

Abstract
Purpose
To determine changes in host gene expression in HSV-1 latent trigeminal ganglia (TG) after hyperthermic stress.
Methods
Scarified corneas of 6-week-old female BALB/c mice were inoculated with either HSV-1 17Syn+ (high phenotypic reactivator) or 17ΔPst(LAT−) (low phenotypic reactivator) at 104 plaque-forming units/eye. At 28 days after infection, viral reactivation was induced in some of the infected mice with hyperthermic stress, and the mice were killed after 1 hour. Heat-treated uninfected mice served as the control. Labeled cRNA derived from TG-isolated total RNA was hybridized to 430 2.0 chips containing 14,000 mouse genes. Gene expression was confirmed by quantitative real-time PCR.
Results
There was no difference in gene expression in the non–heat-treated mice. Gene expression in the TG of each of the heat-treated mouse groups (17Syn+, 17ΔPst(LAT−) and uninfected) yielded upregulation of more than twofold of a group of the same genes, designated as heat stress–induced gene expression. Twenty-nine genes (0.2%) were significantly upregulated (2- to 17-fold) when the heat stress–induced gene expression was subtracted from the gene expression of 17Syn+ latent TG relative to 17ΔPst(LAT−) latent TG 1 hour after mouse hyperthermic stress. Nine host adaptive immunity genes comprising Ig molecules, CD83, CD8A, ADA, and CCL8 were the largest subset upregulated, and all were confirmed by real-time PCR. Others identified included genes involved in hypothalamic-pituitary gland functions.
Conclusions
Hyperthermic stress–induced reactivation of the HSV-1 high phenotypic reactivator can upregulate gene expression involved in B-cell function and in T-cell function. CD83 is implicated in HSV-1 latency, suggesting it could also be involved in immune-mediated mechanisms of viral reactivation.
The HSV genome forms an episome in neuronal nuclei from which virtually no detectable viral transcription occurs during latency, except that of the abundantly transcribed latency-associated transcript (LAT).1–3 The major products of LAT are 2- and 1.5-kb RNAs, which are localized to the neuronal nucleus. LAT overlaps the infected cell protein 0 (ICP0), an immediate-early regulatory gene, and is transcribed off the opposite DNA strand. One proposed function of the LAT is the suppression of nearby lytic phase transcripts ICP0, g34.5 virulence gene, and infected cell protein 4 through unknown mechanisms, thereby promoting the establishment and maintenance of latency.3,4 ICP0 is important for productive HSV-1 replication5 and is thought to have a role in reactivation from latency.6 The observations described here support the hypothesis that LAT could act to suppress immediate-early genes. Apart from putative LAT-mediated suppression of lytic transcripts during latency, it is possible that host molecular/immune mechanisms, which contribute to the establishment of latency,7 play a functional role in association with the LAT-ICP0 locus of HSV in suppressing/regulating viral reactivation.
Reactivation of the latent HSV-1 genome has been linked to a reactivation-critical region (rcr) that includes the LAT core promoter through the LAT 5′ exon/enhancer, since recombinants lacking this region display greatly reduced reactivation phenotypes.3,8–11 Reactivation from latent infected sensory ganglia neurons can lead to peripheral shedding of infectious virus which could manifest in the eye as debilitating recurrent ulcerative and/or stromal keratitis through corneal scarring, thinning, and neovascularization.12
Humans are reservoirs of HSV-1, and asymptomatic shedding is a major factor in the spread of the virus.13–15 The majority of the adult population harbor latent HSV-1 in their neural tissue and are seropositive,16,17 although the percentage of the total population that develops herpetic lesions is only approximately 25% to 35%. Recent human data have documented a significantly high frequency of shedding of HSV-1 DNA in tears, saliva, and genital secretions.13,15,18–20 Some strains of HSV-1 exhibit high reactivation rates in animal models.21 Also, reactivation of HSV-1 can result in recurrent herpes labialis lesions (RHLs) and in oral shedding of virus. Most patients with RHLs shed viral DNA both before and after the appearance of clinical lesions.22 These variations in shedding and disease could be caused in part by the genetics and phenotype of the virus and genetic makeup of the host.23
A question that has not been fully addressed concerns the specific host mechanisms that influence the LAT-ICP0 locus of HSV during reactivation. To unravel the associated potential molecular events during reactivation of high phenotypic reactivation strains, we used gene expression to evaluate potential differences in host responses by comparing phenotypic differences between the LAT-positive 17Syn+, a high phenotypic reactivator,24–26 and an LAT promoter deletion virus, 17ΔPst-(LAT−). The 17ΔPst(LAT−) has been demonstrated to be a low phenotypic reactivation strain in both mice and rabbits,24,25,27 although the copy numbers of viral DNA recovered were equivalent to those of its parent 17Syn+ in the rabbit trigeminal ganglia (TG).24
Gene expression in vivo in latency studies suggests a clear pattern of genes belonging to specific functional groups indicating active biological activity during latency. We recently analyzed gene expression in the rabbit TG harboring latent HSV-1 strain McKrae using new and novel custom oligonucleotide rabbit arrays (Agilent Technologies, Palo Alto, CA) and showed significant alterations of genes in host defense and immune response, protein and carbohydrate metabolism, cell adhesion, and apoptosis.28
The HSV genome remains quiescent with no detectable viral lytic genes, thereby establishing a state of latency. During this latency phase, the viral genome persists and is stable for long periods unless interrupted by stimuli to reactivate. Interruption of latency by stimuli such as stress occurs by the induction of the lytic cycle with the subsequent appearance of clinically apparent lesions or asymptomatic shedding of virus at or near the primary site of infection. Previous studies have shown that HSV-1 reactivation leading to shedding of virus does not occur within the majority of neurons; rather, it is an isolated event triggered in only a few neurons within the sensory ganglia.29–31 Hyperthermic stress30,32,33 and immunosuppression34–37 are two stimuli that can be used to interrupt latency to induce HSV reactivation.
