

Abstract
Asb-4 is a gene that is specifically expressed in the hypothalamic energy homeostasis-associated areas and is down regulated in the arcuate nucleus of fasted Sprague Dawley and obese Zucker rats. It has two functional domains, the ankyrin repeat and the SOCS box. The function of Asb-4 is unclear. We used yeast two hybridization to search for protein(s) that interact with Asb-4. With Asb-4 minus its SOCS box (Asb-4/Δsb) as a bait, we screened mouse testis and arcuate nucleus cDNA libraries and identified G-protein pathway suppressor 1 (GPS1, also known as CSN1) as an Asb-4 interacting protein. GPS1 co-immunoprecipitated with Asb-4 both in vitro and in human HEK293 cells. When Asb-4 and GPS1 were co-transfected into HEK293 cells, expression of Asb-4 reduced the protein level of GPS1. Deletion of the SOCS box (Asb4/Δsb) did not abolish the inhibitory effect of Asb-4 on GPS1, indicating that the SOCS box was not needed for its inhibitory effect. In NIH 3T3 L1 cells, expression of GPS1 enhanced c-Jun NH2-terminal kinase (JNK) activity. Co-expression of Asb-4 with GPS1 inhibited JNK activity. Treatment of the cells with insulin (20 nM) stimulated JNK activity. Expression of GPS1 potentiated the stimulatory effect of insulin, whereas co-expression of Asb-4 along with GPS1 inhibited JNK activity. In HEK293 cells expression of GPS1 elevated phosphorylation of insulin receptor substrate 1 (IRS-1) at serine 307, co-expression of Asb-4 with GPS1 reduced the IRSser307 phosphorylation. The present study demonstrates that Asb-4 interacts with GPS1 and inhibits JNK activity.
Keywords: Asb-4, G-protein pathway suppresser 1(GPS1), COP9 signalosome, subunit 1 CSN1, c-Jun NH2-terminal kinase (JNK), Insulin receptor substrate 1 (IRS-1)
1. Introduction
Asb-4 is a member of the ankyrin repeat and SOCS box containing protein family (Asb-1 to Asb-18). All representatives of the Asb protein family have two functional domains: an ankyrin repeat region where specific protein-protein interactions occur and a SOCS box region which serves as a generic adapter directing the degradation of proteins targeted by the ankyrin repeat region [1]. The 18 Asb proteins have varying forms and numbers of ankyrin repeats and other novel regions, suggesting they bind differing target proteins. Asb-4 was originally identified by database searches using a suppressor of cytokine signaling (SOCS) box consensus sequence and was found to be expressed in the testis. Asb-4 has nine ankyrin repeats N-terminal to its SOCS box [2]. Both the interacting protein(s) and the function of Asb-4 remain to be elucidated.
Using microarray analysis, we have previously reported that Asb-4 mRNA is expressed in the hypothalamic arcuate nucleus (ARC) of the rat and is down-regulated by fasting [3]. Further studies showed that Asb-4 mRNA was also down-regulated in the ARC of the genetically obese Zucker rat, another model of energy disequilibrium. In situ hybridization revealed that expression of Asb-4 mRNA was restricted to neuroanatomical areas in the hypothalamus and amygdala associated with energy homeostasis. Double in situ hybridization showed that Asb-4 mRNA is differentially expressed in two types of ARC neurons critical to energy homeostasis, pro-opiomelanocortin (POMC) and neuropeptide Y (NPY) neurons. These findings implied that Asb-4 might have a role in the regulation of energy homeostasis. In the present work, we used yeast two hybridization to search for proteins that interact with Asb-4 and identified G-protein pathway suppressor 1 (GPS1) as an Asb-4 interacting protein.
