

Abstract
The product of gene At3g16450.1 from Arabidopsis thaliana is a 32 kDa, 299-residue protein classified as resembling a myrosinase-binding protein (MyroBP). MyroBPs are found in plants as part of a complex with the glucosinolate-degrading enzyme, myrosinase, and are suspected to play a role in myrosinase-dependent defense against pathogens. Many MyroBPs and MyroBP-related proteins are composed of repeated homologous sequences with unknown structure. We report here the three-dimensional structure of the At3g16450.1 protein from Arabidopsis, which consists of two tandem repeats. Because the size of the protein is larger than that amenable to high-throughput analysis by uniformly 13C/15N labeling methods, we used our stereo-array isotope labeling (SAIL) technology to prepare an optimally 2H/13C/15N-labeled sample. NMR data sets collected with the SAIL-protein enabled us to assign 1H, 13C and 15N chemical shifts to 95.5% of all atoms, even at the low concentration (0.2 mM) of the protein product. We collected additional NOESY data and solved the three-dimensional structure with the CYANA software package. The structure, the first for a MyroBP family member, revealed that the At3g16450.1 protein consists of two independent, but similar, lectin-fold domains composed of three β-sheets.
Keywords: lectin, NMR structure, stereo-array isotope labeling, structural genomics
Introduction
The flowering plant Arabidopsis thaliana is an important model system for identifying plant genes and determining their functions. Analysis of the completed Arabidopsis thaliana genome revealed the presence of 25,498 genes encoding proteins from 11,000 families, including many new protein families [1]. To investigate the biological importance of these proteins, the Center for Eukaryotic Structural Genomics (CESG) established platforms for protein structure determination by X-ray crystallography and NMR spectroscopy, with protein production both by conventional heterologous gene expression in E. coli and by automated cell-free technology [2]. To date targets for NMR analysis have been limited to proteins < 25 kDa, because this is the conventional size limit for high-throughput structure determination by NMR spectroscopy [2].
One of the motivations at CESG for choosing to develop a cell-free protein production platform was to be able to take advantage of the emerging new technology of optimal isotopic labeling for protein NMR spectroscopy. This approach, named stereo-array isotope labeling (SAIL), utilizes the incorporation of amino acids labeled with 2H, 13C, and 15N so as to minimize spectral complexity and spin diffusion within the protein while allowing the detection of all connectivities required for sequence-specific assignments and the determination of sufficient constraints needed for high-resolution solution structures [3]. The SAIL approach requires cell-free incorporation of the amino acids, because the labeling patterns built into the amino acids would become scrambled if they were incorporated in a cellular system [3]. As its first target for investigation by the SAIL approach, CESG chose A. thaliana gene At3g16450.1, which codes for a 32 kDa, 299-residue protein with unknown structure.
At3g16450.1 is classified as a myrosinase-binding protein (MyroBP)-like protein. Myrosinase is a glucosinolate-degrading enzyme [4], and MyroBP has been identified as a component of high-molecular-mass myrosinase complexes in extracts of Brassica napus seed[5]. The presence of three myrosinase genes and several putative MyroBPs have been reported in A. thaliana [6-8]. The myrosinase/glucosinolate system is involved in plant defense against insects and pathogens [4], and hence MyroBP is implicated in this defense system, although experimental data supporting this notion is still lacking [9]. Many of the MyroBPs and MyroBP-related proteins have a repetitive structure containing two or more homologous sequences [10, 11]. The homologous domains also have sequence similarity to some plant lectins, and, because seed MyroBP from B. napus has been found to bind to p-aminophenyl-α-D-mannopyranoside and to some extent to N-acetylglucosamine, the protein has been reported to possess lectin activity [10]. However, in spite of its functional importance, no three-dimensional structure has been determined for any domain of the MyroBP family.
We report here the three-dimensional structure of the At3g16450.1 protein, which consists of two homologous MyroBP-type domains. The structure, which was determined by NMR spectroscopy from a relatively low quantity of SAIL protein (∼60 nmol = 300 μl of 0.2 mM protein), revealed that At3g16450.1 consists of tandem lectin-like domains corresponding to the two homologous sequences (residues 1–144, 153–299). To explore the sugar binding activity of At3g16450.1, we investigated interactions between immobilized At3g16450.1 protein and fluorescently labeled (pyridylaminated, PA) sugars by frontal affinity chromatography (FAC) [12]. Of the carbohydrates tested, only a few PA-sugars showed significant affinity for the immobilized At3g16450.1. This result is discussed in light of the plausible biological function of this protein. This study demonstrates the power of the SAIL approach in determining the structure of a larger protein by semi-automated means and with a minimal amount of material. It also shows how an NMR structure can be the springboard for easily performed functional investigations.
Results
Preparation of SAIL At3g16450.1
At3g16450.1 is a 299-residue protein with a molecular weight of 32 kDa. In our earlier work [13], we assigned the backbone resonances of At3g16450.1 using samples labeled uniformly with 13C/15N or 2H/13C/15N. However, further progress toward a structure determination was impeded by the problems of spectral crowding and broadened signals, as commonly seen in the NMR spectra of uniformly 13C/15N labeled large proteins. In the present study, we employed the SAIL technique [3] to address these problems. As an initial step, we optimized the conditions for E. coli cell-free production of At3g16450.1, with regard to the reaction temperature, incubation time, and expression vector. For comparison purposes, [U-13C, U-15N]-At3g16450.1 (UL-At3g16450.1) was prepared by using an E. coli in vivo expression system.