In the present study, we sought to determine whether there are differences in the host gene responses of a wild-type HSV-1, strain 17Syn+, in relation to the LAT-promoter deletion, 17ΔPst(LAT−),24 after heat stress-induced reactivation. The conditions were similar to the hyperthermic stress-induced in vivo model used by Higaki et al.33 (inbred strain of mouse and the same type of heat-stress stimulus), except the mice were killed at the earlier time point of 1 hour. Molecular events known to take place during HSV-1 reactivation place into perspective the comparison of gene expression at 1 hour after hyperthermic stress. Sawtell and Thompson38 have summarized the processes involved in reactivation by analyzing neurons for expression and localization of geminin, cyclin-dependent kinase (cdk)2 and -4, and cytochrome c and consequent changes in morphology, virus production, and DNA fragmentation in the mouse model. Latently infected TG excised and cultured in vitro (explanted) to reactivate were compared to reactivation occurring in latently infected TG in vivo after hyperthermic stress. Selection of these proteins was based on the fact that their expression in neurons reflects a major shift in the physiological state of the neuron. From 2 hours to as long as 72 hours after hyperthermic stress, no changes were revealed in any of the attributes evaluated; there were no detectable alterations in geminin involved in blocking the initiation of inappropriate DNA replication in dividing cells and cell cycle-regulatory cdk2 and -4 and the cytoplasmic release of cytochrome c, an intermediate in apoptosis.38 TUNEL-positive neurons were also not detected in vivo after hyperthermic stress at any point.38 Explanted tissue exhibited significant alterations in all the attributes evaluated with the exception of virus production.38 These results were largely consistent with an earlier report.39 Also, numerous in vitro studies have shown functional ICP0 to be essential for efficient HSV reactivation.5 In vivo mechanisms were deduced as subtle and appear to take place very early during the reactivation or viral induction phase. Therefore, important molecular events could be missed during viral induction. Second, degradation and decreased expression of important genes could not be distinguished from each other during molecular events associated with viral induction. Thus, we pursued genes patterns of expression that were highest or peaked at 1 hour. The differences in host gene activation between 17Syn+ and 17ΔPst(LAT−) will provide crucial information about the contribution of the LAT and the viral phenotype in regulating host gene expression during the early phase of reactivation.
Materials and Methods
Mice, Viruses, and Infection
All animal procedures adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the National Institutes of Health Principles of Laboratory Animal Care, using a protocol approved by the LSU Health Sciences Center Institutional Animal Care and Use Committee. Identical groups of 6-week-old female BALB/c mice were anesthetized with ketamine hydrochloride (1 mg/kg) and xylazine (0.5 mg/kg; Vedco Inc., St. Joseph, MO). Scarified corneas were inoculated with a suspension of either HSV-1 17Syn+ or 17ΔPst-(LAT−)—104 plaque-forming units (pfu)/eye in 5 μL. Slit lamp examinations and ocular swab collections were performed 2 and 3 days postinfection (pi) to verify 100% corneal infection. At 28 days pi, ocular swabs were cocultured with primary rabbit kidney cells and tested negative for infectious virus.
Five groups of mice were studied: (1) 17Syn+ latently infected heat-treated, (2) 17ΔPst(LAT−) latently infected heat-treated, (3) 17Syn+ latently infected non–heat-treated, (4) 17ΔPst(LAT−) latently infected non-heat-treated, and (5) uninfected heat-treated. Hyperthermic stress consisted of placing the mice in a constant-temperature H2O bath (43°C) for 10 minutes.30,40 The mice were killed 1 hour later. TG were aseptically removed and stored in RNA/later at −80°C until used. Forty-five mice (90 TG) with 9 mice (18 TG) per group were used.
Total RNA Extraction
Total RNA was extracted from the TG (RNeasy kit; Qiagen, German-town, MD). RNA was treated with DNase (Ambion, Austin, TX), according to the manufacturer’s protocol. RNA was purified and concentrated (Cleanup kit; Qiagen) and assayed spectrophotometrically. No significant differences were detected in spectral purity, rate of degradation, or yield of RNA from the TG among the groups. Microfiltration (2100 Bioanalyzer; Agilent Technologies, Santa Clara, CA) indicated that the quality of RNA samples had 18s/28s >1 and RNA Integrity Number values >7.
DNA Microarray Experiments
Purified RNA samples (6 μg) were reverse transcribed by T7-(dT)24 oligomer, reverse transcriptase (Superscript II; Invitrogen, Carlsbad, CA), and DNA Polymerase I (Invitrogen) for first- and second-strand cDNA with a cDNA synthesis kit (Superscript; Invitrogen) and purified (Genechip sample cleanup module; Affymetrix Inc., Santa Clara, CA). The cDNA was used for the synthesis of biotin-labeled antisense cRNA (target) in an in vitro transcription (IVT) reaction (Bioarray HighYield RNA Transcript labeling kit; Enzo, Farmingdale, NY). The IVT-cRNA was repurified and then fragmented with fragmentation buffer (Affymetrix Inc.) containing 20 μg in 40 μL. Denatured cRNA was pelleted to obtain a condensation fragment and was dissolved in hybridization buffer to a final concentration of 1.1 μg/μL. The cRNA cocktail was incubated at 99°C and 45°C for a total of 10 minutes and hybridized for 16 hours to mouse expression array 430 2.0 chips (Affymetrix, Inc.), representing 14,000 genes. Duplicate chips were used for each of the individual samples. The array chips were washed, stabilized, and stained according to the manufacturer’s recommendations. Each chip was scanned in the phycoerythrin filter with a confocal laser scanner. Data analysis was performed (Microarray suite 5.0; Affymetrix, Inc.) to generate an absolute analysis for each chip.
The relative abundance of individual genes is based on the signal intensities of the corresponding probe sets that were analyzed (ArrayAssist 4.0; Stratagene, La Jolla, CA). Data from the experiments were individually normalized by using robust multiarray analysis with correction for GC content (gcRMA).41 The quality of all microarray experiments was assessed by the housekeeping gene probe, mouse β-actin, to measure the consistency of the hybridization signals from its 3′, middle, and 5′ fragment of the mRNA coding regions.42 A Student’s t-test was performed to find genes with a P < 0.05. These genes were then filtered to find genes with a twofold change between the composite data from 17Syn+ latent infected TG and 17ΔPst(LAT−) latent TG 1 hour after mouse hyperthermic stress. Probe ontology of the genes complemented and annotations on the microarray are accessible through the NetAffx Analysis Center (Affymetrix, Inc.). Genes were clustered according to information available at the National Center for Biotechnology Information (NCBI), National Library of Medicine, National Institutes of Health (Bethesda, MD).