2. Material and Methods
2.1. Antibodies and reagents
Goat anti-Myc and anti-HA antibody-conjugated agarose (for immunoprecipitation) and rabbit anti-GPS1 (CSN1) antibody ( purified IgG against human and mouse GPS1) were purchased from Bethyl Laboratories (Montgomery, TX). Mouse monoclonal anti-Myc and rabbit anti-HA antibodies were purchased from BD Clontech (Palo Alto, CA). Anti-phospho-IRS-1(Ser 307) was purchased from Upstate ( Lake Placid, NY). Anti-FLAG-M2 antibody-conjugated agarose was purchased from Sigma-Aldrich (St. Louis, MO). Goat anti POMC and NPY polyclonal antibodies were purchased from Santa Cruz Biotechnology, Inc ( Santa Cruz, CA). Rabbit polyclonal antibodies against the ankyrin repeat domain of Asb-4 (244–258aa of the Asb-4 protein sequence) were generated and purified by affinity chromatography with custom service by Invitrogen (Carlsbad, CA). Specificity of the antibodies was characterized using purified GST-Asb-4 fusion protein. pCMV-Myc, pCMV-HA and pEGPF-N1 mammalian expression vectors were purchased from BD Clontech. CMV-FLAG-JNK vector was provided by Dr. Deepak Nihalani (Department of Internal Medicine, University of Michigan). Myc-tagged ubiquitin cDNA, HA-IRS1 and SOCS1 were provided by Dr. Liangyou Rui (Department of Molecular & Integrative Physiology, University of Michigan ).
2.2. Yeast two hybridization
The Matchmaker GAL4 Two-Hybrid System 3 (BD Clontech) was used to identify Asb-4 interacting proteins. Asb-4 minus its SOCS box region (Asb-4/Δsb) was fused to the C-terminal of the GAL4 DNA binding domain of the pGBKT7 vector to generate the bait. Mouse testis and ARC cDNA libraries were constructed and screened according to the manufacture’s instruction. The resulting transformants were plated onto selective medium lacking tryptophan, leucine, histidine, adenine (quadruple dropout medium, QDO) and incubated at 30°C until colonies formed. Ade+/His+ colonies were selected after 10 days and streaked onto QDO with X-α-gal to identify expression of α-galactosidase. Library plasmids were rescued from the Ade+/His+/MEL+ clones and transformed into Escherichia coli XL10Gold, DH5α under ampicillin selection to yield prey plasmids. Prey plasmids were sequenced and the nucleotide and in-frame amino acid sequences were analyzed using the GenBank data base with the BLAST program.
Specificity of interactions between candidate prey plasmids and Asb-4/Δsb were assayed by co-transforming prey plasmids with either pGBKT7-Asb-4/Δsb bait plasmid or pGBKT7 empty plasmid into yeast and plating on QDO with X-α-gal. Prey plasmids that interacted with the pGBKT7 empty plasmid were excluded from further analysis. A total of eight specific interacting clones were identified.
2.3. In vitro co-immunoprecipitation (Co-IP)
Myc-tagged Asb-4/Δsb and HA-tagged GPS1(C-terminal aa 221–471) were transcribed, translated and labeled with [35S]methionine in vitro via T7 promoters in the pGBKT7-Asb-4/Δsb bait plasmid and pGADT7-GPS1 prey plasmid using a TNT-coupled reticulocyte lysate system (Promega, Madison, WI). A BD Matchmaker Co-IP kit ( BD Clontech ) was used. Briefly, the epitope tagged bait and prey proteins were mixed and incubated at room temperature for 1 hr. c-Myc monoclonal antibody or HA polyclonal antibody was added and incubated at room temperature for another hour. Protein A beads were used to precipitate the immunocomplex. After extensive washing, beads were resuspended in SDS-PAGE loading buffer and heated at 80°C for 5 min. Proteins were resolved with SDS-PAGE gel, and the gel was fixed, dried and exposed with X-ray film.
2.4. Co-IP in mammalian cell line
The full length coding region of mouse Asb-4 cDNA was cloned by RT-PCR from the mouse hypothalamic neuronal cell line, GT1-1, and subcloned into the pCMV-Myc vector at EcoR1 and Not I sites. Mouse GPS1 cDNA with full length coding region was cloned by RT-PCR from a testis cDNA library and subcloned into the pCMV-HA vector at Bgl II and Not I sites. HEK293 cells were grown in Dulbecco’s modified Eagles medium supplemented with 10% fetal bovine serum and 100 units/ml of penicillin and 100 μg/ml of streptomycin in a 37°C incubator supplemented with 5% CO2. Cells were transfected with 2.5 μg (100 mm dish) of pCMV-Myc-Asb-4 plasmid DNA along with 2.5 μg of pCMV-HA-GPS1 plasmid DNA using lipofectamine 2000 as described by the manufacturer (Invitrogen). Forty hours after transfection, cells were disrupted by sonication in lysis buffer [150 mM NaCl, 20 mM Na2HPO4, pH 7.4, 1% Triton X-100 supplemented with protease inhibitor cocktail (Sigma)] and centrifuged at 14,000 × g for 10 min at 4° C. Myc-Asb-4 or HA-GPS1 were immunoprecipitated using anti-Myc or anti-HA antibody conjugated agarose. After extensive washing the beads were resuspended in SDS-PAGE loading buffer and heated at 80°C for 5min. The protein was resolved with SDS-PAGE gel and transferred to polyvinylidene membranes. The membranes were blotted with either anti-Myc, anti-HA, anti-Asb-4 or anti-GPS1 antibody.