Comparison of NMR spectra of SAIL- and UL-At3g16450.1
Although the concentration of the SAIL-protein was lower than that of the UL-protein by a factor of three (SAIL: 0.2 mM; UL: 0.6 mM), the NMR spectra of SAIL-At3g16450.1 exhibited higher signal-to-noise than those of UL-At3g16450.1. The 1H-13C constant-time HSQC spectrum of SAIL-At3g16450.1 was less crowded and better resolved than that of UL-At3g16450.1 (Fig. 1a, b). The extensive stereo- and regio-specific deuteration of the SAIL-protein led to diminished overlaps and sharpened peaks, particularly in the methylene region, without compromising essential structural information (Fig. 1c, d). In the methyl region, the regio-specifically labeled methyl resonances from the SAIL sample were much less crowded (Fig. 1e, f). By virtue of these striking spectral improvements, it became possible to use established methods [14] to assign 95.5% of the resonances of SAIL-At3g16450.1. The chemical shifts of SAIL-At3g16450.1 were deposited in the Biological Magnetic Resonance Data Bank (BMRB) [15] with accession number 15607. In addition, 93% of the backbone carbonyl 13C shifts had been assigned earlier using uniformly 13C, 15N labeled protein [13]. These assigned chemical shifts were used as input to the TALOS program [16] to obtain dihedral angle constraints.
Fig. 1.

Comparison of 1H-13C constant-time HSQC NMR spectra of 0.6 mM of UL-At3g16450.1 and 0.2 mM of SAIL-At3g16450.1. (a) Full spectrum of UL-At3g16450.1. (b) Full spectrum of SAIL-At3g16450.1. (c) Methylene region of UL-At3g16450.1. (d) Methylene region of SAIL-At3g16450.1. (e) Methyl region of UL-At3g16450.1. (f) Methyl region of SAIL-At3g16450.1. Spectra were recorded at 27.5 °C at 1H frequency of 800 MHz. In the case of the SAIL-protein, 2H decoupling was applied during the 13C chemical shift evolution.
Solution structure of SAIL At3g16450.1
The assignment of the NOE peaks of At3g16450.1 and the structure determination were accomplished by use of the CYANA program [17, 18]. The structural statistics are summarized in Table 1. The 20 conformers representing the structures of At3g16450.1 could be superimposed closely within each individual domain (residues 1–144 or residues 153–299) but not both together (Fig. 2a, b). Residues 16–21 and 45–47 exhibited severe line broadening, probably arising from dynamics on the intermediate time scale for chemical shifts. As a result, these are the least well defined regions of the N-terminal domain. The C-terminal domain yielded reasonably well converged structures, including the side-chain conformations of residues in its core (Fig. 2c, d).
Table 1.
NMR constraints and structure calculation statistics for At3g16450.1a
| Completeness of the chemical shift assignments (%): | |
| All | 95.5 |
| Backbone | 97.8 |
| Side-chain | 93.3 |
| NOE distance constraints: | |
| Total | 1982 |
| Short-range, |i − j| ≤ 1 | 1192 |
| Medium-range, 1 < |i − j| < 5 | 111 |
| Long-range, |i − j| ≥ 5, intra-molecular | 679 |
| Maximal violation (Å) | 0.18 |
| Torsion angle constraints: | |
| φ | 138 |
| ψ | 136 |
| Maximal violation (°) | 2.6 |
| Restrained hydrogen bonds | 124 |
| CYANA target function value (Å2) | 1.77 ± 0.56 |
| AMBER energies (kcal/mol): | |
| Total | -7508 ± 21 |
| van der Waals | -2239 ± 30 |
| Ramachandran plot statistics (%) [35]: | |
| Residues in most favored regions | 89.0 |
| Residues in additional allowed regions | 9.5 |
| Residues in generously allowed regions | 1.0 |
| Residues in disallowed regions | 0.5 |
| RMSD from the averaged coordinates (Å): | |
| Backbone atoms of residues 2–144 (N-domain) | 1.12 ± 0.19 |
| Heavy atoms of residues 2–144 (N-domain) | 1.65 ± 0.16 |
| Backbone atoms of residues 153–297 (C-domain) | 0.69 ± 0.10 |
| Heavy atoms of residues 153–297 (C-domain) | 1.08 ± 0.09 |
The completeness of the 1H, 13C, 15N chemical shift assignments was evaluated for the aliphatic, aromatic, backbone amide, and Asn/Gln/Trp side-chain amide nuclei, excluding the carbon and nitrogen atoms not bound to 1H. Where applicable, the value given corresponds to the average over the 20 energy-refined conformers that represent the solution structure. CYANA target function values were computed before energy refinement.
Fig. 2.