Quantitative Real-Time Polymerase Chain Reaction
Reverse-transcribed total RNA for 10 genes was prepared from the TG of the heat-treated groups of 17Syn+ and 17ΔPst(LAT−) latent infected mice. The nine adaptive immunity genes were analyzed to confirm the relative quantitative expression levels. One other gene was also analyzed by this method as a random appraisal of the expression levels pattern in the remaining 20 genes. Primer pairs used were specifically designed and synthesized by Integrated DNA Technologies (IDT; San Diego, CA). These were immunoglobulin κ chain variable 32 (IgK-V32), forward primer, 5′-TGT CTG CAT CCC TTG GAG ACA CAA-3′, reverse primer, 5′-ACT AAA CCT TGA TGG GAC GCC TGT-3′; immunoglobulin λ chain, variable 1 (IgL-V1), forward primer, 5′-TGT TCG GTG GAG GAA CCA AAC TGA-3′, reverse primer, 5′-ACA CCA GTG TGG CCT TGT TAG TCT-3′, IgL-V1 homologue, forward primer, 5′-ACA GTC ACA CTC ACT TGT CGC TCA-3′, reverse primer, 5′-TCT CCA ATC AGG GAG CCT GAG AAT-3′; immunoglobulin heavy chain 4 (IgH-4 or serum IgG1), forward primer, 5′-AGC CAG CGG AGA ACT ACA AGA ACA-3′, reverse primer, 5′-TGC TCT TCT GCA CAT TGA GCT TGC-3′; CD83 antigen (CD83), forward primer, 5′-ATG AGC TCC ATC CTC AGA TGG CAA-3′, reverse primer, 5′-AAA GCA CTC ACG AGG TTG ACC AGA-3′; CD8 antigen alpha chain (CD8A), forward primer, 5′-TCC TGG GAT CAC CAG CAT GCT TTA-3′, reverse primer, 5′-TTT GAC AGT CAG CGT CTT CCT CCA-3′; chemokine (C-C motif) ligand 8 (CCL8), forward primer, 5′-GTG CTT CTT TGC CTG CTG CTC ATA-3′, reverse primer, 5′-AGA CAT ACC CTG CTT GGT CTG GAA-3′; adenosine deaminase (ADA), forward primer, 5′-GGG AGA GCA AGC ATT TGG CAT CAA-3′, reverse primer, 5′-AGC CAC CAC GGT CTT CTG ATT GTA-3′; RIKEN 2010309G21 gene immunoglobulin lambda chain complex (2010309G21RIKIgL), forward primer, 5′-ATG TAC TTT GCG ACG ACC TGG GAT- 3′, reverse primer, 5′-ACC AGT GCG ATA CAC CTA GAC GTT-3′; and otospiralin (OTOS) forward primer, 5′-AAG AGT AGC CGG AAC ATT GGC AGT-3′, reverse primer, 5′-TCA GTT CAC TGA GGA CCA CCT TGT-3′. The real-time PCR for β-actin was performed concurrently: forward primer, 5′-GTG GGC CGC TCT AGG CAC CAA-3′, reverse primer, 5′-CTC TTT GAT GTC ACG CAC GAT TTC-3′. Reverse transcription was primed with T7-Oligo (dT) primer (Affymetrix, Inc.). Real-time PCR reactions were performed in a 20-μL volume containing a solution of 1× supermix (iQ SYBR Green; Bio-Rad, Hercules, CA), 0.5 μM forward primer, 0.5 μM reverse primer, and 1 μL cDNA or 20 ng reverse-transcribed total RNA. A four-step protocol was used: denaturation, 3 minutes at 95°C; amplification and quantification, 40 cycles for 15 seconds at 95°C and for 30 seconds at 60°C; melting curve, 60 to 95°C with a heating rate of 0.5°C per second; followed by cooling (MyiQ Single Color Real-Time PCR Detection System; Bio-Rad). A single peak melting curve was observed for each gene product. Relative quantitative expression levels were determined for each gene. All results are displayed as an expression ratio of the 17Syn+ latent TG to the 17ΔPst(LAT−) latent TG after 1 hour of mouse heat treatment, normalized against β-actin expression levels using the 2−ΔΔCT method.43
Results
Heat-Stress–Induced Gene Expression
There was no difference in gene expression in the 17Syn+ latent TG relative to 17ΔPst(LAT−) latent TG of non–heat-treated mice (data not shown). The signal intensity values data between the 17Syn+ latent TG relative to the 17ΔPst(LAT−) latent TG 1 hour after mouse hyperthermic stress were normalized (Microarray Suite 5; Affymetrix, Inc.) resulting in 29 upregulated genes (2-fold change; P < 0.05). Since gcRMA normalization is currently recommended as the method of choice by some investigators,44 it was subsequently applied to normalize the data of all experiments. There was no change in the number of genes significantly upregulated based on this method. Gene expression in 17Syn+ latent TG, 17ΔPst(LAT−) latent TG and uninfected TG 1 hour after mouse hyperthermic stress exhibited similar levels of increase above twofold of a group of genes designated as heat-stress–induced gene expression: heat shock chaperone DNAJB1(HSP40) homologue subfamily B member 1, DNAJB1(HSP40); 60-kDa heat shock chaperone (HSP60); stress-activated c-Jun kinase 3; oxidative stress-induced protein; cyclooxygenase 2 (COX-2); and manganese superoxide dismutase precursor 2 (SOD-2) (data not shown). This heat-stress–induced gene expression in our study provides further evidence that such genes are expressed in response to hyperthermic stress.33,40
Subtractive Gene Expression
Subtractive gene expression showed that CD83 and other immune response genes were upregulated during viral induction to reactivate. Twenty-nine of 14,000 (0.2%) genes broadly categorized into functional activities (Fig. 1) were identified as significantly expressed after subtraction of the heat-stress–induced gene expression from gene expression of the 17Syn+ latent TG relative to the LAT negative recombinant, 17ΔPst latent TG 1 hour after mouse hyperthermic stress (Fig. 2; Supplementary Table S1, online at http://www.iovs.org/cgi/content/full/50/6/2855/DC1). This process of using heat stress for evaluating gene expression specifically linked to viral induction is designated subtractive gene expression. These 29 genes exhibited a 2- to 17-fold increased expression in the 17Syn+ latent TG. DNAJB1 (HSP40) was included in the original 29 genes because the expression of this gene in the latent infected TG of heat-treated mice was threefold higher than the uninfected TG of heat-treated mice.
