1 proteins contained XREs (Fig. 1a). Since AhR/ARNT heterodimers are known to recognize these sites, it seemed possible that the AD1 domain pulled down these duplexes indirectly, by binding AhR/ARNT heterodimer. The two different sequences of the pulled-down biotin-labeled DNA probes were GGGGATCGCGTGACAACCC (sense-direction) and GATCAGGCGCGCACGCAGGCCTCG (antisense-direction). The former sequences are consensus XRE sequences (underlined), identified and described in TRASFEC (http://www.biobase-international.com) and the latter sequences have been experimentally confirmed to bind AhR/ARNT [21].
Fig. 1.
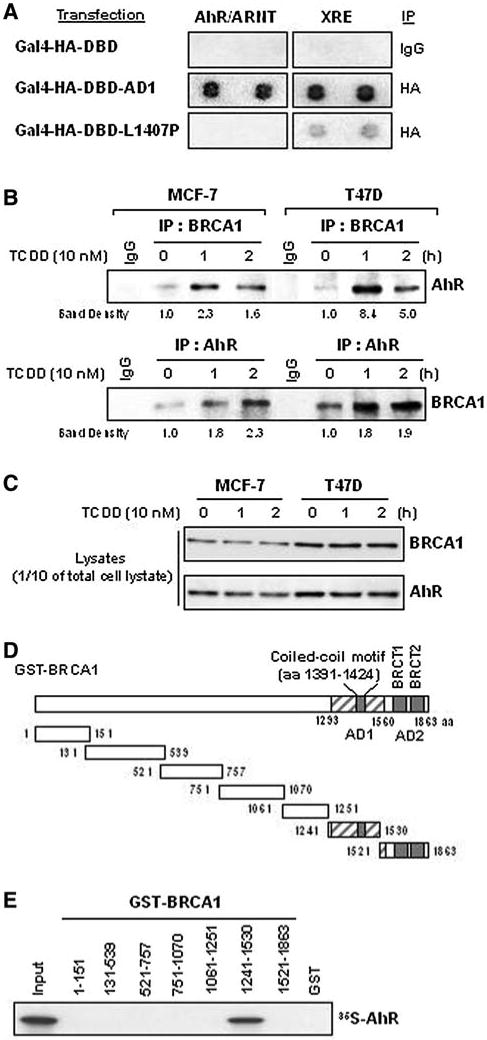
BRCA1 interacts with AhR through its AD1 domain. a The AD1 domain interacts with DNA duplexes that can bind AhR/ARNT. A mixture of biotin-labeled DNA duplexes, each able to bind a specific transcription factor(s), was mixed with nuclear extracts from cells transfected with expression vectors for Gal4-HA-DBD-AD1 (wild-type amino acids) or Gal4-HA-DBD-L1407P (an AD1 mutant), incubated and IP'd with anti-HA antibody or mouse IgG (the negative control). The pulled-down biotin-labeled DNAs were identified by hybridization to an ordered array of specific complementary nucleotides and identifying the hybrids by using streptavidin-HRP as the reporter, as described in “Materials and methods”. The results presented are representative of three independent experiments. b The in vivo interaction between BRCA1 and AhR increases following TCDD treatment. Whole cell lysates from MCF-7 and T47D cells, treated with DMSO or TCDD (10 nM) for 1 or 2 h, were IP'd with anti-BRCA1 antibodies and analyzed by WB for endogenous AhR. Similar lysates were also IP'd with an anti-AhR antibody and then analyzed for endogenous BRCA1 by WB analysis. c Ten percent of the total cell lystates used for the IP-WBs was run on 6% SDS-PAGE and the amounts of BRCA1 and AhR protein were determined by WB analysis. d Diagram of the relative locations of AD1, AD2, the coiled-coil motif and various deletion mutants of GST-BRCA1. e GST pull-down assays using the BRCA1 deletion mutants described in d. The GST pull-down assays and the 35S-IVT-AhR probe preparations are described in “Materials and methods” and previously (49). The results shown are representative of three independent GST pull-down assays for each combination
Physical interactions between BRCA1 and AhR
The results shown in Fig. 1a suggest that full-length wild-type BRCA1 protein might also form a complex with AhR, ARNT or the AhR/ARNT heterodimer, at least when the heterodimer is bound to an XRE element. We previously reported that BRCA1 can interact with ARNT, although via a different region of BRCA1 (the N-terminal region) [9]. In contrast, the results of the immunoprecipitation-Western blot (IP-WB) analysis and GST pull-down assays shown here (Fig. 1b–e) suggest a completely different interaction, an interaction between the AD1 domain of BRCA1 and AhR. Since AhR become activated by its ligand binding, we determine to use TCDD, the most powerful AhR ligand identified so far, to determine whether TCDD-activated AhR can interacts with BRCA1. When lysates of cells treated with DMSO or 10 nM of TCDD for 1 or 2 h were IP'd with an anti-BRCA1 antibody and then used for WB analysis, AhR was detected under both conditions (Fig. 1b). Similarly, BRCA1 was found in complexes IP'd with an anti-AhR antibody under both conditions. These results suggest that endogenous BRCA1 and AhR can interact in vivo. In addition, short treatments of TCDD enhanced this interaction without significantly altering the total amounts of either BRCA1 or AhR protein in either MCF-7 or T47D cells (Fig. 1c). We then used GST pull-down assays to determine if this interaction can occur in vitro, using 35S-labeled in vitro transcribed/translated (IVT) AhR, and if BRCA1 regions other than AD1 might also be involved (see Fig. 1d for a BRCA1 deletion map). Only one of the tested GST-BRCA1 fragments (aa 1241–1530) captured the 35S-labeled IVT AhR (Fig. 1e). These results suggest that the AD1 domain of BRCA1 is sufficient, both in vivo and in vitro, for interacting with AhR and that this interaction could be direct protein–protein binding.