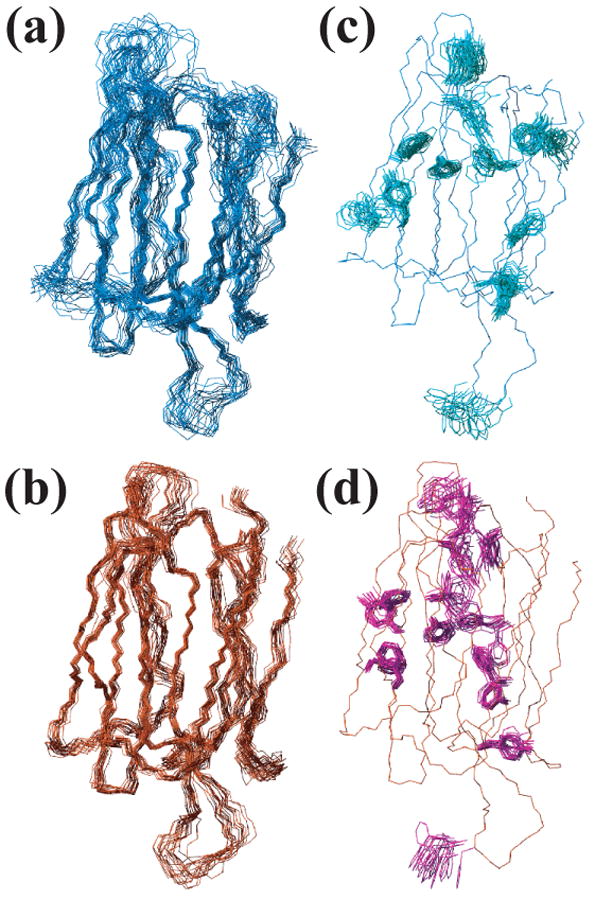
Three-dimensional NMR structure of At3g16450.1. (a) Superposition of the 20 energy-minimized conformers that represent the three-dimensional solution structure of the N-terminal domain. (b) Superposition of conformers representing the C-terminal domain. (c) Aromatic side chains (light green) and one backbone trace (blue) of the NMR structures for the N-terminal domain. (d) Aromatic side chains (magenta) and one backbone trace (red) of the NMR structure of the C-terminal domain.
Residues 145–152 in the linker region between the two domains are highly disordered. In addition, a careful search failed to reveal any interdomain NOE peaks. Thus the relative orientations of the two domains appear not to be fixed, and the overall structure of At3g16450.1 is best described as two tandem structural domains connected by a flexible linker (Fig. 3a). The secondary structural elements of At3g16450.1, extracted from the coordinates of the three-dimensional structure by the DSSP algorithm [19], showed that each domain has a similar structure consisting of three β-sheets related by pseudo three-fold symmetry (Fig. 3b).
Fig. 3.
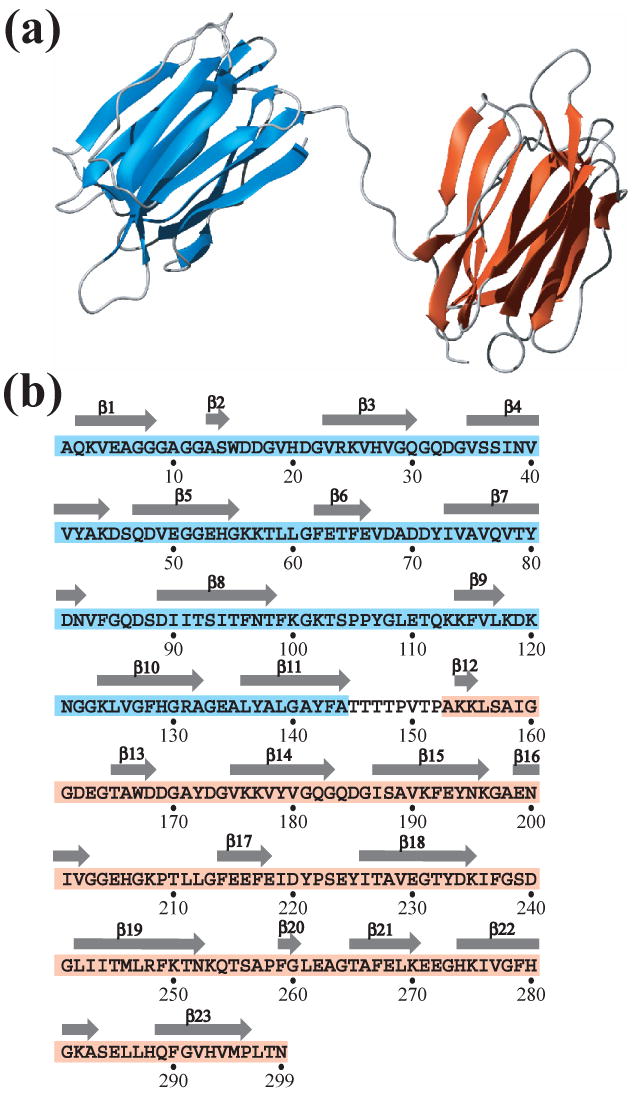
Secondary structure of At3g16450.1. (a) Ribbon representation of the NMR structure of At3g16450.1. These figures were prepared with MOLMOL (25). Due to the lack of NOEs, the relative orientation between the N- and C-terminal domains could not be defined. (b) Primary sequence of At3g16450.1. The sequences that correspond to the N-terminal (residues 1-144) and C-terminal (residues 153-299) structural domains are highlighted in cyan and pink, respectively, and β–strands are depicted as arrows above the sequence.