2.5. Western blotting
Equal amounts of protein were resolved on SDS-PAGE gel, transferred to polyvinylidene membranes and immunoblotted with primary antibodies. Horseradish peroxidase conjugated secondary antibodies were used to detect antigen-antibody complexes via the ECL detection system (Amersham Biosciences, Piscataway, NJ).
2.6. EGFP tagging and immunohistochemistry
The distribution of Asb-4 in GT1-1 and HEK 293 cells was visualized with an Asb-4-EGFP fusion protein. Mouse Asb-4 cDNA with full length coding region was subcloned into pEGFP-N1 vector at Hind III and BamH I sites (fused to the N-terminal of enhanced green fluorescent protein, EGFP). The vector was transfected into the mouse hypothalamic neuronal cell line GT1-1, and HEK293 cell. Two days after transfection, cells were fixed with 4% paraformaldehyde for 1 hr and stained with rabbit anti-GPS1 antibody (1:400 dilution)[4]. Normal rabbit IgG with the same dilution factor was used as a negative control. Rhodamine(red)-conjugated goat anti-rabbit secondary antibody was used to visualize GPS1. Rat brain was fixed in 4% paraformaldehyde and coronal sections of 14 μ were cut on a cryostat. The sections were rinsed in phosphate buffered saline, 0.5% triton X-100 and blocked in 10% serum, and then incubated with primary antibodies [ rabbit anti-Asb-4 antibody (1:150) and goat anti-POMC or NPY antibody (1:100 respectively)] overnight at 4°C. For Asb-4 staining, biotinylated horse anti- rabbit secondary antibody was used and then visualized with FITC488(green) -conjugated avidin. To visualized POMC or NPY neuron, rhodamine (red)-conjugated donkey anti-goat secondary antibody was used. Nuclei were stained with 4′, 6-diamidino-2-phenylindole, dihydrochloride (DAPI, Invitrogen).
2.7. JNK assay
The JNK immunocomplex kinase assay was performed as previously described [5]. Plasmid (2.5μg) encoding Flag-tagged JNK1 was transfected into NIH3T3 L1 cells in 100 mm dishes along with 2.5μg of GPS1 and 2.5μg Asb-4. The total amount of plasmid DNA in each transfection was equalized with empty vectors. Forty hours after transfection, cells were lysed in PK buffer [50 mM HEPES, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1mM Na3VO4, 50 mM NaF, 1% Triton X-100, 10% glycerol, and protease inhibitor cocktail (Sigma)]. Equal amounts of lysate proteins from each transfection were incubated with M2 monoclonal antibody conjugated to agarose for 18 hr at 4°C. The beads were washed twice with PK lysis buffer and twice with kinase buffer (25 mM HEPES, pH 7.4, 20 mM MgCl2, 0.5 mM EGTA, 12.5 mM β-glycerophosphate, 0.1 mM orthovanadate, 0.5 mM NaF). The complex was incubated for 30 min at 30°C in 30 μl of kinase buffer containing 20 μM ATP, 5μCi [γ-32P ]ATP, and 2μg GST-c-Jun (1–79). The reaction was terminated by the adding 10 μl 4 × SDS loading buffer and heating at 80°C for 5 min. The reaction mixture was subjected to SDS-PAGE electrophoresis, transferred to polyvinylidene membrane and exposed to X-ray film.
2.8. Densitometry and Statistical analysis
Bands density was analyzed and normalized with internal control using the Kodak Gel Logic 440 Imaging System. Values were expressed as mean ± SEM of 3 separate experiments. Student’s t-test was used for statistical analysis and p< 0.05 was adopted as significance.