AhR and AhR ligands stimulate AD1 domain-dependent transcription
Since AhR binds AD1, we then asked whether this binding could affect the transcription function of AD1 and whether an AhR ligand, TCDD, would also have an effect. For this test, HEK293 cells were co-transfected with the Gal4-Luc reporter gene and one or more expression vectors (Gal4-HA-DBD-AD1, Gal4-HA-DBD-AD2) and either AhR or JunB (for the AD1-containing cells), and then treated (or not) with TCDD. (The diagram in Fig. 1d shows the relative positions of AD1, AD2 and the coiled-coil motif in AD1.) We found that overexpressed AhR stimulated AD1-dependent transcription but not AD2-dependent transcription. TCDD treatment enhanced this AhR-stimulated AD1 activity but had no ability to affect either negative control, overexpression of the AD2 or DBD (Fig. 2a) in HEK293 cells. We found similar effects in a human breast cancer cell line, MCF-7, although the effects are pronounced (Fig. 2b). As expected [6], JunB overexpression also stimulated AD1-dependent transcription under our conditions. These results suggest that both AhR and activated AhR (AhR bound to a ligand) might stimulate AD1-driven gene expression under normal physiological conditions. We then tested several other known AhR agonists for their ability to stimulate this AD1-driven reporter gene expression. All of the AhR agonists tested [two dioxins, TCDD and 3-MC; one chemopreventive agent, indole-3-caribinol (I3C), and two chemotherapeutic drugs, DF 203 and 5F 203] stimulated AD1-dependent transcription (data not shown). This stimulation was further enhanced by overexpressing AhR (Fig. 2c).
Fig. 2.

The capacity of the AD1 domain to stimulate reporter activity is increased by exogenous AhR and AhR ligands. a, b Cells (HEK293 and MCF-7) were co-transfected with a reporter plasmid and various combinations of expression vectors (see text). AhR significantly stimulated Gal4-Luc reporter activity in cells co-transfected with Gal4-HA-DBD-AD1 (**P < 0.005 for comparisons of cells transfected with and without AhR). A positive control, JunB, also significantly stimulated AD1 activity (*P < 0.05 or **P < 0.005 for comparisons of cells transfected with and without JunB expression vector). After overnight incubation, cells transfected with AhR were treated with 10 nM of TCDD or DMSO for 24 h as indicated when reporter activity was measured. AhR increased Gal4-DBD-AD1 transcription activity by six to sevenfold (HEK293) or by 1.6–1.7-fold (MCF-7). TCDD treatment enhanced AhR-stimulated AD1-driven reporter activity by approximately twofold in HEK293 cells. Expression vectors for Gal4-DBD-AD2 and Gal4-DBD were used as the negative controls. Luciferase activity was normalized to the values obtained with the Gal4-DBD expression vector alone. c Cells (HEK293) co-transfected with Gal4-HA-DBD-AD1 and Gal4-Luc (and either an empty vector or an AhR expression vector) and incubated with various AhR agonists for 24 h were assayed for luciferase activity. The values presented have been corrected for the background values (with the empty vector); 2282, 8119, 3316, 2916, 6082, and 3016, from left to right, respectively. Significant differences were found between DMSO-treated and AhR agonist-treated samples (**P < 0.005). a–c The experiments were performed at least three times independently. The values presented, mean ± SEM, were calculated from one representative experiments, each done in quadruplicate (N = 4). In addition, a pCMV-βgal expression vector (50 ng) was included in all transfections and was used to normalize for differences in transfection efficiencies as previously described [9]
AhR ligand-stimulated AD1 domain-dependent transcription is repressed by AhR antagonists
The transcription-stimulating effect of activated AhR can be blocked with either a chemical antagonist (α-NF) [19] or by overexpressing naturally occurring AhR repressors, either human (HAhRR) [15] or a fish, F. heteroclitus, AhRR (FAhRR) [11]. We used these agents to test whether the stimulation of AD1 domain-driven transcription observed following AhR agonist treatments requires activated AhR. α-NF reduced TCDD stimulated AD1 domain-dependent transcription, either in the absence or presence of the enhancement observed by overexpressing AhR (Fig. 3a). Similar results were obtained when expression vectors for either AhRR or FAhRR were co-transfected with the Gal4-HA-DBD-AD1 expression vectors (Fig. 3b, c). This reduction is more clearly demonstrated when AhR is also overexpressed or when cells are also treated with TCDD (Fig. 3b, c). These finding suggest that TCDD-stimulated AD1 domain-dependent transcription requires AhR and possibly activated AhR.
Fig. 3.