The coordinates of the 20 energy-refined conformers that represent the solution structure of At3g16450.1 were deposited in the Protein Data Bank with accession code 2JZ4. A structural homology search using the program DALI at EMBL [20, 21] yielded as the closest structure the agglutinin from Maclura promifera (PDB code: 1JOT), a plant lectin. The RMSD values for the N- and C-terminal domains vs. the agglutinin are 2.2 and 2.0Å, respectively. Thus each of the two domains of At3g16450.1 adopts a lectin fold. The relative orientation of the N-terminal domain to the C-terminal domains could not be defined owing to the absence of inter-domain NOEs. To confirm the molecular organization of the tandem arrangement, expression vectors were constructed that separately encoded the N-terminal half (residues 1–153) and the C-terminal half (residues 151–299) of At3g16450.1, and these were used to prepare 15N-labeled samples of each domain. The 1H-15N HSQC spectrum of each domain was well dispersed and, when overlaid, closely approximated the spectrum of full-length of At3g16450.1 (Fig. 4a, b). This result confirms the structural arrangement of At3g16450.1 as two independent, tandem structural domains.
Fig. 4.

Comparison of the NMR spectra of full-length At3g16450.1 and its isolated N- and C-terminal halves. (a) 1H-15N HSQC spectrum of full-length (residues 1–299) SAIL-At3g16450.1. (b) Overlay of 1H-15N HSQC spectra of the N-terminal (residues 1–153, blue) and the C-terminal (residues 151–299, red) halves of [U-15N]-At3g16450.1. These spectra were acquired at 27.5 °C, pH 6.8 on a Bruker DRX600 NMR spectrometer. The pattern of the overlaid spectra is almost identical to that of full length construct, showing that the two domains of At3g16450.1 are largely independent.
Interaction analysis of At3g16450.1 with sugars
Because each structural domain of At3g16450.1 was found to adopt a lectin fold, we assayed At3g16450.1 for possible sugar-binding activity. We utilized thirteen different fluorescence-labeled oligosaccharides (PA-sugars) as candidates. Among them, four PA-sugars eluted more slowly than control PA-sugars from a column of immobilized At3g16450.1 (Fig. 5a, b; Table 2). On the basis of the elution profiles, the Kd values of the four PA sugars to At3g16450.1 were estimated to be weak, at most 10-4 M. To further examine the observed interaction, we acquired 1H-15N HSQC spectra of 15N-labeled At3g16450.1 in the presence and absence of (Glcα1-4Glc)3. However, addition of the (Glcα1-4Glc)3 did not cause any perturbation of NMR resonances, even when the concentration of the sugar was ten times higher than that of the protein (data not shown). By contrast, NMR titration of At3g16450.1 with (Glcα1-4Glc)3-PA led to distinct chemical shift changes for certain NMR resonances (Fig. 5c), whereas addition of PA as the ligand resulted only in limited, subtle changes. These results suggest that both the PA and the (Glcα1-4Glc)3 elements contribute to the observed interactions. Residues in both, the N- and C-terminal domains of At3g16450.1 were affected by the presence of PA-sugar (Fig. 5c, blue and red boxes). Taken together, these binding analyses suggest that At3g16450.1 has a potential to bind PA-sugar with specificity for the sugar structure, although none of the various sugars tested exhibited a strong affinity.
Fig. 5.
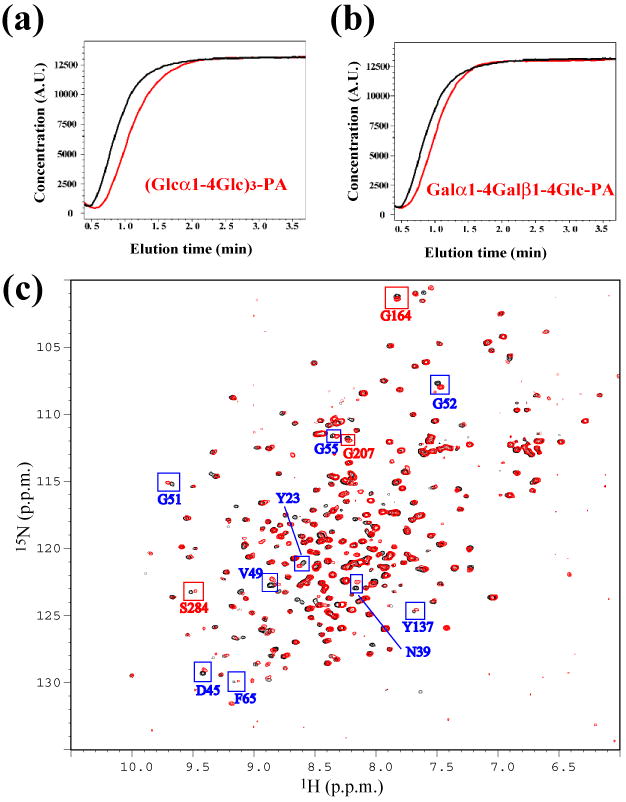
Investigation of sugar binding properties of At3g16450.1. (a) Elution profile of the FAC binding assay for (Glcα1-4Glc)3-PA (red) and control sugar (black). (b) FAC binding assay for Galα1-4Galβ1-4Glc-PA (red) and control PA-sugar (black). (c) Overlay of the 1H-15N HSQC spectra of uniformly 15N labeled At3g16450.1 in the absence (black) and presence (red) of (Glcα1-4Glc)3-PA. Assignments and boxes (blue for N-terminal domain; red for C-terminal domain) label some of the perturbed resonances.
Table 2.