3. Results
3.1. Yeast two hybridization
One of the clones that demonstrated specific interaction with Asb-4/Δsb in yeast-two hybridization contained the C terminal 250 amino acid residues of mouse G protein pathway suppressor 1 (GPS1). Repeated culture experiments were performed to assure specificity of this interaction. Colonies grew and demonstrated X-α-Gal blue coloration on QDO plates when the pGADT7-GPS1 prey vector was co-transformed with the pGBKT7-Asb-4/Δsb bait vector. Colony growth did not occur when the pGADT7-GPS1 prey vector was co-transformed with the empty pGADT7 vector, indicating that GPS1 specifically interacted with Asb-4/Δsb.
3.2. Co-immunoprecipitation(Co-IP)
In order to further confirm the interaction between Asb-4/Δsb and GPS1 (aa220–471), both Myc-Asb-4/Δsb and HA-GPS1 (aa220–471) fusion proteins were synthesized. In vitro Co-IP was performed with either anti-Myc or anti-HA antibody. Figure 1 demonstrates that Myc-Asb-4/Δsb co-immunoprecipitated with GPS1 (aa220–471), as illustrated by interactions with the anti-HA antibody (left lane), and that HA-GPS1 (aa220–471) co-immunoprecipitated with Asb-4/Δsb when anti-Myc antibody was used (right lane). Next, we performed Co-IP in human HEK293 cells. Myc-tagged full length Asb-4 (Myc-Asb-4) and HA-tagged full length GPS1 (HA-GPS1) were co-expressed in human HEK293 cells and co-immunoprecipitated using either anti-Myc or anti-HA antibody-conjugated agarose. As shown in Figure 2, HA-GPS1 co-precipitated with Myc-Asb-4, detected by anti-Myc antibody (Figure 2A), and Myc-Asb-4 co-precipitated with HA-GPS1 detected by anti-HA antibody (Figure 2B).
Fig. 1. In vitro co-immunoprecipitation of GPS1 with Asb4/Δsb.
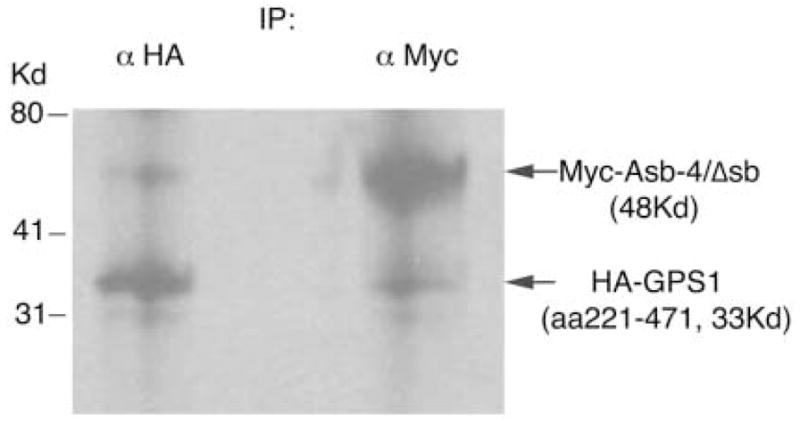
Myc-Asb4/Δsb and HA-GPS1 were translated and labeled with [35S]methionine in vitro. Products were then combined and precipitated with either anti-HA antibody (left) or anti-Myc antibody (right).
Fig. 2. Co-immunoprecipitation of HA-GPS1 with Myc-Asb-4 in HEK293 cells.