The effect of AhR on AD1-dependent transcription is reduced by AhR antagonists. a HEK293 co-transfected with the Gal4-Luc reporter and the Gal4-HA-DBD-AD1 expression vector (with or without an AhR expression vector) were incubated with or without TCDD and α-NF (a chemical antagonist for AhR) as indicated, and luciferase activity was measured as described in the legend to Fig. 2. The highest value obtained in each of the experiments presented in Fig. 3 was set at 100 and the other values were then normalized to this value. The effect of α-NF is significant (*P < 0.05) for the basal, un-induced reporter activity or highly significant (**P < 0.005) for TCDD alone, AhR alone or AhR plus TCDD. b Cells (HEK293) were co-transfected with the Gal4-Luc reporter and expression vectors for Gal4-HA-DBD-AD1 and AhRR as indicated in the figure. The effect of HAhRR was highly significant (**P < 0.005) under all conditions tested. c Cells (HEK293) were co-transfected with the Gal4-Luc reporter and expression vectors for Gal4-HA-DBD-AD1 and FAhRR as indicated in the figure. FAhRR had no significant effect on basal reporter activity but a significant (*P < 0.05) or a highly significant effect (**P < 0.005) on TCDD- or exogenous AhR-stimulated reporter activity that was driven by AD1 overexpression. The data presented is representative of three experiments, each done in quadruplicate
A role for the coiled-coil motif in AD1 for its physical and functional interactions with AhR
Previous studies have shown that transcriptional activation by the AD1 domain requires its coiled-coil motif [5, 6]. We asked if this is also true when AD1-dependent transcription is stimulated by AhR. To do this we examined the effects of point mutations in this coiled-coil region, some of which are known to disrupt the coiled-coil motif and which also inactivate AD1-dependent transcription. In a previous study, L1407P, L1407A, L1414A and I1405V point mutants significantly disrupted the interaction of AD1 and JunB and inactivated the ability of AD1 to regulate the transcription regulation activity of JunB. Several full-length HA-tagged BRCA1 constructs (wild-type or carrying various single point mutations, L1407P, L1407A, L1414A, I1405V, H1402Y and H1421Y) and HA-tagged AD1 domain (Gal4-HA-DBD-AD1) expression vectors with the same point mutants were used for this experiment [6]. The AD1 coiled-coil motif contains a heptad repeat whose seven positions are labeled from (a) to (g). The (a) and (d) positions are hydrophobic and form the helix interfaces, while (b), (c), (e), (f) and (g) are hydrophilic, forming the solvent-exposed surface of the coiled-coil motif. Previous studies showed that the wild-type leucine residues at positions (a) and (d) are critical for maintaining the coiled-coil structure [13, 14]. Several point mutations, including three substitutions at these two leucines (L1407P, L1407A and L1414A) were previously examined for their effect on the coiled-coil structure [6]. We tested point mutations at six positions in this motif for their effects on the AD1–AhR interaction. Our positive control confirmed that the wild-type AD1 domain and AhR can interact in HEK293 cells. For our test HEK293 cells were co-transfected with expression vectors for GFP-AhR and mutant coiled-coil AD1 domains, then IP'd with an anti-GFP antibody followed by WB analysis using an anti-HA antibody. We found that four of the six different AD1 point mutants tested interacted less with AhR than the wild-type AD1 domain (Fig. 4a), including the three leucine-containing mutants (L1407P, L1407A and L1414A) and one isoleucine substitution in the coiled-coil region (I1405V). The two mutants containing histidine substitutions interacted with AhR as strongly as the wild-type AD1 domain. Similar results were obtained when HA-tagged full-length BRCA1 proteins (wild-type or carrying mutations in the AD1 domain) were co-transfected with GFP-AhR (Fig. 4b). We then asked if the extent of these physical interactions with the AD1 domain-only constructs (as measured in Fig. 4a) correlated with the relative amounts of AD1 functional activity, as measured with the Gal4-Luc reporter plasmid, and found a strong positive correlation (Fig. 4c). However, histidine substitutions may have a greater relative effect in the functional, reporter activity test, than in the physical AhR–AD1 interaction, co-IP test. The results of these AD1 mutant tests offer additional evidence that AD1 and AhR can interact and that this interaction can influence the ability of BRCA1 to activate transcription via its AD1 domain.
Fig. 4.