Summary of the FAC binding assay for At3g16450.1 with various PA-sugars
| PA sugars that showed affinity for At3g16450.1 | Major natural location |
|---|---|
| (Glcα1-4Glc)3 maltohexaose | Starch of higher plants |
| (Glcα1-6Glc)3 isomaltohexaose | Starch of higher plants |
| Galα1-4Galβ1-4Glc | Glycolipid |
| GalNAcα1-3(Fucα1-2)Galβ1-3(Fucα1-4)GlcNAcβ1-3Galβ1-4Glc | Glycolipid |
| PA sugars that did not show affinity for At3g16450.1 | Major natural location |
| Galβ1-3(Fucα1-4)GlcNAcβ1-3Galβ1-4Glc | Glycolipid |
| Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4Glc | Glycolipid |
| (GlcNAcβ1-4GlcNAc)3 | Chitohexaose (insects and crustaceans) |
| (Glcβ1-4Glc)3 | Cellohexaose (cell walls of higher plants) |
| (Glcβ1-3Glc)3 | Laminarihexaose (pachyman of Poria cocos) |
| Man9GN2 (high-mannose type)(Code no. M9.1) | N-glycan |
| GlcNAcβ1-2Manα1-6(GlcNAcβ1-2Manα1-3)Manβ1-4GlcNAcβ1-4(Fucα1-6)GlcNAc (Code no.210.1) | N-glycan |
| Galβ1-4GlcNAcβ1-2Mana1-6(Galβ1-4GlcNAcβ1-2Manα1-3)Manβ1-4GlcNAcβ1-4(Fucα1-6)GlcNAc (Code no.210.4) | N-glycan |
| GlcNAcβ1-2Manα1-6(GlcNAcβ1-2Manα1-3)Manβ1-4(Xylβ1-2)GlcNAcβ1-4(Fucα1-3)GlcNAc (Code no.210.1FX) | N-glycan |
Discussion
In this study, we determined the solution structure of 32 kDa At3g16450.1 protein from A. thaliana by the SAIL-NMR method. This is the first application of SAIL-NMR in a structural genomics effort. It provided the first structure for a member of the hitherto structurally unexplored MyroBP family.
At3g16450.1 consists of two tandem domains, each composed of three β-sheets. The fold of each domain is nearly identical to that of the agglutinin (PDB code: 1JOT), which shares sequence identities of 26 and 33% with the N- and C-terminal domains of At3g16450.1, respectively. Sequence similarity searches performed by PSI-BLAST [22] identified other MyroBP s and MyroBP-like proteins from A. thaliana and B. napus with sequence identities to the At3g16450.1 domains ranging from 30% to 70%. The most highly conserved regions correspond to the β-strands (Fig. 6). The N- and C-terminal domains of At3g16450.1, with 51% sequence identity to each other, are superimposed with RMSDs of 1.3 Å for the backbone of the β-strands and 1.7 Å including the loop regions, indicating that all of these family members adopt a similar fold.
Fig. 6.

Alignment of MyroBP-related sequences. Sequences of the N-terminal and C-terminal domains of At3g16450.1 are aligned with those of MyroBP from B. napus and of MyroBP-like proteins from A. thaliana (At1g52030, At3g16400, At3g16440, At3g16470, At3g21380). Asterisks and vertical bars indicate identical and similar residues, respectively. β-strands of At3g16450.1 are depicted as arrows above the sequence.
It has been reported that seed MyroBP from B. napus possesses lectin activity by binding to p-aminophenyl-α-D-mannopyranoside and to some extent to N-acetylglucosamine [10]. Because myrosinase contains potential N-linked sugar binding sites [23], the sugar binding activity of MyroBP is suspected to be implicated in the binding to myrosinase. In the case of At3g16450.1, the protein did not show a significant affinity for sugar structures specific to N-linked glycan, but rather showed weak affinity for starch or glycolipid, raising the possibility that the lectin activity of the MyroBP family is also involved in an interaction between a myrosinase complex and other molecules. It is also noteworthy that a UniGene database search [24] suggested that At3g16450.1 is expressed in leaf and root. Because myrosinases also have been shown to be expressed in A. thaliana leaf [6, 8], it may be suspected that At3g16450.1 forms a complex with myrosinase, thereby guiding the myrosinase complex to a damaged site in leaf via weak interactions with starch in leaf or glycolipid from foreign pathogens. However, it is obvious that further study will be required to determine the biological importance of MyroBP-sugar interactions.
Many MyroBP and MyroBP-related proteins contain tandem lectin domains as shown in Fig. 6. As commonly seen in carbohydrate binding proteins, it is possible that the tandem domains present in other MyroBP family members engage in multivalent sugar binding, as suggested for At3g16450.1 by the NMR chemical shift perturbation experiments (Fig. 5c). It is also probable that each homologous domain of the MyroBP family possesses different ligand binding properties, thereby providing a broad binding specificity for foreign organisms. In some proteins containing tandem homologous domains, inter-domain interactions fix the relative orientation of the domains in a specific multi-domain structure that is essential for biological function. Other proteins with tandem domains contain a flexible linker, and a specific structure may be adopted only when a target is bound. The present study, suggests that At3g16450.1 belongs to the latter category.