Figure 1.
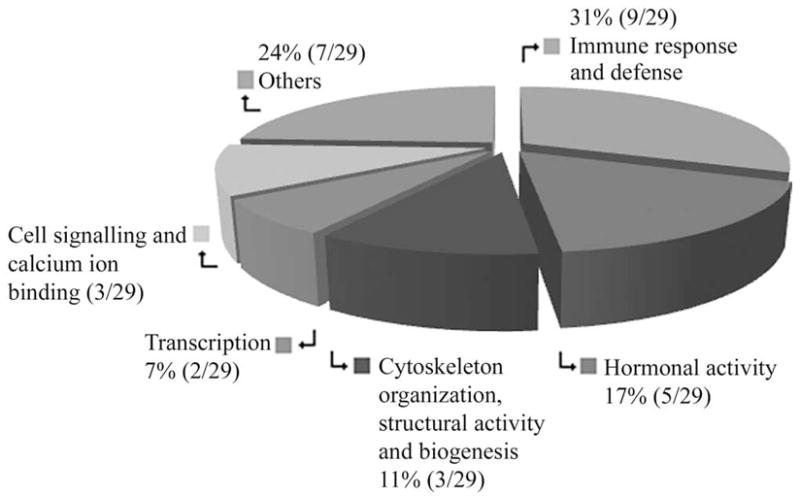
Broad categories of expressed gene functional activities in the 17Syn+ high phenotypic reactivator latent TG relative to a LAT− recombinant, 17ΔPst low phenotypic reactivator latent TG 1 hour after hyperthermic stress in the mouse.
Figure 2.

Normalized intensities of the 29 genes that were expressed at more than twofold in the 17Syn+ high phenotypic reactivator latent TG relative to an LAT− recombinant, 17ΔPst low phenotypic reactivator latent TG 1 hour after mouse hyperthermic stress.
The 29 upregulated genes (Fig. 2; Supplementary Table S1) were host immune response and defense (31% or 9/29), hormones of the hypothalamuspituitary glands and a natural signaling neuropeptide (17% or 5/29), transcription (7% or 2/29), cell signaling and calcium ion binding (10% or 3/29), cytoskeleton organization, structural activity, and biogenesis (11% or 3/29) and others comprising proteins, heat shock chaperone, ATP binding, mitochondrial transport, and coagulation (24% or 7/29; Fig. 1).
Association of CD83 Upregulation in Viral Induction with the Presence of the LAT-ICP0 Locus of HSV
The largest subgroup that was upregulated by three- to eightfold consisted of nine adaptive immunity genes: IgK-V32; IgL-V1; IgL-V1 homologue; IgH-4 (serum IgG1); 2010309G21RIKIgL; CD8A; CD83; ADA; and CCL8 (Table 1; Fig. 3; Supplementary Table S1). ADA, uncoupling protein 1 (UCP1), and activating transcription factor 3 (ATF3) constituted genes in three pathways that were significantly upregulated (Supplementary Table S1). Hormone genes with a three- to fourfold upregulation of expression were four of the hypothalamic-pituitary genes: glycoprotein hormones alpha subunit (CGA), corticotrophin-releasing hormone-binding protein (CRHBP), growth hormone (GH), luteinizing hormone β (LHB) and one neuropeptide, galanin (GAL) (Table 2; Supplementary Table S1). In addition, the expression of other genes was significantly upregulated. Transcription: apart from ATF3, FBJ osteosarcoma oncogene B (FOSB); cell signaling and calcium ion binding: δ-like 1 homologue (DLK1), pleckstrin (PLEK), and OTOS; cytoskeleton reorganization: keratin complex 2 basic gene 4 (KRT2-4) and its homologue and loricrin (LOR), DNAJB1 (HSP40) and its homologue; and ATP binding: translocated promoter region (TPR) and coagulation factor C homologue (COCH) (Supplementary Table S1).
Table 1.
Changes in Host Immune Response and Defense Genes Upregulated in the HSV-1 17Syn+ Latent TG Relative to 17ΔPst(LAT−) Latent TG 1 Hour after Mouse Hyperthermic Stress
| Change (x-Fold) | |||
|---|---|---|---|
| Affymetrix Probe Number* | Gene Symbol (Name) | Microarray | Real-Time PCR |
| 1427837_at | IgK-V32 (immunoglobulin kappa chain variable 32 (V32)) | +7.9 | +1.51 |
| 1424931_s_at | IgL-VI (immunoglobulin lambda chain, variable 1) | +3.6 | +13.59 |
| 1430523_s_at | IgL-VI homologue (immunoglobulin lambda chain, variable 1) | +3.4 | +2.17 |
| 1425324_x_at | IgH-4 (serum IgG1) (immunoglobulin heavy chain 4) | +2.9 | +8.40 |
| 1416111_at | CD83 (CD83 antigen) | +3.3 | +17.39 |
| 1444078_at | CD8A (CD8 antigen, alpha chain) | +2.9 | +2.95 |
| 1419684_at | CCL8 [chemokine (C-C motif) ligand 8] | +3.0 | +3.05 |
| 1417976_at | ADA (adenosine deaminase)† | +5.3 | +9.65 |
| 1428719_at | 2010309G21RIKIgL (RIKEN cDNA 2010309G21 gene immunoglobulin lambda chain complex) | +3.0 | +1.04 |
Changes in expression of nine genes related to host immune response and defense based on microarrays and quantitative real-time PCR in the 17Syn+ high phenotypic reactivator latent TG relative to a LAT− recombinant, 17ΔPst low phenotypic reactivator latent TG 1 hour after mouse hyperthermic stress. Relative quantitative expression levels were determined for each gene. All results are expressed as an expression ratio of the 17Syn+ latent TG to the 17ΔPst(LAT−) latent TG of heat-treated mice, normalized against β-actin expression levels using the 2−ΔΔCT method.43
Affymetrix, Inc., Santa Clara, CA.