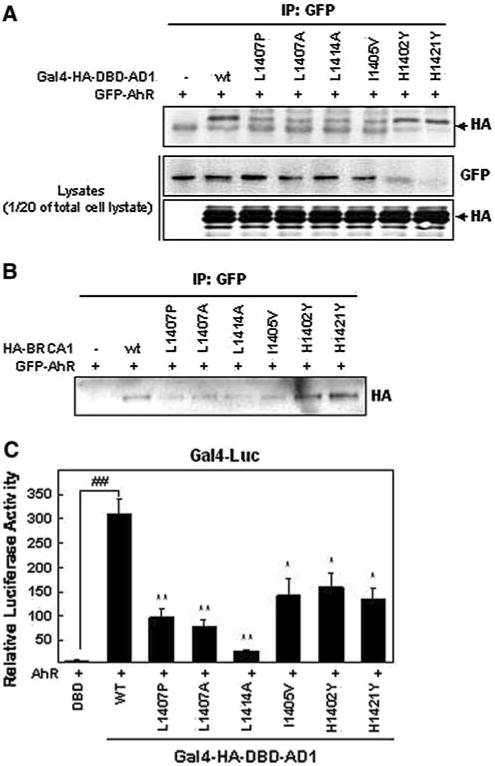
AD1 point mutants affect both physical and functional BRCA1–AhR interactions. a Physical interactions between AhR and AD1 domain mutants using an AD1 domain-only expression vector. HEK293 cells co-transfected with GFP-AhR and Gal4-HA-DBD-AD1 expression vectors (wild-type and mutants) were used for our standard IP-WB analysis: whole cell lysates were IP'd with an anti-GFP antibody and the relative amounts of AD1 co-IP'd were detected with anti-HA antibody. b Physical interactions between AhR and AD1 domain mutants using full-length BRCA1. HEK293 cells were co-transfected with full-length HA-BRCA1 (wild-type and mutants) and GFP-AhR. IP-WB analysis was performed as described in a. The WBs shown in panels a and b are representative of three independent experiments. c Functional interactions between AhR and AD1 mutants using AD1 domain-only expression vectors. Cells were co-transfected for 24 h with the Gal4-Luc reporter and expression vectors for Gal4-HA-DBD-AD1 (wild-type or mutant) and AhR. The values presented have been adjusted in two steps. First, the background values were subtracted and then these values were normalized to the Gal4-DBD value (=1). The background values subtracted were, from left to right 913, 1800, 1422, 1608, 1762, 2409, 4452 and 4223. Significant (*P < 0.05) or very significant (**P < 0.005) differences were found between wild-type AD1 reporter activity and all of the mutants (*P < 0.05, **P < 0.005) or between the wild-type AD1 domain and the DBD control (##P < 0.005). The values presented, mean ± SEM, were calculated from three independent experiments each done in quadruplicate (N = 4)
The wild-type AD1 domain of full-length BRCA1 is required for maximum activation of an XRE-driven reporter gene
We previously reported that overexpression of wild-type full-length BRCA1 enhances TCDD-induced expression from a reporter plasmid carrying the XRE from the promoter of the CYP1A1 gene [9]. In order to test whether single point mutants in the AD1 domain of overexpressed full-length BRCA1 affect this TCDD-induced reporter plasmid expression, we co-transfected cells with the previously used reporter plasmid p(XRE.1A1)-Luc and full-length BRCA1, encoding either the wild-type AD1 domain or six different AD1 domain point mutants. We found, as expected, reduced reporter activity following TCDD treatment in all of the AD1 mutants in both cell lines tested, HEK293 and MCF-7 (Fig. 5a, b).
Fig. 5.
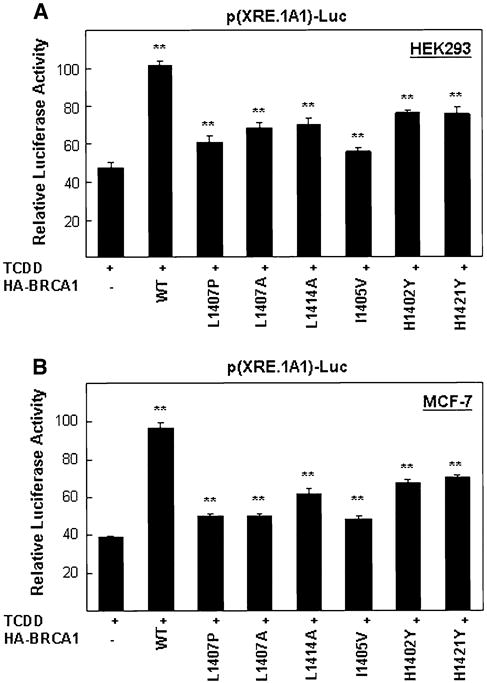
AD1 point mutants are less responsive to TCDD treatments than wild-type AD1 in a reporter plasmid assay. a HEK293 or b MCF-7 cells were co-transfected for 24 h with the p(XRE.1A1)-Luc reporter gene and either an empty expression vector or this expression vector encoding various full-length HA-tagged BRCA1 proteins (wild-type or AD1 point mutants), and then incubated with TCDD for an additional 24 h when reporter activity was measured as described in the legend to Fig. 2. The values presented have been corrected by subtracting background values (empty expression vector-transfected cells). The background values for a were, from left to right, 1401, 2629, 1764, 1576, 1651, 1604, 2355 and 2091. The background values for b were, from left to right, 1088, 2037, 1278, 1227, 1296, 1396, 1895 and 1745. In addition, a pCMV-βgal expression vector (50 ng) was included in all transfections and was used to normalize for differences in transfection efficiencies as discribed in Fig. 1 legend. These values were then normalized to the average value obtained for the HA-wild-type BRCA1 samples, which is set at 100. Note that the controls in this experiment are TCDD treated cells that are not overexpressing BRCA1 (either wild-type or mutant). Significant (*P < 0.05) differences were found between four AD1 mutants (L1407P, L1407A, L1414A and I1405V) and control empty vector (pCDNA3 plus TCDD). Very significant (**P < 0.005) differences were found between AD1 (wild-type, H1402Y and H1421Y) and control. The values presented, mean ± SEM, were calculated from three independent experiments each done in quadruplicate (N = 4)