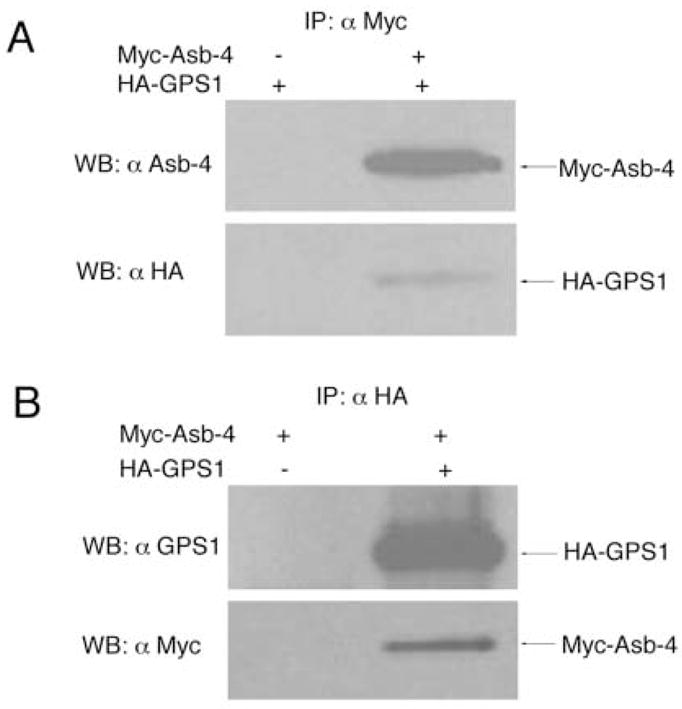
A. pCMV-HA-GPS1 (full length) vector was co-transfected with either pCMV empty vector or pCMV-Myc-Asb-4 vector in HEK293 cells. Cell lysates were immunoprecipitated with anti-Myc antibody-conjugated agarose and blotted with either anti Asb-4 antibody or anti HA antibody. B. pCMV-Myc-Asb-4 vector was co-transfected with either pCMV empty vector or pCMV-HA-GPS1 (full length) vector in HEK293 cells. Cell lysates were immunoprecipitated with anti-HA antibody-conjugated agarose and blotted with either anti GPS1 antibody or anti-Myc antibody.
3.3. Localization of Asb-4 with GPS1
EGFP-tagging of Asb-4 and immunostaining of GPS1 demonstrated that Asb-4 is predominantly localized in the cytoplasmic compartment, and GPS1 is localized in nuclear and the cytoplasmic compartments in both GT1-1 cell and HEK293 cell (Figure 3A). In addition, immunostaining of Asb-4 with anti-Asb-4 antibody in rat arcuate nucleus also showed that Asb-4 protein is localized in cytoplasmic compartment in both POMC and NPY neurons (Figure 3B). These observations suggest that interactions of Asb-4 with GPS1 occur within the cytosolic cellular compartment.
Fig. 3. Co-localization of Asb-4 with GPS1 and distribution of Asb-4 in neurons.
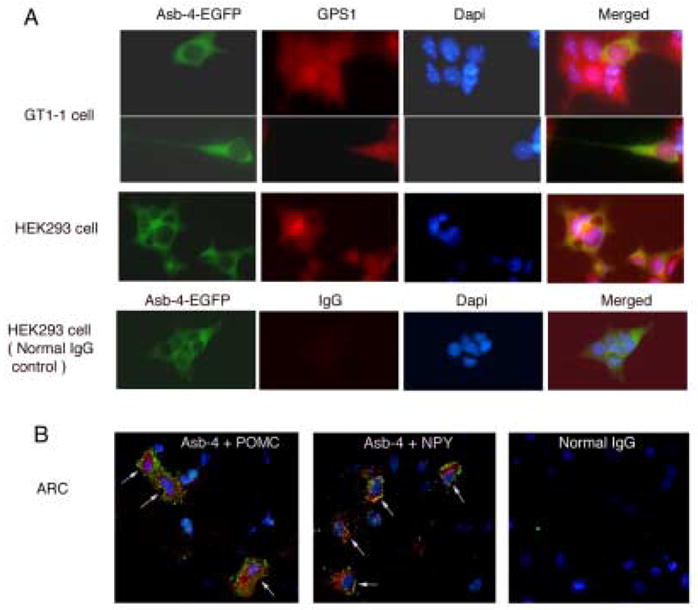
A. Green represents Asb-4-EGFP fusion protein and red represents GPS1. Yellow, merged images represent co-localization of Asb-4-EGFP with GPS1. B. Confocal photomicrographs of dual stainings of Asb-4 (green ) and POMC or NPY (Red). Normal IgG was used in negative control. Nuclei are stained with DAPI.
3.4. Effect of Asb-4 on the level of GPS1
We next examined the effect of Asb-4 expression on GPS1 level. When Myc-Asb-4 and HA-GPS1 were co-expressed in HEK293 cells, Asb-4 expression reduced GPS1 protein level dose-dependently (Figure 4A). In contrast, Asb-4 expression had no effect on the level of enhanced fluorescent protein (EGFP, Figure 4B). Deletion of the SOCS box region (Asb4/Δsb) did not abolish the inhibitory effect of Asb-4 on GPS1 levels, indicating that the SOCS box of Asb-4 is not needed for suppression of GPS1 levels by Asb-4 (Figure 4C).
Fig 4. Effect of Asb-4 on the protein level of GPS1 in HEK293 cells.
