

Abstract
Experiments performed with Holtzman rats demonstrated that brain iron (Fe) was lower by postnatal day 13 (P13) in pups born and nursed by dams that began copper-deficient (−Cu) treatment at embryonic day 7. Transcardial perfusion of P24– P26 males and females to remove blood Fe contamination revealed that brain Fe was still 20% lower in −Cu than +Cu rats. Estimated blood content of brain for −Cu rats was greater than for +Cu rats; for all groups, values ranged between 0.43 and 1.03%. Using group-specific data and regression analyses, r = 0.99, relating blood Fe to hemoglobin, brain Fe in non-perfused rats in a replicate study was lower by 33% at P13 and 39% at P24 in −Cu rats. Brain extracts from these rats and from P50 rats from a post-weaning model were compared by immunobloting for transferrin receptor (TfR1). P24 brain −Cu/+Cu TfR1 was 3.08, suggesting that brains of −Cu rats were indeed Fe deficient. This ratio in P13 rats was 1.44, p < 0.05. No change in P50 −Cu rat brain TfR1 or Fe content was detected despite a 50% reduction in plasma Fe. The results suggest that brain Fe accumulation depends on adequate Cu nutriture during perinatal development.
Keywords: brain, copper, copper-deficient rats, iron, transferrin receptor
Proper brain development requires a balance of essential nutrients to ensure that hyperplasia and hypertrophy produce the correct number of optimally differentiated cells in a timely manner. Even micronutrient imbalance, such as iron and zinc (Zn) deficiency, can impact on behavioral development (Wauben and Wainwright 1999). Another essential metal, copper (Cu), is also necessary for proper brain development. Ewes grazing on pastures low in Cu content produce ataxic lambs with brain deformities (Bennetts and Chapman 1937). Laboratory rodents (guinea pigs, rats and mice) fed Cu-deficient diets during gestation produce offspring exhibiting hypomyelination, and focal and widespread necrosis (Everson et al. 1967; Carlton and Kelly 1969; Prohaska and Smith 1982). Recent studies in rats have indicated that recovery from perinatal Cu deficiency may be impossible, as persistent sensory motor dysfunction has been observed in rats repleted with a generous supply of Cu for over 6 months (Prohaska and Hoffman 1996; Penland and Prohaska 2004).
Biochemical mechanisms for the altered brain neurochemistry and behavior associated with Cu deficiency have not been elucidated. Mammals express about a dozen Cu-dependent enzymes. Changes in these cuproenzymes due to Cu restriction may be connected to neuropathology (O'Dell and Prohaska 1983). Cu deficiency is associated with lower brain cytochrome c oxidase activity (Gallagher et al. 1956). This could alter mitochondrial function and brain energy metabolism. Lower brain Cu, Zn-superoxide dismutase activity may render the brain susceptible to reactive oxygen toxicity (Prohaska and Wells 1974). Reduction in brain norepinephrine due to lower dopamine-β-monooxygenase activity in Cu deficiency could impact on brain development (Prohaska and Wells 1974). Lower activity of peptidyl α-amidating monooxygenase in Cu-deficient brain might limit neuropeptide activation (Prohaska et al. 1995). However, none of these hypotheses have been thoroughly tested. Other factors may be responsible for the neuropathology and behavioral changes of copper deficiency.
One such possibility is iron (Fe) limitation. It is well established that Fe deficiency in perinatal development has long-lasting behavioral effects even after dietary Fe supplementation (Beard and Connor 2003). Iron deficiency results in hypomyelination (Beard et al. 2003), alters monoamine metabolism (Youdim and Green 1976), and is associated with lower cytochrome oxidase activity (de Deungria et al. 2000). Thus, there are many similarities between perinatal Cu and Fe deficiencies.
Previous observations showed that brains of Cu-deficient rat pups had significantly lower concentrations of Fe, but not zinc, compared with Cu-adequate pups (Prohaska and Wells 1975). Since Cu-deficient rat pups are anemic and the previous brain Fe data were on tissue that contained blood, it was believed that most, if not all, of the apparent difference in brain Fe content could be explained by hemoglobin differences between controls and Cu-deficient rat pups. Anemia is a hallmark feature of both dietary Cu and Fe deficiency (Fox 2003). A recent study confirmed the reduction in rat pup brain Fe following perinatal Cu deficiency and also reported that this deficit persisted following 6 months of recovery (Penland and Prohaska 2004). Furthermore, the lower brain Fe levels of Cu-repleted rats occurred in animals with normal hemoglobin content, ruling out the possibility of blood contamination. Perinatal Fe deficiency also results in a persistent reduction in Fe content upon repletion (Dallman et al. 1975).
The purpose of the current studies was to evaluate brain Fe accumulation during perinatal Cu deficiency, to analyze the contribution of blood Fe in non-perfused rat brain, and to determine whether the cell perceived the putative changes in the brain by measuring transferrin receptor expression, a protein whose expression is influenced by brain Fe status (Siddappa et al. 2003).
Materials and methods
Animal care and diets
Holtzman sperm-positive rats, purchased commercially (Harlan Sprague Dawley, Indianapolis, IN, USA), received one of two dietary treatments, Cu-deficient or Cu-adequate, consisting of a Cu-deficient modified AIN-76 A diet (Teklad Laboratories, Madison, WI, USA) and either low-Cu drinking water or Cu-supplemented drinking water, respectively. The purified diet contained 0.34 mg Cu/kg and 48 mg Fe/kg by chemical analysis. This diet contains approximately one-tenth of the optimal dietary Cu level for rats. Offspring and dams on the Cu-deficient treatment drank deionized water, whereas Cu-adequate treatment groups drank water that contained 20 mg Cu/L by adding CuSO4 to the drinking water. Rats were given free access to diet and drinking water.