JunB and AhR compete with each other for binding to AD1
A previous report demonstrated a specific interaction between JunB and the AD1 domain and that this interaction required the wild-type coiled-coil motif of AD1 [6]. Since the AD1–AhR interaction also requires the coiled-coil motif in AD1, we asked if JunB can compete with AhR for binding to AD1. To test this possibility, HEK293 cells were transfected with two different amounts of a JunB expression vector (0.5 and 1 μg) and fixed amounts of both Gal4-HA-DBD-AD1 and GFP-AhR expression vectors. After 24 h, the exogenous AD1 in whole cell lysates was IP'd with an anti-HA antibody and the relative amounts of overexpressed GFP-AhR that was co-IP'd determined by WB analysis. Relatively more AhR was detected in the absence of exogenous JunB than when JunB was overexpressed. This result suggests that JunB and AhR can compete with each other for binding to AD1, possibly competing for the same or overlapping surfaces on the coiled-coil motif of AD1 (Fig. 6a). Next, we asked if this AhR–JunB competition can also occur under conditions of xenobiotic-induced stress (i.e., TCDD treatment). TCDD treatments will increase the amount of activated endogenous AhR. Cells transfected with a constant amount of Gal4-HA-DBD-AD1 and pCMV-JunB were treated with TCDD (0, 6 and 24 h) when cell lysates were IP'd with anti-HA antibody. Relatively more JunB was co-IP'd with HA in the absence of TCDD than its presence. The TCDD treatment decreased the AD1–JunB interaction in a time-dependent manner, without affecting the total amount of either overexpressed protein (Fig. 6b), suggesting that activated AhR is a better competitor than non-activated AhR for the AD1 site recognized by both AhR and JunB. Since overexpression of JunB can stimulate AD1-dependent transcription on Gal4-Luc reporter [6] and may interfere the interaction of the AD1-activated AhR, we asked whether overexpression of JunB could reduce the ability of exogenously expressed to stimulate AD1-dependent transcription in Gal4-Luc reporter in the presence of TCDD. As expected, overexpression of only AhR strongly stimulated reporter plasmid activity in the presence of TCDD. However, when the AhR, AD1 overexpressing, TCDD treated cells that were also over-expressing JunB, significantly less reporter activity was produced and this JunB effect was dose-dependent (Fig. 6c). These results suggest that activated AhR and JunB compete for binding to AD1 and that the interaction between AD1 and activated AhR by TCDD is a more potent stimulator of transcription from the reporter plasmid used in this test than JunB. Thus, JunB may sequester AD1 away from the kinds of transcription-promoting interactions AD1 can have with AhR or activated AhR, at least for some XRE-containing promoters.
Fig. 6.
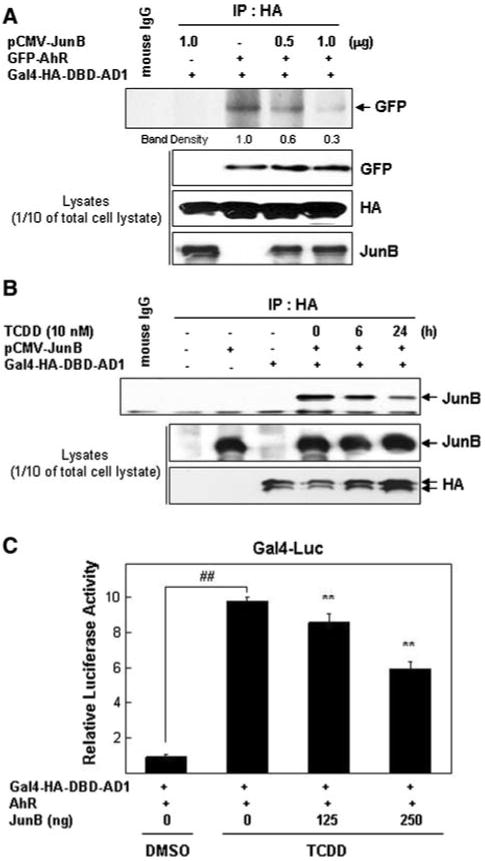
JunB and AhR compete for binding to AD1. a JunB disrupts the interaction between AhR and AD1. HEK293 cells were co-transfected for 24 h as indicated in the figure with expression vectors for Gal4-HA-DBD-AD1 and GFP-AhR ± pCMV-JunB (0.5 or 1.0 μg), whole cell lysates prepared and IP'd with an anti-HA antibody. The presence of exogenous GFP-AhR in IP'd complexes was detected by WB analysis using an anti-GFP antibody. Mouse IgG was used as the negative IP control. Relative levels of exogenous GFP-AhR, AD1 and JunB proteins were determined by WB analysis using appropriate antibodies. These WBs are representative of three independent experiments. b TCDD disrupts the interaction between JunB and AD1. HEK293 cells co-transfected with expression vectors for Gal4-HA-DBD-AD1 ± pCMV-JunB were treated with TCDD for 6 or 24 h, whole cell lysates prepared and IP'd with an anti-HA antibody. The presence of exogenous JunB in IP'd complexes was detected by WB analysis using an anti-JunB antibody. Mouse IgG was used as the negative control. Expression levels of exogenously expressed JunB and AD1 were determined by WB analysis using appropriate antibodies. This experiment was performed three times. The one shown is representative. c JunB reduces the capacity of AhR, AD1 and TCDD to stimulate reporter gene expression. HEK293 cells co-transfected with the Gal4-Luc reporter plasmid and expression vectors for Gal4-HA-DBD-AD1, pCMV-JunB and GFP-AhR were treated with 10 nM of TCDD for 24 h when luciferase activity were determined as described in Fig. 2 legend. TCDD treatment significantly increased Gal4-Luc activity in cells containing exogenous Gal4-HA-DBD-AD1 and AhR compared to the DMSO treated cells (##P < 0.005). Overexpression of JunB (either 125 or 250 ng) significantly decreased reporter activity (**P < 0.005) comparing to the no JunB transfected group