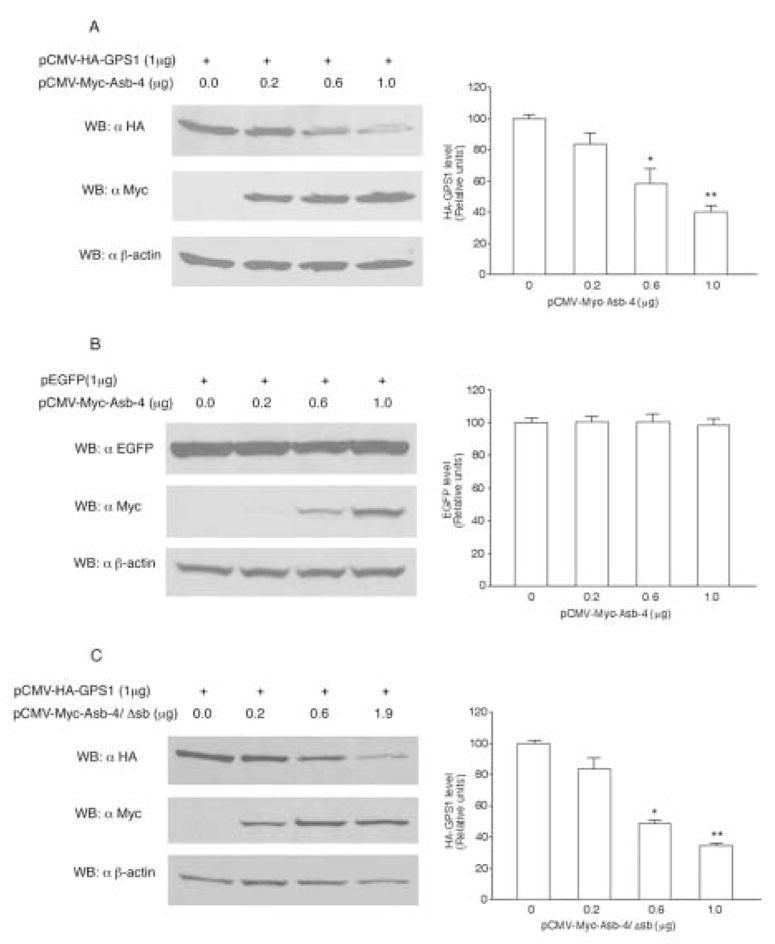
A. HEK293 cells were co-transfected with 1 μg (35 mm dish) of pCMV-HA-GPS1 vector along with differing amounts of pCMV-Myc-Asb-4 vector. Cell lysates were blotted with anti-HA antibody. B. The HEK293 cells were co-transfected with 1 μg pEGFP vector along with differing amounts of pCMV-Myc-Asb-4 vector. Cell lysates were blotted with anti EGFP antibody. C. HEK293 cells were co-transfected with 1 μg of pCMV-HA-GPS1 vector along with differing amounts of pCMV-Myc-Asb-4/Δsb vector. Cell lysates were blotted with anti-HA antibody. The total amount of vector DNA in each transfection was equalized with pCMV empty vector. The membranes were stripped and reblotted with anti-Myc and anti-β-actin antibodies respectively. *p < 0.05, **p < 0.01 vs. control.
The general model of action of Asb proteins involves ubiquitination of proteins targeted by the ankyrin repeat region of the molecule. We did not observe ubiquitination of GPS1 directed by Asb-4 or Asb-4/Δsb (Figure 5, Lane 4 and 5). As a positive control, insulin receptor substrate 1 (IRS-1) was shown to be ubiquitinized by suppressor of cytokine signaling 1 (SOCS1) (Figure 5, Lane 2) [6]. These results suggest that the ankyrin repeat domain of Asb-4 mediates the interaction with GPS1 and the reduction of GPS1 protein levels, but that a novel mechanism, not requiring ubiquitination is involved.
Fig. 5. GPS1 was not ubiquitinized by Asb-4.