Pregnant dams were placed on the Cu-deficient treatment 7 days after they were identified as sperm-positive and 2 days following parturition, litter size was adjusted to 10 pups. Offspring in the perinatal model were sampled at various ages during the suckling period. In total, 24 litters (12 Cu-adequate and 12 Cu-deficient) were studied in a set of three separate experiments. An additional postnatal study was conducted in which male weanling rats were maintained on the two treatments (n = 5) for 30 days from postnatal age 20 (P20) to P50. All animals were maintained at 24°C with 55% relative humidity on a 12 h light cycle (07.00–19.00 hours). All protocols were formally approved by the University of Minnesota Animal Care Committee.
Rats were killed by decapitation and brains were removed, weighed and processed for biochemical analysis. Brains were dissected into three parts on a chilled glass plate. The regions were cerebellum, medulla oblongata + pons, and the remainder (largely forebrain). For P3 pups, whole brains were analyzed, as were whole brains of P24–P26 rats that were perfused.
Transcardial perfusion
Male and female P24–P26 rats in two separate studies were perfused after being deeply anesthetized with halothane. Pups were flushed via transcardial perfusion with heparinized (4 U/mL) phosphate-buffered saline, pH 7.4, for 2 min. Brains were then removed and transferred to acid-washed flasks for metal analysis. Perfusate was analyzed for Cu and Fe content. Levels were less than digestion blanks.
Biochemical analyses
Hemoglobin was determined spectrophotometrically as cyanmethemoglobin. Plasma was obtained from microhematocrit tubes following centrifugation, and the activity of the cuproprotein ceruloplasmin (EC 1.16.3.1) was measured following oxidation of o-dianisidine at 37°C. Plasma Fe was determined by atomic absorption spectroscopy (AAS) after treatment with hot (85°C) trichloroacetic acid as suggested in the instrument protocols (Perkin-Elmer, Wellesley, MA, USA). Protein levels were measured by a modified Lowry procedure using bovine albumin as reference.
Copper and iron analyses
Brains, aliquots of whole blood, liver, and 1 g portions of diet were wet-digested with 4 mL concentrated HNO3 (Trace Metal Grade, Fischer Scientific, Pittsburg, PA, USA) and the residue brought to 4.0 mL with 0.1 m/L HNO3. Samples were then analyzed for total Cu and Fe by flame AAS (Perkin Elmer). The method was checked with a certified bovine liver standard (1577, U.S. National Bureau of Standards and Technology, Gaithersburg, MD, USA).
Western immunoblots
Western blotting analysis was performed by size fractionation of proteins on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels and electroblot transfer to 0.2 μm nitrocellulose membranes (Protran, Schleicher & Schuell). Membranes were stained with Ponceau S (Sigma, St. Louis, MO, USA) to verify equal loading of protein and then used in immunoblotting as described elsewhere (Prohaska and Brokate 2001). Some membranes were reprobed after incubation of membranes with buffer containing 2-mercaptoethanol and SDS at 55°C for 30 min.
Tranferrin receptor (TfR1) protein was detected using a monoclonal mouse anti-human antibody (13-6800, Zymed Laboratories, South San Francisco, CA, USA) at a 1 : 500 dilution (1 μg/mL).
Glucose transporter one (GLUT1) protein was detected using a previously characterized GLUT1 rabbit anti-serum at a 1 : 5000 dilution, generously supplied by Dr Lester R. Drewes (McCall et al. 1996). Secondary species-specific antibodies were diluted 1 : 5000. SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL, USA) was used to detect selected proteins. Chemiluminescence was captured using high-speed blue X-ray film (Lake Superior X Ray Inc., Duluth, MN, USA), and densitometry was carried out using the FluorChem system (Alpha Innotech, San Leandro, CA, USA). The sizes of the immunoreactive bands were estimated from regression analysis using standard peptides (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Hemoglobin and blood Fe data were analyzed by linear regression. Specific mean comparisons were tested by Student's t-test, α = 0.05. Data were analyzed using statistical software (Statview 4.5, Abacus Concepts, Berkeley, CA, USA).
Results
Relationship between blood iron and hemoglobin
Previous work on Fe levels in the brains of Cu-deficient rats was deemed inaccurate because the brain tissue contained blood. We estimate that the ratio of rat blood Fe (300 μg/g) to unperfused rat brain Fe (10 μg/g) is about 30. The volume of blood in unperfused brain is about 0.5–1%, resulting in a 15–30% error in estimating total brain Fe that contains blood. Therefore, to evaluate whether brain Fe content was really altered by Cu status, corrections for blood contamination were needed. Perfusion of every rat brain prior to metal analysis would be time consuming, potentially leading to contamination, and would prevent the use of organs, including brain, for metabolic studies or biochemical assays.
One indirect approach was to estimate and correct for blood Fe in the brain by determination of hemoglobin (Hb). Four P24 Cu-deficient male (− CuM) and female (− CuF) rats were compared with four Cu-adequate male (+ CuM) and female (+ CuF) rats (Fig. 1). Each rat was from a separate litter. One aliquot of blood was used to measure Hb concentration and another aliquot was digested with nitric acid to determine total blood Fe. There was an excellent correlation between these two variables in both experiments and for both dietary treatments (Fig. 1). There was sufficient difference between males and females that each regression equation was used separately in subsequent experiments. These regression data imply that measuring blood Hb could be used to predict blood Fe with reasonable accuracy.