JunB represses the enhancing activity of AD1 on TCDD (AhR)-mediated XRE-containing gene expression
Previously we demonstrated that full-length wild-type BRCA1 can enhance TCDD-induced p(XRE.1A1)-Luc reporter activity [9]. We now show that overexpression of the AD1 domain by itself can also enhance reporter activity in a dose-dependent manner in the presence of TCDD and under two other conditions; in the presence of exogenous AhR and in the absence of either TCDD or exogenous AhR (Fig. 7a). These dose-dependent effects appear to be greater when AhR is overexpressed than when 10 nM of TCDD is used or when only the empty expression vector is used. Finally, we tested whether JunB affects reporter plasmid activity in a dose-dependent manner under various conditions. As expected, exogenous JunB significantly reduced the enhancing effects of exogenous AhR, either alone (Fig. 7b) or in combination with overexpressed AD1 domain (Fig. 7b) or overexpressed full-length wild-type BRCA1 (Fig. 7d). Exogenous JunB also reduced, in a dose-dependent manner, the TCDD-alone driven stimulation of reporter plasmid activity (Fig. 7c, e) or TCDD in combination with overexpressed AD1 domain (Fig. 7c) or overexpressed full-length wild-type BRCA1 (Fig. 7e).
Fig. 7.
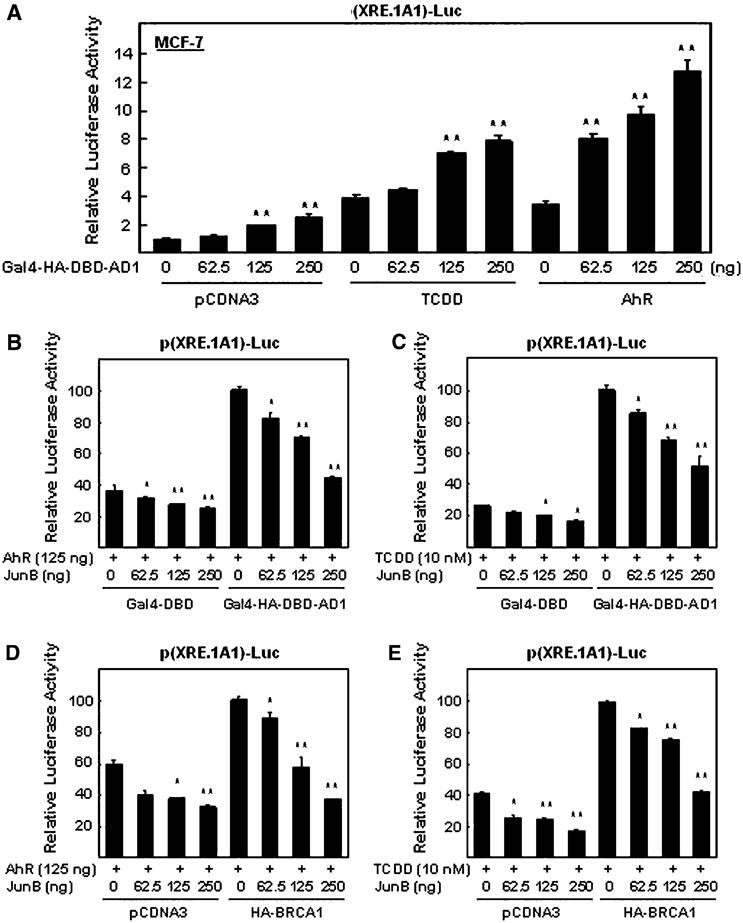
Exogenous JunB reduces the ability of BRCA1 to stimulate the p(XRE.1A1)-Luc reporter activity observed after co-transfection with an AhR expression vector or treating with TCDD using MCF-7 cells. a Overexpression of the AD1 domain increases, in a dose-dependent manner, the ability of TCDD treatment or overexpressed AhR to enhance p(XRE.1A1)-Luc reporter activity. Cells co-transfected with increasing amounts of Gal4-HA-DBD-AD1 and a constant amount of p(XRE.1A1)-Luc were also co-transfected with either an AhR expression vector or empty vector (and treated with DMSO) or treated with TCDD-alone. Lysates were prepared and used to measure luciferase activity as described in Fig. 2 legend. b, c Increasing doses of exogenous JunB increasingly reduce the enhanced AD1-dependent p(XRE.1A1)-Luc reporter activity resulting from overexpressing AhR (b) or treating with TCDD (c). d, e Increasing doses of JunB increasingly reduce the enhanced BRCA1 (full-length wild-type)-dependent p(XRE.1A1)-Luc reporter activity resulting from overexpressing AhR or treating with TCDD. The data is presented as bar graphs showing the mean ± SEM of quadruplicate wells from three independent experiments. The luciferase activities are normalized to the values obtained in the presence of BRCA1 (or AD1) and AhR (or TCDD), but in the absence of exogenous JunB (column 5 in b–e)
Discussion
Here we have identified an additional transcription factor, AhR that can interact with the AD1 domain of BRCA1 and have begun to characterize the functional consequences of this interaction, including showing that JunB can compete with AhR for binding to AD1 and that this competition has functional consequences. We found that (1) AhR interacts with BRCA1, possibly directly; (2) the AD1 domain of BRCA1 may be the only region of BRCA1 that can by itself interact with AhR; thus, AD1 is both necessary and sufficient for the BRCA1–AhR interaction detected under our conditions; (3) the coiled-coil motif of the AD1 domain is critical for this interaction; (4) a xenobiotic stressor, TCDD, increases this interaction in vivo; (5) overexpressed AhR increases the capacity of the endogenous AD1/BRCA1 domain to up-regulate transcriptional activation; (6) the coiled-coil motif of the AD1 domain is critical for acquiring this increased capacity; (7) various additional xenobiotic stressors also enhance AhR-stimulated AD1 transcriptional activation; (8) AD1 domain mutations reduce the capacity of full-length BRCA1 to enhance TCDD-induced activity of a reporter plasmid carrying a XRE relevant to normal xenobiotic stress responses; (9) AhR and JunB compete with one another for interacting with BRCA1, presumably at or near the coiled-coil motif in the AD1 domain; (10) overexpressed JunB reduces BRCA1-mediated xenobiotic-induced expression from an XRE-containing promoter, presumably by competing with activated AhR for binding to AD1.