The major problems with structural genomics by NMR are low solubility and molecular weight limitations [2]. As shown by this study, the SAIL-NMR method provides a promising approach to overcoming both of these problems. One important aspect of the SAIL technology is that the signal intensities for the SAIL protein are several times stronger than for the corresponding UL sample [3], thus making it possible to perform structure determination of proteins even at a low concentration. In this study, the structure was determined with a 0.2 mM sample of SAIL-At3g16450.1. The SAIL-NMR method offers prospects for determining structures of proteins with low solubility or poor yield. The SAIL method can also accelerate the process of structural analysis. Simplification of the NMR spectra by the SAIL method (Fig. 1), raises the possibility of semi- or fully-automated analysis of NMR data leading to structure determination with minimal human intervention. We are developing a platform of software that exploits the benefits of the SAIL method [25-27]. Finally, the SAIL method is expected to enable functional investigations of larger proteins.
Experimental procedures
Plasmid construction
The construction of pET15b (Novagen) harboring At3g16450.1 was described previously [13]. The vector used for cell-free production of At3g16450.1 was constructed according to a strategy described previously [28]. Genes corresponding to the N-terminal histidine tag derived from the pET15b vector (Novagen) and At3g16450.1 were subcloned into pIVEX2.3d (Roche) between the NcoI/NdeI and NdeI/BamHI sites, respectively. Silent mutations were introduced into the N-terminal sequence to enhance the expression rate [28]. Expression vectors coding for the N-terminal (residues 1–153) and the C-terminal (residues 151-299) domains of At3g16450.1 were constructed by cloning the corresponding target sequence into the NdeI and BamHI sites of pET15b.
Preparation of labeled proteins
[U-15N]- and [U-13C, U-15N]-proteins were produced by culturing Escherichia coli BL21 (DE3) strain harboring the corresponding expression vector in M9 medium containing 15NH4Cl and/or [U-13C]-glucose as the sole nitrogen and carbon sources. Cells were cultured at 30 °C with shaking. Expression was induced by the addition of IPTG with a final concentration of 1 mM, and cells were harvested 6.5 h after the induction.
SAIL At3g16450.1 was produced by E. coli cell-free expression. A total of 110 mg of SAIL amino acid mixture was used, with the amount of each individual SAIL amino acid proportional to the amino acid composition of At3g16450.1. A home-made E. coli S30 extract was used, and the reaction was performed as previously reported [25, 28]. The volumes of the inner and outer solutions were 10 ml and 40 ml, respectively. The reaction was carried out at 30 °C for 15 h with shaking. To prevent degradation of the produced protein, a protease inhibitor cocktail (Roche) was added to the reaction. The At3g16450.1 protein was purified as described previously [13].
NMR Spectroscopy
The NMR sample used for the structure determination contained 0.2 mM SAIL At3g16450.1 protein in 20 mM bis-Tris-d19 (Cambridge Isotope Laboratories), 100 mM KCl, 10% D2O, pH 6.8. NMR spectra were recorded on a Bruker Avance 600 MHz spectrometer equipped with a 5 mm TXI CryoProbe, and on a Bruker Avance 800 MHz spectrometer at 27.5 °C. The spectra were processed with the programs XWINNMR version 3.5 (Bruker) or NMRPipe [29], and analyzed using the program SPARKY (University of California, San Francisco). Backbone and β-CH resonances were assigned using 2D HSQC, and 3D HN(CO)CACB and HBHA(CO)NH spectra. Side-chain resonances were assigned using 3D H(CCCO)NH, (H)CC(CO)NH, HCCH-TOCSY, ct-HCCH-COSY, 13C-edited NOESY and 15N-edited NOESY spectra. 15N- and 13C-edited NOESY spectra were recorded with a mixing time of 75 ms, and the interproton distance constraints were obtained from the NOESY peaks, which were picked and manually filtered using Sparky.
Collection of conformational constraints, structure calculation and refinement
Automated NOE cross-peak assignments [30] and structure calculations with torsion angle dynamics were performed using the program CYANA version 2.2 [31]. Backbone torsion angle constraints obtained from database searches with the program TALOS [16] were incorporated into the structure calculation. Simulated annealing with 20000 torsion angle dynamics time steps per conformer was performed in the CYANA structure calculations. In the final cycle of the CYANA protocol, 100 conformers were generated and further refined using the AMBER 9 software package [32] with a full-atom force field [33]. The refinement comprised three stages, initial minimization, molecular dynamics, and final minimization. Minimization and molecular dynamics consisted of 1500 steps and 20 ps duration respectively. A generalized Born implicit solvent model was used to account for the solvent effects [34]. The force constants for distance and torsion angle constraints were 50 kcal/(mol·Å2) and 200 kcal/(mol·rad2) respectively. From the resulting structures of this first AMBER refinement, we extracted backbone hydrogen bond constraints in the regular secondary elements that were present in more than 75% of the 100 conformers. With these as additional constraints, we repeated the refinement. From the conformers that did not significantly violate experimental constraints, we selected the 20 lowest energy structures for analysis. The structural quality was evaluated with PROCHECK-NMR [35]. The program MOLMOL [36] was used to visualize the structures. The coordinates of the 20 energy-refined CYANA conformers of At3g16450.1 have been deposited in the Protein Data Bank (accession code 2JZ4). The chemical shifts of At3g16450.1 have been deposited in the BioMagResBank (accession code 15607).