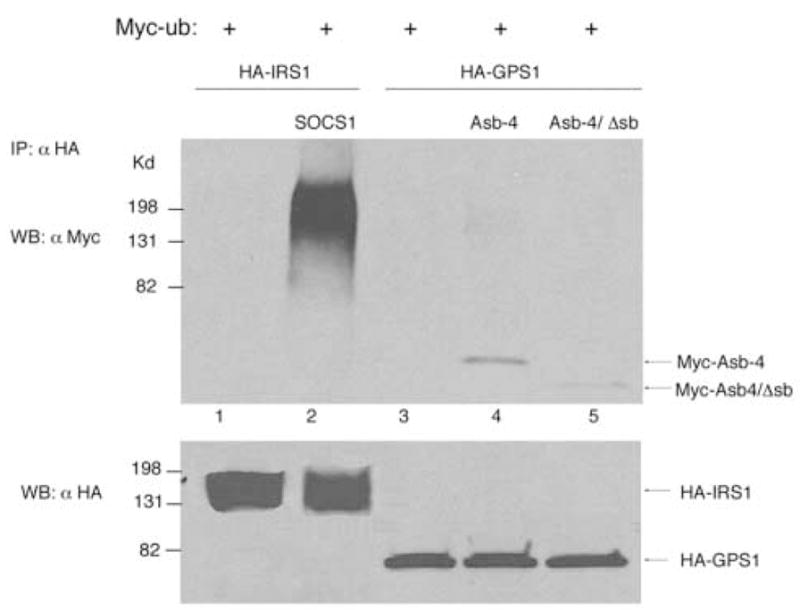
Lane 1: HEK293 cells were co-transfected with Myc-ub and HA-IRS1. Lane 2: HEK293 cells were co-transfected with Myc-ub, HA-IRS1 and SOCS1. Lane 3-5:HEK293 cells were co-transfected with Myc-ub, HA-GPS1, Myc-Asb-4 or Asb-4/Δsb as indicated. Total amount of DNA in each transfection was equalized with empty vector. Cell lysates were mmunoprecipitated with anti-HA antibody and blotted with anti-Myc antibody. Membranes were stripped and reblotted with anti-HA antibody.
Since Asb-4 contains nine ankyrin repeats we next asked which repeats are required for the inhibitory effects of Asb-4 on GPS1. Constructs representing serial ankyrin repeat deletions were formed and co-expressed with GPS1. As shown in Figure 6, deletion of ankyrin repeat 9 or 8 and 9 (1–8 or 1–7 retained) abolished the inhibitory effects of Asb-4 on GPS1 levels. In accordance with this observation, deletion of ankyrin repeats 6 through 9 or 4 through 9 also abolished the inhibitory effects of Asb-4 on GPS1 levels (data not shown). Ankyrin repeats 8–9 (deletion of 1–7) did not show any inhibitory effects on GPS1. These data showed that at least one of the first seven repeats and the 9th repeat are necessary for the inhibitory effect of Asb-4 on GPS1.
Fig. 6. Deletion of the ankyrin repeats abolished the inhibitory effect of Asb-4 on GPS1.
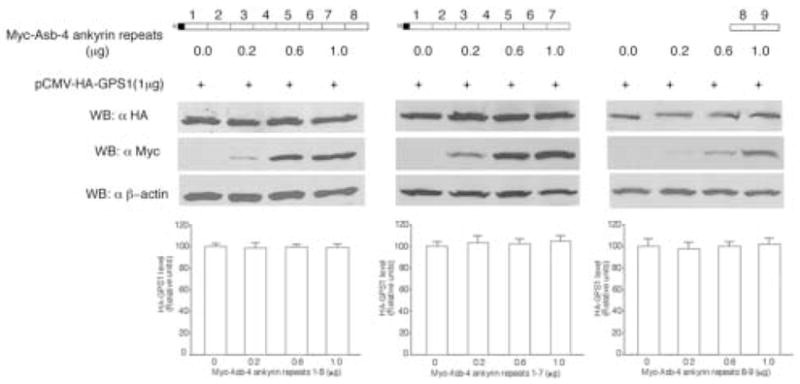
HEK293 cells were co-transfected with 1 μg of pCMV-HA-GPS1 vector along with different amounts of pCMV-Myc-ankyrin repeat variants. The total amount of DNA in each transfection was equalized with empty vector. Cell lysates were blotted with anti-HA antibody and reblotted with anti-Myc and anti-β-actin antibodies.