Fig. 1.
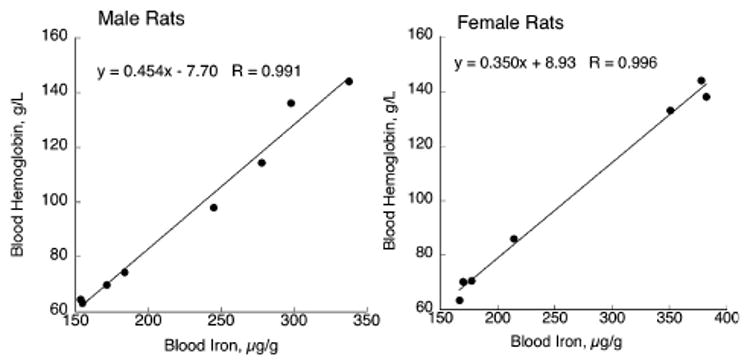
Correlation between blood hemoglobin and blood iron in P24–P26 male and female Holtzman rats. Following perinatal Cu deficiency, weanling rats (four Cu-deficient and four Cu-adequate) were decapitated and blood was analyzed. One sample from a Cu-adequate female was spilled prior to iron measurement. Linear regression was used to fit data.
Estimation of brain iron following perfusion and calculation of contaminating blood iron
Additional littermates from the two experiments following perinatal Cu deficiency were used to evaluate brain Fe content (Table 1). Dietary Cu deficiency reduced brain Cu content by 83% and 85% in non-perfused − CuM and − CuF compared with + CuM and +CuF rats, respectively. Transcardial perfusion did not alter brain Cu content. Hb levels in the non-perfused − CuM and − CuF rats were significantly lower than + CuM and + CuF controls (Table 1). Using the regression equations for each gender (Fig. 1), blood Fe was estimated for all four groups of rats (Table 1).
Table 1.
Characteristics of 24–26-day-old rats following perinatal copper deficiency
| Males | Females | |||
|---|---|---|---|---|
| Characteristics | Cu-adequate | Cu-deficient | Cu-adequate | Cu-deficient |
| NP-Brain Cu, μg/g | 2.24 ± 0.06 | 0.376 ± 0.039 | 2.34 ± 0.041 | 0.352 ± 0.013 |
| P-Brain Cu, μg/g | 2.19 ± 0.10 | 0.377 ± 0.030 | 2.13 ± 0.050 | 0.372 ± 0.014 |
| NP-Hemoglobin, g/L | 160 ± 2.7 | 97.5 ± 9.6 | 139 ± 2.4 | 72.6 ± 4.8 |
| Est–Blood Fe, μg/g | 370 ± 6.0 | 232 ± 21 | 395 ± 15 | 179 ± 10.9 |
| P-Blood in brain, % | 0.43 | 0.77 | 0.73 | 1.03 |
Values are means ± SEM (n = 4). Rats were born to, and nursed by Cu-deficient or Cu-adequate dams. Treatment began 2 weeks prior to parturition. Pups were maintained on the same treatment as their dams until killing. Littermates were either killed by decapitation and not perfused (NP), or perfused transcardially to remove blood from brain (P). Blood iron was estimated from hemoglobin concentration and regression analysis (Fig. 1). Blood content in brain was estimated by determining the amount of iron lowered by perfusion (μg/g) (Fig. 2) relative to the total iron in blood for non-perfused littermates. Dietary treatment effects were tested by unpaired Student's t-test. Compared with Cu-adequate rats, Cu-deficient rats had lower brain copper, hemoglobin and estimated blood iron concentrations, p < 0.05. Perfusion did not lower brain copper content, p > 0.05.
The fraction of blood present in the brain in non-perfused rats was estimated by calculating the difference in brain Fe after transcardial perfusion (Fig. 2), in μg/g, divided by the estimate of blood Fe (Table 1), since the perfused rats were littermate treatment-matched in all cases.
Fig. 2.
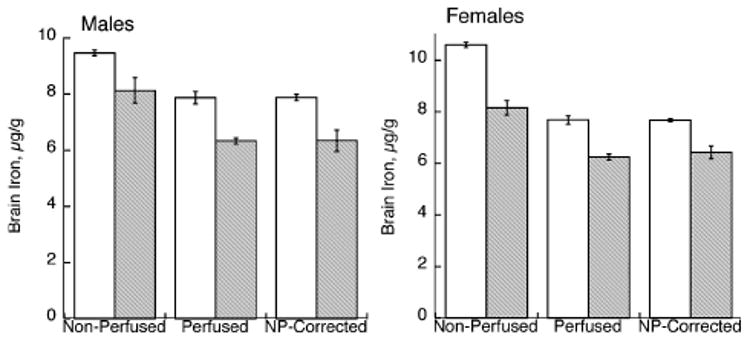
Effect of perinatal Cu deficiency on brain iron concentration in P24–P26 male and female rats. Iron was determined by flame atomic absorption spectroscopy in non-perfused and perfused rats. Each bar represents the mean ± SEM (n = 4). Open bars represent values for Cu-adequate rats and shaded bars for Cu-deficient rats. Perfusion lowered the brain iron concentration in both Cu-adequate and Cu-deficient rats of both genders, p < 0.05. Brain iron was determined in another set (n = 4) of non-perfused littermates but was corrected for blood Fe contamination after measurement of hemoglobin and estimation of blood iron. These values were not different from those measured in perfused littermates.