Frontal affinity chromatography
M9.1 (The code numbers and structures of pyridylaminated (PA-)oligosaccharides refer to the GALAXY web site at www.glycoanalysis.info/ENG/index.html [37]), 210.1, 210.4, and 210.1FX were purchased from Seikagaku Kogyo Co. GalNAcα1-3(Fucα1-2)Galβ1-3(Fucα1-4)GlcNAcβ1-3Galβ1-4Glc-PA and Neu5Acα2-6Galβ1-4GlcNAcβ1-2Manα1-6(Neu5Acα2-3Galβ1-3(Neu5Acα2-6)GlcNAcβ1-4(Neu5Acα2-6Galβ1-4GlcNAcβ1-2)Manα1-3)Manβ1-4GlcNAcβ1-4GlcNAc-PA were from Takara Bio. Inc. Other PA-glycans were prepared by amination of the commercial oligosaccharides with 2-aminopyridine [38]. Galβ1-3(Fucα1-4)GlcNAcβ1-3Galβ1-4Glc and Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4Glc were purchased from Calbiochem. Cellohesaose, chitohesaose, isomaltohexaose, laminarihesaose, and maltohexaose were from Seikagaku Kogyo Co.
At3g16450.1 containing the N-terminal His-tag was dissolved in 10 mM HEPES buffer, pH 7.6 containing 150 mM NaCl, 1 mM CaCl2 and bound to Ni-NTA Agarose. After immobilization, the agarose beads were packed into a stainless steel column (4.0 × 10 mm, GL Sciences).
FAC analysis was carried out as described previously [39]. PA-oligosaccharides were dissolved at a concentration of 10 nM in 10 mM HEPES, pH 7.6 containing 150 mM NaCl, 1 mM CaCl2 and applied onto the At3g16450.1 column at a flow rate of 0.25 ml/min at 20 °C. The elution profile was monitored by the fluorescence intensity at 400 nm (excitation at 320 nm). Tetra-sialyl PA-glycan, Neu5Acα2-6Galβ1-4GlcNAcβ1-2Manα1-6(Neu5Acα2-3Galβ1-3(Neu5Acα2-6)GlcNAcβ1-4(Neu5Acα2-6Galβ1-4GlcNAcβ1-2)Manα1-3)Manβ1-4GlcNAcβ1-4GlcNAc-PA, was used as a control sugar to determine the elution volume of the unbound oligosaccharide.
NMR chemical shift perturbation mapping
NMR samples were prepared containing free [U-15N] At3g16450.1 (0.1 mM protein, 10 mM HEPES, pH 7.6, 150 mM KCl, 1 mM CaCl2) and its complex with PA-sugar (same solvent composition plus 0.5 mM PA-(Glcα1-4Glc)3). 1H‐15N HSQC spectra of the isolated and titrated samples were acquired at 27.5 °C on a Bruker Avance 600 MHz NMR spectrometer.
Acknowledgments
This work was supported by the Technology Development for Protein Analyses and Targeted Protein Research Program of the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), the Core Research for Evolutional Science and Technology (CREST) of the Japan Science and Technology Agency (JST), a Grant-in-Aid for Scientific Research of the Japan Society for the Promotion of Science (JSPS), the National Institutes of Health, Protein Structure Initiative through grants P50 GM64598 and U54 GM074901, and by the Volkswagen Foundation.
Abbreviations
- FAC
frontal affinity chromatography
- HSQC
hetero-nuclear single quantum correlation spectroscopy
- MyroBP
myrosinase-binding protein
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- PA
pyridylamine
- RMSD
root mean square deviation
- SAIL
Stereo-array isotope labeling
- UL
uniformly labeled
References
- 1.The Arabidopsis Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 2.Vinarov DA, Loushin Newman CL, Markley JL. Wheat germ cell-free platform for eukaryotic protein production. FEBS J. 2006;273:4160–4169. doi: 10.1111/j.1742-4658.2006.05434.x. [DOI] [PubMed] [Google Scholar]
- 3.Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Ono AM, Güntert P. Optimal isotope labelling for NMR protein structure determinations. Nature. 2006;440:52–57. doi: 10.1038/nature04525. [DOI] [PubMed] [Google Scholar]
- 4.Rask L, Andréasson E, Ekbom B, Eriksson S, Pontoppidan B, Meijer J. Myrosinase: gene family evolution and herbivore defense in Brassicaceae. Plant Mol Biol. 2000;42:93–113. [PubMed] [Google Scholar]
- 5.Lönnerdal B, Janson JC. Studies on myrosinases. II. Purification and characterization of a myrosinase from rapeseed (Brassica napus L.) Biochim Biophys Acta. 1973;315:421–429. [Google Scholar]
- 6.Xue J, Jørgensen M, Pihlgren U, Rask L. The myrosinase gene family in Arabidopsis thaliana: gene organization, expression and evolution. Plant Mol Biol. 1995;27:911–922. doi: 10.1007/BF00037019. [DOI] [PubMed] [Google Scholar]
- 7.Takechi K, Sakamoto W, Utsugi S, Murata M, Motoyoshi F. Characterization of a flower-specific gene encoding a putative myrosinase binding protein in Arabidopsis thaliana. Plant Cell Physiol. 1999;40:1287–1296. doi: 10.1093/oxfordjournals.pcp.a029517. [DOI] [PubMed] [Google Scholar]
- 8.Capella AN, Menossi M, Arruda P, Benedetti CE. COI1 affects myrosinase activity and controls the expression of two flower-specific myrosinase-binding protein homologues in Arabidopsis. Planta. 2001;213:691–699. doi: 10.1007/s004250100548. [DOI] [PubMed] [Google Scholar]