Pathway for Ada upregulated in the purine nucleotide metabolism.
Figure 3.
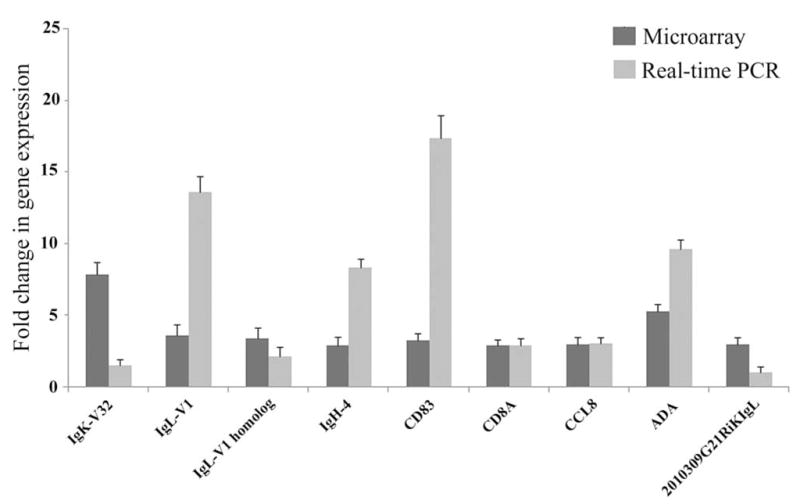
The change (x-fold) in gene expression of microarray versus real-time polymerase chain reaction of the nine adaptive immunity genes IgK-V32, Ig-V1, Ig-V1 homologue, IgH-4(serum IgG1), CD83, CD8A, CCL8, ADA, and 2010309G21RIKIgL under the two detection methods in the 17Syn+ high phenotypic reactivator latent TG relative to the LAT− recombinant, 17ΔPst low phenotypic reactivator latent TG 1 hour after mouse hyperthermic stress.
Table 2.
Changes in Hormonal Activity Genes Upregulated in the HSV-1 17Syn+ Latent TG Relative to 17ΔPst(LAT−) Latent TG 1 Hour after Mouse Hyperthermic Stress
| Affymetrix Probe Number* | Gene Symbol (Name) | Change (x-Fold) Microarray |
|---|---|---|
| 1418549_at | CGA (glycoprotein hormones, alpha subunit) | +3.8 |
| 1436127_at | CRHBP (corticotropin releasing hormone binding protein) | +3.8 |
| 1450795_at | LHB (luteinizing hormone beta) | +2.8 |
| 1437522_x_at | GH (growth hormone) | +2.7 |
| 1460668_at | GAL (galanin) | +2.8 |
Changes in expression of five genes related to hormone activity based on microarrays in the 17Syn+ high phenotypic reactivator latent TG relative to a LAT negative recombinant, 17ΔPst low phenotypic reactivator latent TG 1 hour after mouse hyperthermic stress.
Affymetrix, Inc., Santa Clara, CA.
Transcript levels of all nine adaptive immunity genes analyzed by real-time PCR confirmed their upregulated expression. IgL-V31, IgH-4 (serum IgG1), CD83, CD8A, CCL8, and ADA exhibited similar or higher increases in expression, whereas this method showed lower increases for IgK-V32, the IgL-V1 homologue, and 2010309G21RIKIgL (Table 1; Fig. 3; Supplementary Table S1). OTOS, randomly selected from the remaining 20 genes also showed increased expression by this method (Supplementary Table S1).
Discussion
This study revealed changes in gene expression in 17Syn+ latent TG relative to 17ΔPst(LAT−) latent TG 1 hour after mouse hyperthermic stress. The expression of 29 genes was increased in the TG from mice latent with 17Syn+, a virus which reactivates in rabbits (Fig. 2; Supplementary Table S1). A subset of nine of the genes upregulated by three- to eightfold (Fig. 1; Table 1) is part of the circulating Ig complex and molecules central to adaptive immunity such as antigen binding and cytotoxicity. Expression of genes including the Ig joining chain, a molecule that protects against invasion by foreign antigens, has been reported in the latent rabbit TG.28 Ig joining chain gene expression implicates B cell function in response to the McKrae strain of HSV which undergoes spontaneous reactivation in rabbits. Gene expression of Granzyme A, an important effector molecule of cytotoxic T cells and natural killer (NK) cells, as well as interferon-inducible and regulatory proteins, has been reported in the HSV-1 F strain latent mouse TG.45 Two factors are important for the absence of Granzyme A upregulation in our findings: there is a significant difference in host responses to the HSV genome during the latency phase in comparison to the viral induction/reactivation phase as was observed in our earlier study,28 and hyperthermic stress in vivo and explant in vitro to induce viral reactivation result in significantly different quantitative and qualitative molecular effects.38 Previous studies suggest the involvement of an active immune response aimed at suppressing the virus during latency.46–50
The expression of IgK-V32, IgL-V1, and IgH-4 (serum IgG1) involved in B-cell function, and CD83, CD8A, CCL8, and ADA involved in T-cell function (Table 1; Fig. 1) suggests a crucial role for adaptive immunity during HSV-1 induction and, in effect, reactivation. Induction of reactivation of the latent HSV-1 genome has been linked to the rcr.3,8–11 The rcr is thought to influence the ICP0 in relation to the proximity of ICP0 to the rcr. CD83 influences ICP0 activity in the establishment of latency.51 Thus, CD83 upregulation in the induction of LAT-positive 17Syn+ associates CD83 activity with ICP0 function during reactivation. CD83, an Ig superfamily member, is strongly upregulated during maturation of human dendritic cells (DCs) which are the most potent antigen-presenting cells of the immune system.52 Sentinel DCs have the unique ability to prime naïve CD4+ and CD8+ T cells and thereby induce a primary immune response.53,54 Expression of CD83 was not coincidental in our findings. A recent study has identified a novel HSV-1 immune escape mechanism involving ICP0.51 CD83 is degraded by ICP0 in mature DCs in the establishment of latency.51 The reactivating virus could employ an escape mechanism involving high expression of CD83 to potentially impair T-cell function. Maturing DCs respond to chemokines,55 and CCL8, an immunoregulatory chemokine, was expressed. The induction of chemokine synthesis in the HSV-1-infected cornea has been reported.56 T cells and NK cells express CD8, and we observed the expression of the alpha chain, CD8A. ADA, which is also involved in the proliferation and differentiation of T cells, was identified.57 Although, it is yet to be adduced whether CD83 or the other immune molecules are directly expressed in neurons, it is possible that immune competent cells in apposition to neurons could be the source. The detection of these immune molecules suggests their efficient penetration into the neurons which is significant. Our report suggests that there is a relationship between viral induction and gene expression of B cell function Ig molecules, as well as the T-cell function molecules CD83, CD8A, ADA, and CCL8. Further, the impairment of T-cell function by CD83 could also be aided by the other immune molecules CD8A, ADA, and CCL8. Alternatively, this phenomenon could indicate a sensitized adaptive immunity to counter viral reactivation. This provides further insight into the ability of the virus to escape elimination by the host and, potentially, to reactivate. The virus could be using multiple mechanisms to exploit the immune system51,58 for the establishment of latency and to reactivate. Gene expression in the cells of the hypothalamus-pituitary axis tissues was also significantly upregulated (Fig. 1; Table 2).