Perfusion lowered brain Fe in both Cu-deficient and Cu-adequate rats. The estimate for blood contamination in the brains of − CuM and − CuF was higher than for + CuM and + CuF. This estimate was then used to correct another set of individual non-perfused littermates in which Hb and total brain Fe were measured. The corrected brain Fe values, after subtracting brain Fe due to blood, matched the values measured in perfused rats of all four groups (two diets and two genders) precisely (Fig. 2). Mean percent ± SEM (n = 4) for corrected versus perfused was 99.3 ± 0.69.
Previous work had suggested that the brain Fe reduction in Cu-deficient rats was observed in cortex and brainstem but not cerebellum (Prohaska and Wells 1975). Most of the current analyses were on whole brain or brain minus the cerebellum. Thus, additional analyses were performed on cerebella of non-perfused P24–P27 male rats following the perinatal model described above. Compared with + CuM rats, − CuM rats were anemic [mean ± SEM (n = 4) Hb values (g/L) were 60.3 ± 1.7 compared with 103 ± 1.8, p < 0.01]. Cerebellar Cu values (μg/g) in − CuM rats were 0.44 ± 0.05, 81% lower than values in + CuM rats, 2.33 ± 0.05, p < 0.01. Cerebellar Fe (μg/g), corrected for blood Fe contamination, in − CuM rats was 6.94 ± 0.28 and 21% lower than values in + CuM rats, 8.83 ± 0.14, p < 0.01.
Effect of Cu deficiency on brain Fe accumulated during suckling and post-weaning
The impact of dietary Cu deficiency on accumulation of brain Cu at P3, P13 and P24 in male pups demonstrates a severe blockage in Cu accretion (Fig. 3). In contrast, brain Fe content was not different at P3, but was significantly lower in − CuM rats at P13 and P24 compared with the +CuM pups. The difference at P24 was greater than at P13 (Fig. 3).
Fig. 3.
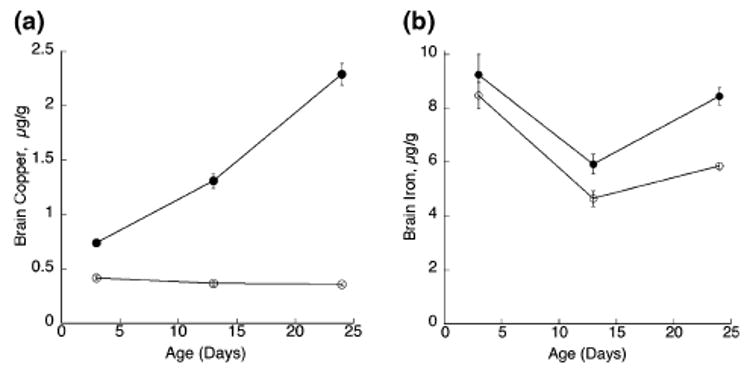
Concentration of brain copper and iron, based on fresh weight, in suckling rats during perinatal Cu deficiency. Each point is the mean ± SEM of four male rats from separate litters. Cu-deficient (open circles) values for both copper and iron were lower than values for Cu-adequate (solid circles) pups except for brain iron at P3, p < 0.05. Brain iron values were corrected for blood iron contamination.
A repeat experiment confirmed the Fe deficit in another set of P13 and P24 − CuM rats (Table 2). These − CuM pups were anemic and smaller than their + CuM controls (Table 2). Brain Cu content changes also reflected dietary treatment, with severe deficits at both P13 and P24. A pooled plasma sample from P24 rats indicated a large reduction in plasma Fe concentration.
Table 2.
Status of Cu-adequate and Cu-deficient male rats following copper deficiency
| Postnatal | Perinatal | Perinatal | ||||
|---|---|---|---|---|---|---|
| Characteristic | Cu+ | Cu− | Cu+ | Cu− | Cu+ | Cu− |
| Age, days | 50 | 50 | 24 | 24 | 13 | 13 |
| Body weight, g | 276 ± 17 | 280 ± 20 | 89 ± 1.1 | 45 ± 1.5* | 38.2 ± 1.9 | 31.6 ± 1.7* |
| Hemoglobin, g/L | 162 ± 1.7 | 112 ± 25 | 102 ± 3.0 | 53.8 ± 3.5* | 98.0 ± 2.5 | 67.6 ± 2.8* |
| Brain Cu, μg/g | 2.55 ± 0.32 | 1.61 ± 0.07* | 2.22 ± 0.07 | 0.29 ± 0.07* | 1.31 ± 0.11 | 0.35 ± 0.04* |
| Brain Fe, μg/g | 11.5 ± 0.32 | 11.3 ± 0.94 | 9.03 ± 0.30 | 5.50 ± 0.38* | 6.11 ± 0.28 | 4.10 ± 0.17* |
| Plasma Fe, μg/mL | 2.29 ± 0.07 | 1.17 ± 0.21* | 3.01 | 0.34 | ND | ND |
Values are means ± SEM. Postnatal rats (n = 3) were maintained on Cu-adequate (Cu+) or Cu-deficient (Cu−) treatment for 30 days after weaning. Perinatal rats (n = 4) were born to, and nursed by, Cu-deficient or Cu-adequate dams. Treatment began 2 weeks prior to parturition. Brain iron concentration was corrected for blood iron contamination. For suckling rats, plasma iron was a pooled estimate and only available for 24-day-old pups. For a given age, treatment means were tested by unpaired Student's t-test,
p < 0.05.
A sample of 7-week-old − CuM rats from the postnatal model was compared with + CuM controls (Table 2). The degree of Cu deficiency was much more modest in this postnatal model, as brain Cu was only reduced by 37%. Body weight, Hb and brain Fe (even after correction for blood Fe contamination) was not altered by Cu deficiency in this model (Table 2). There was a 49% reduction in plasma Fe concentration, likely reflecting the loss of ceruloplasmin ferroxidase activity (data not shown).