Our results suggest that AD1 domain interactions with AhR or activated AhR could contribute to normal physiological responses to xenobiotic insults. However, this is not the only role for BRCA1 in these responses: we previously showed that the N-terminus of BRCA1 stabilizes ARNT, potentially allowing up-regulation of AhR/ARNT-driven transcription [9]. These two possible roles for BRCA1 in xenobiotic responses could function independent or they could work together, perhaps creating a positive feedback loop. For example, a xenobiotic binds to and activates AhR which then activates BRCA1 (via the AD1 domain); BRCA1 then activates AhR/ARNT (by stabilizing ARNT) and finally stabilized AhR/ARNT might further activate BRCA1.
The existence of two BRCA1-dependent mechanisms for amplifying AhR/ARNT-driven transcription suggests that cells may want the ability to carefully calibrate the strength of their responses to xenobiotics that can activate AhR and/or may want to link this BRCA1-centered ability to one or more other regulatory functions of BRCA1. For example, the role of BRCA1 in ARNT stability might be easily regulated. The competition between JunB and AhR for binding to the AD1 domain could be used to regulate AD1-driven transcription. Additional experiments will be needed to test these and other possibilities.
In both our previous and in this study we found that BRCA1 forms a complex with a component of the AhR/ARNT transcription factor and regulates the ability of these proteins to stimulate transcription. Although our current study suggests that the AhR–AD1 interaction could be physiologically relevant for regulating transcription during xenobiotic stress, our data does not directly address the question of whether this interaction also has biological significance in the context of tumorization or tumor suppression. However, several other studies have shown that constitutive activation of AhR is associated with induction or promotion of tumorization [1, 16]. Thus, in view of these studies our data suggest that the activated AhR–BRCA1 complex could contribute to breast cancer formation and/or progression after exposure to environmental agents that can activate AhR, perhaps by altering the spectrum of BRCA1 regulated gene expression.
Acknowledgments
Dr. Bae has been supported, in part, by the National Institute of Environmental Health Science, NIH (ES01440-01) and the Susan G. Komen Foundation for the Cure (BCTR119906 and FAS0703858). Drs. Li and Hu have been supported by CA93506 (R. L) and CA118578 (Y. H), respectively. We appreciate Drs. Albert J. Fornace Jr. and Thomas L. Mattson for helpful discussions.
Abbreviations
- ARNT
Aryl hydrocarbon receptor nuclear translocator
- AhR
Aryl hydrocarbon receptor
- BRCA1
Breast cancer susceptibility gene-1
- DMEM
Dulbecco's modified eagles' medium
- DMSO
Dimethyl sulfoxide
- FBS
Fetal bovine serum
- IP
Immunoprecipitation
- SEM
Standard error of mean
- WB
Western blot
- AD1
Activation domain 1
Contributor Information
Hyo Jin Kang, Department of Oncology, Lombardi Comprehensive Cancer Center, Georgetown University, 3970 Reservoir Road, NW, Washington, DC 20057, USA.
Hee Jeong Kim, Department of Oncology, Lombardi Comprehensive Cancer Center, Georgetown University, 3970 Reservoir Road, NW, Washington, DC 20057, USA.
Chi-Heum Cho, Department of Obstetrics and Gynecology, School of Medicine, Keimyung University, Daegu, South Korea.
Yanfen Hu, Department of Molecular Medicine, Institute of Biotechnology, University of Texas, Health Science Center at San Antonio, 15355 Lambda Drive, San Antonio, TX 78245, USA.