Real-time PCR analysis was conducted on all the nine adaptive immunity genes to confirm the host gene transcription levels observed in the microarray experiment. Although some of the levels of the upregulated genes are different between the two methods of detection, we always observed the same pattern of upregulation for IgK-V32, Ig-V1, Ig-V1 homologue, IgH-4 (serum IgG1), CD83, CD8A, CCL8, ADA, and 2010309G21RIKIgL gene expression (Fig. 3), suggesting that, overall, the microarray data are accurate. The very high transcript level of CD83 indicates that it is the likely candidate of ICP0-dependent degradation51 as the virus reactivates and the host may be expressing more to compensate for the CD83 loss. Alternately, the high CD83 transcript level could represent viral modification to interfere with T-cell function.
Our study provides insight into the potential role of adaptive immunity genes in the LAT-ICP0 locus of HSV. The action surrounding the regulation of HSV latency and potential reactivation resides, in large part, in the LAT-ICP0 region of the HSV genome. Functionally, adaptive immunity could interpolate the opposing and, at the same time, complementary relationship between ICP0 and LAT. A proteolytic shed soluble isoform of CD83 (sCD83)59,60 puts into perspective the role of the T cell in viral-mediated activity during reactivation from latency. The effect of sCD83 was analyzed in vivo by using the murine experimental autoimmune encephalomyelitis model. sCD83 was found to be very effective in a prophylactic and a therapeutic application, underlying its high immunosuppressive potential also in vivo.61 sCD83 has been reported not to be shed after infection with HSV-1.51 Thus, our focus will be to directly evaluate T-cell activity during latency using sCD83 to test whether T-cell function is distorted by the virus to reactivate. Further, we will evaluate synthetic compounds that have a reactive immunoregulatory conformation to interfere with T-cell proliferation in acute, latent, and reactivation models of HSV-1 mouse infection. All evidence considered, there is an impetus for immune intervention/therapy and gene therapy for the treatment of HSV-1 infection, blocking of viral reactivation, and the reduction of recurrent ocular disease.
Supplementary Material
Acknowledgments
Supported in part by National Institutes of Health Grants EY006311 (JMH), EY002622 (HEK), and EY02377 (LSU Eye Center Core Grant for Vision Research); an unrestricted research grant from LSU Health Sciences Center (PSB); a Research to Prevent Blindness Senior Scientific Investigator Award (JMH); and an unrestricted grant to the Louisiana State University Eye Center from Research to Prevent Blindness, New York, New York.
The authors thank Doan Nguyen and Patrick Byrne for assistance in microarray research.
Footnotes
Disclosure: C. Clement, None; P.S. Bhattacharjee, None; H.E. Kaufman, None; J.M. Hill, None
References
- 1.Roizman B, Knipe DM, Whitley RJ. Herpes simplex viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Baltimore, MD: Lippincott Williams & Wilkins; 2007. pp. 2503–2602. [Google Scholar]
- 2.Bloom DC. HSV LAT and neuronal survival. Int Rev Immunol. 2004;23:187–198. doi: 10.1080/08830180490265592. [DOI] [PubMed] [Google Scholar]
- 3.Bloom DC. HSV-1 latency and the roles of the LATs. In: Sandri-Goldin RM, editor. Alpha Herpesviruses: Molecular and Cellular Biology. Norwich, UK: Caister Academic Press; 2006. pp. 325–342. [Google Scholar]
- 4.Chen SH, Kramer MF, Schaffer PA, Coen DM. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1997;71:5878–5884. doi: 10.1128/jvi.71.8.5878-5884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Preston CM. Repression of viral transcription during herpes simplex virus latency. J Gen Virol. 2000;81:1–19. doi: 10.1099/0022-1317-81-1-1. [DOI] [PubMed] [Google Scholar]
- 6.Everett RD, Boutell C, Orr A. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J Virol. 2004;78:1763–1774. doi: 10.1128/JVI.78.4.1763-1774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller CS, Danaher RJ, Jacob RJ. Molecular aspects of herpes simplex virus 1 latency, reactivation and recurrence. Crit Rev Oral Biol Med. 1998;9:541–562. doi: 10.1177/10454411980090040901. [DOI] [PubMed] [Google Scholar]
- 8.Bloom DC, Hill JM, Devi-Rao G, Wagner EK, Feldman LT, Stevens JG. A 348-base-pair region in the latency-associated transcript facilitates herpes simplex virus type 1 reactivation. J Virol. 1996;70:2449–2459. doi: 10.1128/jvi.70.4.2449-2459.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill JM, Sedarati F, Javier RT, Wagner EK, Stevens JG. Herpes simplex virus latent phase transcription facilitates in vivo reactivation. Virology. 1990;174:117–125. doi: 10.1016/0042-6822(90)90060-5. [DOI] [PubMed] [Google Scholar]
- 10.Jarman RG, Loutsch JM, Devi-Rao GB, et al. The region of the HSV-1 latency-associated transcript required for epinephrine-induced reactivation in the rabbit does not include the 2.0-kb intron. Virology. 2002;292:59–69. doi: 10.1006/viro.2001.1265. [DOI] [PubMed] [Google Scholar]
- 11.Leib DA, Bogard CL, Kosz-Vnenchak M, et al. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol. 1989;63:2893–2900. doi: 10.1128/jvi.63.7.2893-2900.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufman HE, Rayfield MA, Gebhardt BM. Herpes simplex viral infections. In: Kaufman HE, Barron BA, McDonald MB, editors. The Cornea. Boston: Butterworth-Heinemann; 1998. pp. 247–277. [Google Scholar]