Evaluation of Fe status by transferrin receptor expression
Reductions in brain Fe at P13 and P24 were correlated with higher transferrin receptor (TfR1) expression (Fig. 4). The most striking result is the greater than twofold increase in TfR1 levels in P24 − CuM rat brain membrane preparations. There was a modest and significant 44% increase at P13 in − CuM rats. TfR1 levels in the brain of P50 rats were not impacted by dietary Cu deficiency, in agreement with the lack of impact on brain Fe levels (Table 2). The immunoblot from P24 rats was repeated for a marker enriched in endothelial cells, glucose transporter 1 (GLUT1). The ratio − CuM /+ CuM for this protein was l.01 compared with a value of 3.08 for TfR1, suggesting little difference in endothelial cell content in − CuM P24 brains based on GLUT1 expression.
Fig. 4.
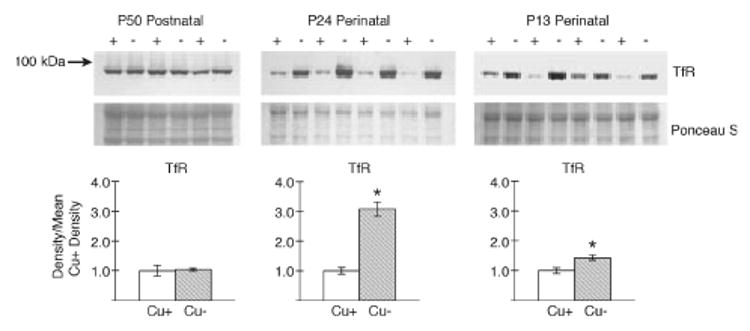
Rat brain expression of transferrin receptor 1 (TfR). SDS– PAGE 10% and western immunoblot analyses of brain membrane extracts from male Cu-adequate (+) (Cu+) and Cu-deficient (−) (Cu−) rats following postnatal (P50) or perinatal (P24 and P13) dietary Cu deficiency were conducted using mouse anti-human TfR antibody. A molecular weight (kDa) marker is shown on the left. Membranes were stained with Ponceau S to illustrate equal loading of protein. Lanes in the P50 membrane contained 15 μg protein, in the P24 membrane 12 μg protein, and in the P13 membrane 15 μg protein. Densities of the TfR bands were determined and compared with the mean values of Cu+ rats. Bars are means ± SEM. Mean Cu-/Cu+ TfR value for P50 was 1.04 and unchanged, whereas for P24 the ratio was 3.08 and for P13, 1.45, * p < 0.05.
Elevation of TfR1 in brain suggests an Fe-deficient state in the perinatal model of Cu deficiency. Plasma Fe was lower in P24 − CuM rats (Table 2) but sufficient plasma was not available at P3 or P13 to measure Fe. Evidence of a Fe-deficient state in the Cu-deficient pups was suggested by liver Fe data. At P3, the − CuF mean ± SEM (n = 8) for liver Fe was lower, at 166 ± 13.5 μg/g, compared with 208 ± 12.8 (n = 8) for +CuF, p < 0.05. At P13, − CuM (n = 4) values of 22.6 ± 0.65 were also lower than + CuM values (n = 4) of 27.6 ± 1.0, p < 0.01. In older Cu-deficient rats, liver Fe is higher than control values. For the P50 − CuM (n = 5) rats, liver Fe was 104 ± 10.5 compared with 70.2 ± 3.4 for the + CuM rats, p < 0.02.
Discussion
Brain iron
Current experiments demonstrate that the accumulation of iron in rat brain during perinatal growth is dependent on adequate copper nutrition of dams. Although this was suggested 30 years ago, results were questionable because those brains contained blood (Prohaska and Wells 1975). The current studies demonstrate that perfusion to remove blood lowers the apparent brain Fe content, but the observed reduction in Fe due to Cu deficiency remains. Since the blood Fe content of a control rat (approximately 300 μg/g) is about 30 times higher than brain Fe (approximately 10 μg/g), a 0.5% blood Fe contamination of the brain represents 15% of the apparent Fe content. Since Cu deficiency is associated with anemia (lower Hb and lower blood Fe), interpretation of brain Fe content requires careful analysis.
Hemoglobin is the major source of Fe in blood. Since the whole blood Fe content is approximately 100 times higher than plasma Fe content, differences in plasma Fe due to Cu deficiency would not influence the correlation between Hb and blood Fe appreciably. In fact, using the estimated Fe content of blood and the specific blood contamination of brain, which ranges between 0.4 and 1.0% for male and female rats, excellent agreement with values obtained by analysis of perfused rats was achieved. Thus, it will be possible to estimate brain Fe content in non-perfused animals if Hb is determined along with total brain Fe by AAS.
Restriction of dietary Cu beginning at the last trimester of gestation in rats resulted in a greatly diminished accumulation of brain Cu in pups as early as P3. This pattern of accumulation and restriction due to Cu deficiency agrees with previous work in rats (Prohaska and Wells 1974). The impact on brain Fe accumulation did not reveal a significant difference until P13. The pattern of Fe accumulation in developing rat brain appears to differ from Cu, with Fe levels at P3 exceeding those at P13 followed by an increase between P13 and P24. This pattern, however, agrees well with previous studies by others (Roskams and Connor 1994).