- 9.Eriksson S, Andréasson E, Ekbom B, Granér G, Pontoppidan B, Taipalensuu J, Zhang J, Rask L, Meijer J. Complex formation of myrosinase isoenzymes in oilseed rape seeds are dependent on the presence of myrosinase-binding proteins. Plant Physiol. 2002;129:1592–1599. doi: 10.1104/pp.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taipalensuu J, Eriksson S, Rask L. The myrosinase-binding protein from Brassica napus seeds possesses lectin activity and has a highly similar vegetatively expressed wound-inducible counterpart. Eur J Biochem. 1997;250:680–688. doi: 10.1111/j.1432-1033.1997.00680.x. [DOI] [PubMed] [Google Scholar]
- 11.Falk A, Taipalensuu J, Ek B, Lenman M, Rask L. Characterization of rapeseed myrosinase-binding protein. Planta. 1995;195:387–395. doi: 10.1007/BF00202596. [DOI] [PubMed] [Google Scholar]
- 12.Kasai K, Oda Y, Nishikawa M, Ishii S. Frontal affinity chromatography: Theory for its application to studies on specific interactions of biomolecules. J Chromatogr. 1986;376:33–47. doi: 10.1016/s0378-4347(00)80822-1. [DOI] [PubMed] [Google Scholar]
- 13.Sugimori N, Torizawa T, Aceti DJ, Thao S, Markley JL, Kainosho M. Letter to the Editor: 1H, 13C and 15N backbone assignment of a 32 kDa hypothetical protein from Arabidopsis thaliana, At3g16450.1. J Biomol NMR. 2004;30:357–358. doi: 10.1007/s10858-005-2324-9. [DOI] [PubMed] [Google Scholar]
- 14.Cavanagh J, Fairbrother WJ, Palmer AG, III, Skelton NJ, Rance M. Protein NMR Spectroscopy. Principles and Practice. 2nd. Academic Press; San Diego, CA: 2006. [Google Scholar]
- 15.Seavey BR, Farr EA, Westler WM, Markley JL. A relational database for sequence-specific protein NMR data. J Biomol NMR. 1991;1:217–236. doi: 10.1007/BF01875516. [DOI] [PubMed] [Google Scholar]
- 16.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 17.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 18.Güntert P. Automated NMR protein structure calculation. Prog Nucl Magn Reson Spectrosc. 2003;43:105–125. [Google Scholar]
- 19.Kabsch W, Sander C. Dictionary of protein secondary structure - pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 20.Holm L, Ouzounis C, Sander C, Tuparev G, Vriend G. A database of protein structure families with common folding motifs. Protein Sci. 1992;1:1691–1698. doi: 10.1002/pro.5560011217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 22.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falk A, Ek B, Rask L. Characterization of a new myrosinase in Brassica napus. Plant Mol Biol. 1995;27:863–874. doi: 10.1007/BF00037015. [DOI] [PubMed] [Google Scholar]
- 24.Schuler GD. Pieces of the puzzle: expressed sequence tags and the catalog of human genes. J Mol Med. 1997;75:694–698. doi: 10.1007/s001090050155. [DOI] [PubMed] [Google Scholar]
- 25.Takeda M, Ikeya T, Güntert P, Kainosho M. Automated structure determination of proteins with the SAIL-FLYA NMR method. Nature Protocols. 2007;2:2896–2902. doi: 10.1038/nprot.2007.423. [DOI] [PubMed] [Google Scholar]
- 26.López-Méndez B, Güntert P. Automated protein structure determination from NMR spectra. J Am Chem Soc. 2006;128:13112–13122. doi: 10.1021/ja061136l. [DOI] [PubMed] [Google Scholar]
- 27.Scott A, López-Méndez B, Güntert P. Fully automated structure determinations of the Fes SH2 domain using different sets of NMR spectra. Magn Reson Chem. 2006;44:S83–S88. doi: 10.1002/mrc.1813. [DOI] [PubMed] [Google Scholar]
- 28.Torizawa T, Shimizu M, Taoka M, Miyano H, Kainosho M. Efficient production of isotopically labeled proteins by cell-free synthesis: A practical protocol. J Biomol NMR. 2004;30:311–325. doi: 10.1007/s10858-004-3534-2. [DOI] [PubMed] [Google Scholar]
- 29.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe - A multidimensional spectral processing system based on Unix pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 30.Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 31.Güntert P. Automated NMR structure calculation with CYANA. Meth Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 32.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 34.Tsui V, Case DA. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers. 2000;56:275–291. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 36.Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi N, Kato K. GALAXY (Glycoanalysis by the three axes of MS and chromatography): a web application that assists structural analyses of N-glycans. Trends Glycosci Glycotechnol. 2003;15:235–251. [Google Scholar]
- 38.Yamamoto S, Hase S, Fukuda S, Sano O, Ikenaka T. Structures of the sugar chains of interferon-γ produced by human myelomonocyte cell line HBL-38. J Biochem (Tokyo) 1989;105:547–555. doi: 10.1093/oxfordjournals.jbchem.a122703. [DOI] [PubMed] [Google Scholar]
- 39.Arata Y, Hirabayashi J, Kasai K. Sugar binding properties of the two lectin domains of the tandem repeat-type galectin LEC-1 (N32) of Caenorhabditis elegans. Detailed analysis by an improved frontal affinity chromatography method. J Biol Chem. 2001;276:3068–3077. doi: 10.1074/jbc.M008602200. [DOI] [PubMed] [Google Scholar]