- 13.Kaufman HE, Azcuy AM, Varnell ED, Sloop GD, Thompson HW, Hill JM. HSV-1 DNA in tears and saliva of normal adults. Invest Ophthalmol Vis Sci. 2005;46:241–247. doi: 10.1167/iovs.04-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mark KE, Wald A, Magaret AS, et al. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J Infect Dis. 2008;198:1141–1149. doi: 10.1086/591913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller CS, Danaher RJ. Asymptomatic shedding of herpes simplex virus (HSV) in the oral cavity. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;105:43–50. doi: 10.1016/j.tripleo.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 16.Xu F, Markowitz LE, Gottlieb SL, Berman SM. Seroprevalence of herpes simplex virus types 1 and 2 in pregnant women in the United States. Am J Obstet Gynecol. 2007;196:43–46. doi: 10.1016/j.ajog.2006.07.051. [DOI] [PubMed] [Google Scholar]
- 17.Xu F, Sternberg MR, Kottiri BJ, et al. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA. 2006;296:964–973. doi: 10.1001/jama.296.8.964. [DOI] [PubMed] [Google Scholar]
- 18.Miller CS, Berger JR, Mootoor Y, Avdiushko SA, Zhu H, Kryscio RJ. High prevalence of multiple human herpesviruses in saliva from human immunodeficiency virus-infected persons in the era of highly active antiretroviral therapy. J Clin Microbiol. 2006;44:2409–2415. doi: 10.1128/JCM.00256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aryee EA, Bailey RL, Natividad-Sancho A, Kaye S, Holland MJ. Detection, quantification and genotyping of herpes simplex virus in cervicovaginal secretions by real-time PCR: A cross sectional survey. Virol J. 2005;2:61. doi: 10.1186/1743-422X-2-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Legoff J, Bouhlal H, Gresenguet G, et al. Real-time PCR quantification of genital shedding of herpes simplex virus (HSV) and human immunodeficiency virus (HIV) in women coinfected with HSV and HIV. J Clin Microbiol. 2006;44:423–432. doi: 10.1128/JCM.44.2.423-432.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berman EJ, Hill JM. Spontaneous ocular shedding of HSV-1 in latently infected rabbits. Invest Ophthalmol Vis Sci. 1985;26:587–590. [PubMed] [Google Scholar]
- 22.Gilbert SC. Oral shedding of herpes simplex virus type 1 in immunocompetent persons. J Oral Pathol Med. 2006;35:548–553. doi: 10.1111/j.1600-0714.2006.00461.x. [DOI] [PubMed] [Google Scholar]
- 23.Decman V, Freeman ML, Kinchington PR, Hendricks RL. Immune control of HSV-1 latency. Viral Immunol. 2005;18:466–473. doi: 10.1089/vim.2005.18.466. [DOI] [PubMed] [Google Scholar]
- 24.Bloom DC, Devi-Rao GB, Hill JM, Stevens JG, Wagner EK. Molecular analysis of herpes simplex virus type 1 during epinephrine induced reactivation of latently infected rabbits in vivo. J Virol. 1994;68:1283–1292. doi: 10.1128/jvi.68.3.1283-1292.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Devi-Rao GB, Bloom DC, Stevens JG, Wagner EK. Herpes simplex virus type 1 DNA replication and gene expression during explant Induced reactivation of latently infected murine sensory ganglia. J Virol. 1994;68:1271–1282. doi: 10.1128/jvi.68.3.1271-1282.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill JM, Wen R, Halford WP. Pathogenesis and molecular biology of ocular HSV in the rabbit. In: Brown MS, MacLean AR, editors. Herpes Simplex Virus Protocols. Totowa, NJ: Humana Press/Wiley; 1998. pp. 291–315. [Google Scholar]
- 27.Wagner EK, Bloom DC. Experimental investigation of herpes simplex virus latency. Clin Microbiol Rev. 1997;10:419–443. doi: 10.1128/cmr.10.3.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clement C, Popp MP, Bloom DC, et al. Microarray analysis of host gene expression for comparison between naive and HSV-1 latent rabbit trigeminal ganglia. Mol Vis. 2008;14:1209–1221. [PMC free article] [PubMed] [Google Scholar]
- 29.McLennan JL, Darby G. Herpes simplex virus latency: the cellular location of virus in dorsal root ganglia and the fate of the infected cell following virus activation. J Gen Virol. 1980;51:233–243. doi: 10.1099/0022-1317-51-2-233. [DOI] [PubMed] [Google Scholar]
- 30.Sawtell NM, Thompson RL. Rapid in vivo reactivation of herpes simplex virus in latently infected murine ganglionic neurons after transient hyperthermia. J Virol. 1992;66:2150–2156. doi: 10.1128/jvi.66.4.2150-2156.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halford WP, Gebhardt BM, Carr DJ. Mechanisms of herpes simplex virus type 1 reactivation. J Virol. 1996;70:5051–5060. doi: 10.1128/jvi.70.8.5051-5060.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sawtell NM. Quantitative analysis of herpes simplex virus reactivation in vivo demonstrates that reactivation in the nervous system is not inhibited at early times postinoculation. J Virol. 2003;77:4127–4138. doi: 10.1128/JVI.77.7.4127-4138.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higaki S, Gebhardt B, Lukiw W, Thompson H, Hill J. Gene expression profiling in the HSV-1 latently infected mouse trigeminal ganglia following hyperthermic stress. Curr Eye Res. 2003;26:231–238. doi: 10.1076/ceyr.26.3.231.14892. [DOI] [PubMed] [Google Scholar]
- 34.Blyth WA, Harbour DA, Hill TJ. Effect of immunosuppression on recurrent herpes simplex in mice. Infect Immun. 1980;29:902–907. doi: 10.1128/iai.29.3.902-907.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cook SD, Paveloff MJ, Doucet JJ, Cottingham AJ, Sedarati F, Hill JM. Ocular herpes simplex virus reactivation in mice latently infected with latency-associated transcript mutants. Invest Ophthalmol Vis Sci. 1991;32:1558–1561. [PubMed] [Google Scholar]