Transferrin receptor
The present data also demonstrate that the 20% reduction in brain Fe in −Cu rats was perceived biochemically as though Fe deficiency was evident. This is suggested by a robust 300% rise in TfR protein expression in weanling rat brain of Cu-deficient pups. Following dietary Fe deficiency, TfR expression in brain is known to increase (Han et al. 2003; Siddappa et al. 2003). Fe deficiency also up-regulates TfR in the brain microvasculature, both in animal studies and in cell culture models (van Gelder et al. 1998; Burdo et al. 2004). The up-regulation of TfR protein is likely mediated by lower tissue Fe and the response of iron regulatory proteins (IRP-1 and IRP-2) that interact with iron regulatory elements (IRE) on TfR mRNA (Siddappa et al. 2003).
The increase in TfR likely represents an increase both in the microvasculature and neurons. It is known that TfR is expressed in neurons and likely not on glial cells or, if so, not abundantly (Moos and Morgan 2004a). The large increase in TfR observed in the membrane immunoblots of − CuM rats could not be accounted for totally by endothelial cell expression, since there is only about a 25% enrichment of TfR expression in brain microvasculature compared with whole brain (Burdo et al. 2004). Even in Fe-deficient samples, the enrichment is only 50%. Thus, the TfR enhancement in − CuM rat brain is most remarkable. Furthermore, if there were increased numbers of capillary endothelial cells due to Cu deficiency, an increase in GLUT1 expression would have been predicted, since this transporter is enriched in capillary endothelial cells (McCall et al. 1996). We did not observe any changes in GLUT1 expression due to Cu deficiency. Increased TfR expression in brains of Cu-deficient rats suggests that the response to ‘Fe deficiency’ was apropos.
Mechanisms for low brain iron during development
A reasonable explanation for lower brain Fe content following dietary Cu deficiency is that supply of Fe in plasma is low. Current data show that plasma Fe is reduced in the − CuM rats that also have lower brain Fe. Fe is taken up by the capillary endothelial cells bound to transferrin via TfR (Moos and Morgan 2004a). In addition to transferrin and TfR, Fe uptake depends on divalent metal transporter (DMT1). Neurons express DMT1, which is needed for endosomal release of Fe taken up by TfR transferrin (Moos and Morgan 2004a). DMT1 may be needed by glia to take up non-transferrin-bound Fe that is released from endothelial cells (Moos and Morgan 2004b). The impact of Cu deficiency on brain DMT1, transferrin and other mediators of uptake needs to be evaluated to fully understand the mechanism for low brain Fe in Cu deficiency.
One Cu protein known to have an impact on brain Fe is ceruloplasmin (Cp), also know as ferroxidase. Humans missing Cp accumulate high levels of Fe in the liver and brain (Harris et al. 1995). Presumably, this is related to the ferroxidase activity of Cp, which depends on Cu. The accumulation of brain Fe in mice lacking Cp does not occur in young animals (Harris et al. 1999; Patel et al. 2002). Rat pups in the present studies have a nearly total loss of plasma Cp activity but lower, rather than higher, brain Fe.
Pups in the present studies were anemic, but this anemia was not due to lack of Cp activity and impaired Fe mobilization, since Fe content of livers was lower in Cu-deficient pups. More likely, the dietary Cu deficiency of dams resulted in impaired Fe transport across the placenta (Danzeisen et al. 2002). Also, milk Fe content is lower in Cu-deficient rats and mice, suggesting impaired Fe transport in mammary tissue as well (Cohen et al. 1985; Prohaska 1989; Gambling et al. 2004). In dietary Cu deficiency, higher liver Fe levels are observed in dams. The reduction in Fe transport from dams to pups is likely mediated by reduction in Cp (ferroxidase) homologs that depend on Cu for Fe oxidation and efflux. Whatever the mechanism, adequate dietary Cu is necessary for dams to transfer Fe to pups so that the brain can accumulate Fe during perinatal development.
Potential consequences
If Fe is limited during gestation and early lactation, repletion of brain Fe pools is not possible (Dallman et al. 1975). Our recent work has shown that brain Fe levels of Cu repleted − CuM rats remain below control values even after 6 months of Cu repletion (Penland and Prohaska 2004). Thus, the consequences of perinatal Cu deficiency may have an impact on CNS function of both Cu- and Fe-mediated processes. As outlined earlier, many alterations of brain biochemistry following Cu deficiency (lower cytochrome oxidase activity, altered monoamine metabolism and hypomyelination) are also observed following perinatal Fe deficiency (O'Dell and Prohaska 1983; Beard and Connor 2003). Even persistent behavioral changes are noted in both Cu- and Fe-deficient pups following dietary recovery (Prohaska and Hoffman 1996; Beard 2003; Penland and Prohaska 2004).
Dietary Cu deficiency in human infants is not well recognized compared with Fe deficiency. However, secondary factors such as excess zinc, premature delivery or genetic susceptibility may influence Cu status. The levels of Cu intake recommended for adults in the USA and Canada, 900 μg per day, are equivalent to a diet containing approximately 1.8 mg Cu/kg (Prohaska and Brokate 2002). This level of dietary Cu in rat dams is insufficient for proper hippocampal development of pups (Hunt and Idso 1995). A level of 2.6 mg/kg fed to rats resulted in pups with lower brain Cu (Hopkins and Failla 1995). The impact of marginal Cu deficiency on brain Fe accumulation needs to be evaluated. In the postnatal model of the present studies, there was no change in brain Fe or TfR expression despite a 50% reduction in plasma Fe. Thus, it is possible that the reduction in brain Fe following perinatal Cu deficiency only occurs when Fe is limiting during early postnatal life. Further research will be necessary to determine the biochemical cause of low brain Fe and its consequences, if any, following dietary Cu deficiency.