Rong Li, Department of Molecular Medicine, Institute of Biotechnology, University of Texas, Health Science Center at San Antonio, 15355 Lambda Drive, San Antonio, TX 78245, USA.
Insoo Bae, Department of Oncology, Lombardi Comprehensive Cancer Center, Georgetown University, 3970 Reservoir Road, NW, Washington, DC 20057, USA, e-mail: ib42@georgetown.edu, Department of Radiation Medicine, Lombardi Comprehensive Cancer Center, Georgetown University, 3970 Reservoir Road, NW, Washington, DC 20057-1469, USA.
References
- 1.Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, Hanberg A, Poellinger L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci USA. 2002;99:9990–9995. doi: 10.1073/pnas.152706299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carver LA, Bradfield CA. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J Biol Chem. 1997;272:11452–11456. doi: 10.1074/jbc.272.17.11452. [DOI] [PubMed] [Google Scholar]
- 3.Dension MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 4.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 5.Hu YF, Miyake T, Ye Q, Li R. Characterization of a novel trans-activation domain of BRCA1 that functions in concert with the BRCA1 C-terminal (BRCT) domain. J Biol Chem. 2000;275:40910–40915. doi: 10.1074/jbc.C000607200. [DOI] [PubMed] [Google Scholar]
- 6.Hu YF, Li R. JunB potentiates function of BRCA1 activation domain 1 (AD1) through a coiled-coil-mediated interaction. Genes Dev. 2002;16:1509–1517. doi: 10.1101/gad.995502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang X, Norman M, Li X. Use of an array technology for profiling and comparing transcription factors activated by TNFalpha and PMA in HeLa cells. Biochim Biophys Acta. 2003;1642:1–8. doi: 10.1016/s0167-4889(03)00080-6. [DOI] [PubMed] [Google Scholar]
- 8.Jiang X, Norman M, Roth L, Li X. Protein-DNA array-based identification of transcription factor activities regulated by interaction with the glucocorticoid receptor. J Biol Chem. 2004;279:38480–38485. doi: 10.1074/jbc.M403948200. [DOI] [PubMed] [Google Scholar]
- 9.Kang HJ, Kim HJ, Kim SK, Barouki R, Cho CH, Khanna KK, Rosen EM, Bae I. BRCA1 modulates xenobiotic stress-inducible gene expression by interacting with ARNT in human breast cancer cells. J Biol Chem. 2006;281:14654–14662. doi: 10.1074/jbc.M601613200. [DOI] [PubMed] [Google Scholar]
- 10.Kang HJ, Kim HJ, Rih JK, Mattson TL, Kim KW, Cho CH, Isaacs JS, Bae I. BRCA1 plays a role in the hypoxic response by regulating HIF-1alpha stability and by modulating vascular endothelial growth factor expression. J Biol Chem. 2006;281:13047–13056. doi: 10.1074/jbc.M513033200. [DOI] [PubMed] [Google Scholar]
- 11.Karchner SI, Franks DG, Powell WH, Hahn ME. Regulatory interactions among three members of the vertebrate aryl hydrocarbon receptor family: AHR repressor, AHR1, and AHR2. J Biol Chem. 2002;277:6949–6959. doi: 10.1074/jbc.M110779200. [DOI] [PubMed] [Google Scholar]
- 12.Kazlauskas A, Sundstrom S, Poellinger L, Pngratz I. The hsp90 haperone complex regulates intracellular localization of the dioxin receptor. Mol Cell Biol. 2001;21:2594–2607. doi: 10.1128/MCB.21.7.2594-2607.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 14.Lupas A. Coiled coils: new structures and new functions. Trends Biochem Sci. 1996;21:375–382. [PubMed] [Google Scholar]
- 15.Mimura J, Ema M, Sogawa K, Fujii-Kuriyama Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999;13:20–25. doi: 10.1101/gad.13.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moennikes O, Loeppen S, Buchmann A, Andersson P, Ittrich C, Poellinger L, Schwarz M. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004;64:4707–4710. doi: 10.1158/0008-5472.CAN-03-0875. [DOI] [PubMed] [Google Scholar]
- 17.Nebert DW, Gonzalez FJ. P450 Genes: structure, evolution, and regulation. Ann Rev Biochem. 1987;56:945–993. doi: 10.1146/annurev.bi.56.070187.004501. [DOI] [PubMed] [Google Scholar]
- 18.Perdew GH, Bradfield CA. Mapping the 90 kDa heat shock protein binding region of the Ah receptor. Biochem Mol Biol Int. 1996;39:589–593. doi: 10.1080/15216549600201651. [DOI] [PubMed] [Google Scholar]
- 19.Swanson HI, Perdew GH. Half-life of aryl hydrocarbon receptor in Hepa 1 cells: evidence for ligand-dependent alterations in cytosolic receptor levels. Arch Biochem Biophys. 1993;302:167–174. doi: 10.1006/abbi.1993.1195. [DOI] [PubMed] [Google Scholar]
- 20.Whitlock JP., Jr Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- 21.Yoo HY, Chang MS, Rho HM. Xenobiotic-responsive element for the transcriptional activation of the rat Cu/Zn superoxide dismutase gene. Biochem Biophys Res Commun. 1999;256:133–137. doi: 10.1006/bbrc.1999.0299. [DOI] [PubMed] [Google Scholar]