- 36.Higaki S, Gebhardt BM, Lukiw WJ, Thompson HW, Hill JM. Effect of immunosuppression on gene expression in the HSV-1 latently infected mouse trigeminal ganglion. Invest Ophthalmol Vis Sci. 2002;43:1862–1869. [PubMed] [Google Scholar]
- 37.Zlotnik I, Smith CE, Grant DP, Peacock S. The effect of immunosuppression on viral encephalitis, with special reference to cyclophosphamide. Br J Exp Pathol. 1970;51:434–439. [PMC free article] [PubMed] [Google Scholar]
- 38.Sawtell NM, Thompson RL. Comparison of herpes simplex virus reactivation in ganglia in vivo and in explants demonstrates quantitative and qualitative differences. J Virol. 2004;78:7784–7794. doi: 10.1128/JVI.78.14.7784-7794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schang LM, Bantly A, Schaffer PA. Explant-induced reactivation of herpes simplex virus occurs in neurons expressing nuclear cdk2 and cdk4. J Virol. 2002;76:7724–7735. doi: 10.1128/JVI.76.15.7724-7735.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill JM, Lukiw WJ, Gebhardt BM, et al. Gene expression analyzed by microarrays in HSV-1 latent mouse trigeminal ganglion following heat stress. Virus Genes. 2001;23:273–280. doi: 10.1023/a:1012517221937. [DOI] [PubMed] [Google Scholar]
- 41.Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J Am Stat Assoc. 2004;99:909–917. [Google Scholar]
- 42.Hubbell E, Liu WM, Mei R. Robust estimators for expression analysis. Bioinformatics. 2002;18:1585–1592. doi: 10.1093/bioinformatics/18.12.1585. [DOI] [PubMed] [Google Scholar]
- 43.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 44.Qin LX, Beyer RP, Hudson FN, Linford NJ, Morris DE, Kerr KF. Evaluation of methods for oligonucleotide array data via quantitative real-time PCR. BMC Bioinformatics. 2006;7:23. doi: 10.1186/1471-2105-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kent JR, Fraser NW. The cellular response to herpes simplex virus type 1 (HSV-1) during latency and reactivation. J Neurovirol. 2005;11:376–383. doi: 10.1080/13550280591002405. [DOI] [PubMed] [Google Scholar]
- 46.Divito S, Cherpes TL, Hendricks RL. A triple entente: virus, neurons, and CD8+ T cells maintain HSV-1 latency. Immunol Res. 2006;36:119–126. doi: 10.1385/IR:36:1:119. [DOI] [PubMed] [Google Scholar]
- 47.Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity. 2003;18:593–603. doi: 10.1016/s1074-7613(03)00112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khanna KM, Lepisto AJ, Hendricks RL. Immunity to latent viral infection: many skirmishes but few fatalities. Trends Immunol. 2004;25:230–234. doi: 10.1016/j.it.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 49.Khanna KM, Lepisto AJ, Decman V, Hendricks RL. Immune control of herpes simplex virus during latency. Curr Opin Immunol. 2004;16:463–469. doi: 10.1016/j.coi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 50.Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med. 2000;191:1459–1466. doi: 10.1084/jem.191.9.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kummer M, Turza NM, Muhl-Zurbes P, et al. Herpes simplex virus type 1 induces CD83 degradation in mature dendritic cells with immediate-early kinetics via the cellular proteasome. J Virol. 2007;81:6326–6338. doi: 10.1128/JVI.02327-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 53.Flores-Romo L. In vivo maturation and migration of dendritic cells. Immunology. 2001;102:255–262. doi: 10.1046/j.1365-2567.2001.01204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 55.Zhou LJ, Tedder TF. CD14+ blood monocytes can differentiate into functionally mature CD83+ dendritic cells. Proc Natl Acad Sci U S A. 1996;93:2588–2592. doi: 10.1073/pnas.93.6.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tumpey TM, Cheng H, Yan XT, Oakes JE, Lausch RN. Chemokine synthesis in the HSV-1-infected cornea and its suppression by interleukin-10. J Leukoc Biol. 1998;63:486–492. doi: 10.1002/jlb.63.4.486. [DOI] [PubMed] [Google Scholar]
- 57.Martin M, Huguet J, Centelles JJ, Franco R. Expression of ecto-adenosine deaminase and CD26 in human T cells triggered by the TCR-CD3 complex: possible role of adenosine deaminase as co-stimulatory molecule. J Immunol. 1995;155:4630–4643. [PubMed] [Google Scholar]
- 58.Clement C, Tiwari V, Scanlan PM, Valyi-Nagy T, Yue BY, Shukla D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J Cell Biol. 2006;174:1009–1021. doi: 10.1083/jcb.200509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hock BD, Haring LF, Steinkasserer A, Taylor KG, Patton WN, McKenzie JL. The soluble form of CD83 is present at elevated levels in a number of hematological malignancies. Leuk Res. 2004;28:237–241. doi: 10.1016/s0145-2126(03)00255-8. [DOI] [PubMed] [Google Scholar]
- 60.Hock BD, Kato M, McKenzie JL, Hart DN. A soluble form of CD83 is released from activated dendritic cells and B lymphocytes, and is detectable in normal human sera. Int Immunol. 2001;13:959 –967. doi: 10.1093/intimm/13.7.959. [DOI] [PubMed] [Google Scholar]
- 61.Zinser E, Lechmann M, Golka A, Lutz MB, Steinkasserer A. Prevention and treatment of experimental autoimmune encephalomyelitis by soluble CD83. J Exp Med. 2004;200:345–351. doi: 10.1084/jem.20030973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.