Acknowledgments
Support for this research was provided by NIH grant HD 39708. The skilful technical assistance of Margaret Broderius and Joshua Pyatskowit is appreciated.
Abbreviations used
- AAS
atomic absorption spectroscopy
- Cp
ceruloplasmin
- Cu
copper
- +CuF
copper-adequate female
- − CuF
copper-deficient female
- +CuM
copper-adequate male
- − CuM
copper-deficient male
- DMT1
divalent metal transporter
- Fe
iron
- GLUT1
glucose transporter one
- Hb
hemoglobin
- P
postnatal day
- TfR1
transferrin receptor
- Zn
zinc
Footnotes
Publisher's Disclaimer: Copyright of Journal of Neurochemistry is the property of Blackwell Publishing Limited and its content may not be copied or emailed to multiple sites or posted to a listserv without the copyright holder's express written permission. However, users may print, download, or email articles for individual use.
References
- Beard J. Iron deficiency alters brain development and functioning. J Nutr. 2003;133:1468S–1472S. doi: 10.1093/jn/133.5.1468S. [DOI] [PubMed] [Google Scholar]
- Beard JL, Connor JR. Iron status and neural functioning. Annu Rev Nutr. 2003;23:41–58. doi: 10.1146/annurev.nutr.23.020102.075739. [DOI] [PubMed] [Google Scholar]
- Beard JL, Wiesinger JA, Connor JR. Pre- and post-weaning iron deficiency alters myelination in Sprague-Dawley rats. Dev Neurosci. 2003;25:308–315. doi: 10.1159/000073507. [DOI] [PubMed] [Google Scholar]
- Bennetts HW, Chapman FE. Copper deficiency in sheep in Western Australia: a preliminary account of the etiology of enzootic ataxia of lambs and an anemia of ewes. Aust Vet J. 1937;13:138–149. [Google Scholar]
- Burdo JR, Simpson IA, Menzies S, Beard J, Connor JR. Regulation of the profile of iron-management proteins in brain microvasculature. J Cereb Blood Flow Metab. 2004;24:67–74. doi: 10.1097/01.WCB.0000095800.98378.03. [DOI] [PubMed] [Google Scholar]
- Carlton WW, Kelly WA. Neural lesions in the offspring of female rats fed a copper-deficient diet. J Nutr. 1969;97:42–52. doi: 10.1093/jn/97.1.42. [DOI] [PubMed] [Google Scholar]
- Cohen NL, Keen CL, Hurley LS, Lonnerdal B. Determinants of copper-deficiency anemia in rats. J Nutr. 1985;115:710–725. doi: 10.1093/jn/115.6.710. [DOI] [PubMed] [Google Scholar]
- Dallman PR, Siimes MA, Manies EC. Brain iron: persistent deficiency following short-term iron deprivation in the young rat. Br J Haematol. 1975;31:209–215. doi: 10.1111/j.1365-2141.1975.tb00851.x. [DOI] [PubMed] [Google Scholar]
- Danzeisen R, Fosset C, Chariana Z, Page K, David S, McArdle HJ. Placental ceruloplasmin homolog is regulated by iron and copper and is implicated in iron metabolism. Am J Physiol. 2002;282:C472–478. doi: 10.1152/ajpcell.00019.2001. [DOI] [PubMed] [Google Scholar]
- de Deungria M, Rao R, Wobken JD, Luciana M, Nelson CA, Georgieff MK. Perinatal iron deficiency decreases cytochrome c oxidase (CytOx) activity in selected regions of neonatal rat brain. Pediatr Res. 2000;48:169–176. doi: 10.1203/00006450-200008000-00009. [DOI] [PubMed] [Google Scholar]
- Everson GJ, Tsai HC, Wang TI. Copper deficiency in the guinea pig. J Nutr. 1967;93:533–540. doi: 10.1093/jn/93.4.533. [DOI] [PubMed] [Google Scholar]
- Fox PL. The copper-iron chronicles: the story of an intimate relationship. Biometals. 2003;16:9–40. doi: 10.1023/a:1020799512190. [DOI] [PubMed] [Google Scholar]
- Gallagher CH, Judah JD, Rees KR. The biochemistry of copper deficiency. I. Enzymological disturbances, blood chemistry and excretion of amino acids. Proc R Soc Lond. 1956;145B:134–150. doi: 10.1098/rspb.1956.0022. [DOI] [PubMed] [Google Scholar]
- Gambling L, Dunford S, McArdle HJ. Iron deficiency in the pregnant rat has differential effects on maternal and fetal copper levels. J Nutr Biochem. 2004;15:366–372. doi: 10.1016/j.jnutbio.2003.12.009. [DOI] [PubMed] [Google Scholar]
- van Gelder W, Huijskes-Heins MI, Cleton-Soeteman MI, van Dijk JP, van Eijk HG. Iron uptake in blood–brain barrier endothelial cells cultured in iron-depleted and iron-enriched media. J Neurochem. 1998;71:1134–1140. doi: 10.1046/j.1471-4159.1998.71031134.x. [DOI] [PubMed] [Google Scholar]
- Han J, Day JR, Connor JR, Beard JL. Gene expression of transferrin and transferrin receptor in brains of control vs. iron-deficient rats. Nutr Neurosci. 2003;6:1–10. [PubMed] [Google Scholar]
- Harris ZL, Takahashi Y, Miyajima H, Serizawa M, MacGillivray RT, Gitlin JD. Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci USA. 1995;92:2539–2543. doi: 10.1073/pnas.92.7.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci USA. 1999;96:10–812. 10–817. doi: 10.1073/pnas.96.19.10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins RG, Failla ML. Chronic intake of a marginally low copper diet impairs in vitro activities of lymphocytes and neutrophils from male rats despite minimal impact on conventional indicators of copper status. J Nutr. 1995;125:2658–2668. doi: 10.1093/jn/125.10.2658. [DOI] [PubMed] [Google Scholar]
- Hunt CD, Idso JP. Moderate copper deprivation during gestation and lactation affects dentate gyrus and hippocampal maturation in immature male rats. J Nutr. 1995;125:2700–2710. doi: 10.1093/jn/125.10.2700. [DOI] [PubMed] [Google Scholar]
- McCall AL, Van Bueren AM, Nipper V, Moholt-Siebert M, Downes H, Lessov N. Forebrain ischemia increases GLUT1 protein in brain microvessels and parenchyma. J Cereb Blood Flow Metab. 1996;16:69–76. doi: 10.1097/00004647-199601000-00008. [DOI] [PubMed] [Google Scholar]
- Moos T, Morgan EH. The metabolism of neuronal iron and its pathogenic role in neurological disease: review. Ann N Y Acad Sci. 2004a;1012:14–26. doi: 10.1196/annals.1306.002. [DOI] [PubMed] [Google Scholar]
- Moos T, Morgan EH. The significance of the mutated divalent metal transporter (DMT1) on iron transport into the Belgrade rat brain. J Neurochem. 2004b;88:233–245. doi: 10.1046/j.1471-4159.2003.02142.x. [DOI] [PubMed] [Google Scholar]
- O'Dell BL, Prohaska JR. Biochemical aspects of copper deficiency in the nervous system. In: Dreosti IE, Smith RM, editors. Neurobiology of the Trace Elements. Humana Press; Clifton NJ: 1983. pp. 41–81. [Google Scholar]
- Patel BN, Dunn RJ, Jeong SY, Zhu Q, Julien JP, David S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J Neurosci. 2002;22:6578–6586. doi: 10.1523/JNEUROSCI.22-15-06578.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penland JG, Prohaska JR. Abnormal motor function persists following recovery from perinatal copper deficiency in rats. J Nutr. 2004;134:1984–1988. doi: 10.1093/jn/134.8.1984. [DOI] [PubMed] [Google Scholar]
- Prohaska JR. Effect of diet on milk copper and iron content of normal and heterozygous brindled mice. Nutr Res. 1989;9:353–356. [Google Scholar]
- Prohaska JR, Brokate B. Dietary copper deficiency alters protein levels of rat dopamine-β-monooxygenase and tyrosine monooxygenase. Exp Biol Med. 2001;226:199–207. doi: 10.1177/153537020122600307. [DOI] [PubMed] [Google Scholar]
- Prohaska JR, Brokate B. The timing of perinatal copper deficiency in mice influences offspring survival. J Nutr. 2002;132:3142–3145. doi: 10.1093/jn/131.10.3142. [DOI] [PubMed] [Google Scholar]
- Prohaska JR, Hoffman RG. Auditory startle response is diminished in rats after recovery from perinatal copper deficiency. J Nutr. 1996;126:618–627. doi: 10.1093/jn/126.3.618. [DOI] [PubMed] [Google Scholar]
- Prohaska JR, Smith TL. Effect of dietary or genetic copper deficiency on brain catecholamines, trace metals and enzymes in mice and rats. J Nutr. 1982;112:1706–1717. doi: 10.1093/jn/112.9.1706. [DOI] [PubMed] [Google Scholar]
- Prohaska JR, Wells WW. Copper deficiency in the developing rat brain: a possible model for Menkes' steely-hair disease. J Neurochem. 1974;23:91–98. doi: 10.1111/j.1471-4159.1974.tb06920.x. [DOI] [PubMed] [Google Scholar]
- Prohaska JR, Wells WW. Copper deficiency in the developing rat brain: evidence for abnormal mitochondria. J Neurochem. 1975;25:221–228. doi: 10.1111/j.1471-4159.1975.tb06956.x. [DOI] [PubMed] [Google Scholar]
- Prohaska JR, Bailey WR, Lear PM. Copper deficiency alters rat peptidylglycine α-amidating monooxygenase activity. J Nutr. 1995;125:1447–1454. doi: 10.1093/jn/125.6.1447. [DOI] [PubMed] [Google Scholar]
- Roskams AJ, Connor JR. Iron, transferrin, and ferritin in the rat brain during development and aging. J Neurochem. 1994;63:709–716. doi: 10.1046/j.1471-4159.1994.63020709.x. [DOI] [PubMed] [Google Scholar]
- Siddappa AJ, Rao RB, Wobken JD, Casperson K, Leibold EA, Connor JR, Georgieff MK. Iron deficiency alters iron regulatory protein and iron transport protein expression in the perinatal rat brain. Pediatr Res. 2003;53:800–807. doi: 10.1203/01.PDR.0000058922.67035.D5. [DOI] [PubMed] [Google Scholar]
- Wauben IP, Wainwright PE. The influence of neonatal nutrition on behavioral development: a critical appraisal. Nutr Rev. 1999;57:35–44. doi: 10.1111/j.1753-4887.1999.tb01776.x. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Green AR. Biogenic monoamine metabolism and functional activity in iron-deficient rats: behavioural correlates. Ciba Found Symp. 1976;51:201–225. doi: 10.1002/9780470720325.ch10. [DOI] [PubMed] [Google Scholar]