3.5. Effect of GPS1 and Asb-4 on JNK activity and IRS-1 phosphorylation at serine 307
In NIH 3T3 L1 cells, expression of GPS1 enhanced JNK activity. Co-expression of Asb-4 with GPS1 inhibited JNK activity (Figure 7A). Treatment of the cells with insulin (20 nM) stimulated JNK activity. Expression of GPS1 potentiated the stimulatory effect of insulin, whereas co-expression of Asb-4 along with GPS1 inhibited JNK activity (Figure 7B). In HEK293 cell expression of GPS1 increased the phosphorylation of IRS-1 at serine307. Co-expression of Asb-4 with GPS1 inhibited the serine307 phosphorylation of IRS-1.
Fig. 7. Effect of GPS1 and Asb-4 on JNK activity and phosho-IRSser307.
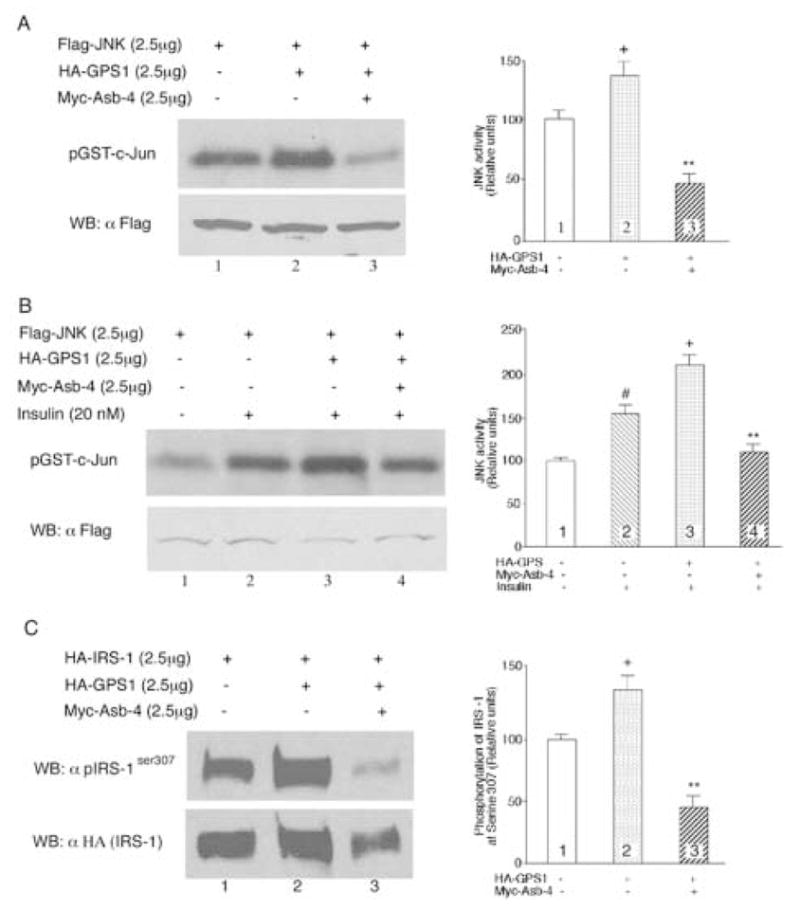
A. 2.5μg of plasmid encoding Flag-tagged JNK1 was transfected into NIH3T3 L1 cells along with 2.5μg of GPS1, 2.5μg Asb-4, or both. The amount of plasmid DNA in each transfection was equalized with pCMV empty vector. JNK was immunoprecipitated with anti-FLAG M2 antibody-conjugated agarose and JNK activity was assayed using an immunocomplex kinase assay with c-Jun (2μg each sample) as substrate. Column 2 vs 1, +p < 0.05; Column 3 vs 2, **P < 0.01. B. JNK activity was assayed in the presence of insulin (20 nM). Column 2 vs 1, #p < 0.05; Column 3 vs 2, +p < 0.01. Column 4 vs 3, **p < 0.01. C. 2.5μg of plasmid encoding HA-tagged IRS-1 was transfected into HEK293 cells along with 2.5μg of GPS1, 2.5μg Asb-4, or both. The cell lysates were blotted with anti-phospho-IRS-1ser307 antibody. Column 2 vs 1, +p < 0.05; Column 3 vs 2, **P < 0.01.
4. Discussion
Using yeast two hybridization we have identified GPS1 (CSN1) as an Asb-4 interacting protein. This interaction was confirmed by repeat hybridization, co-precipitation in vitro and in transfected human HEK293 cells. Only the ankyrin repeat portion of Asb-4 was used as bait in the yeast two hybridization system, and the inframe protein sequence in the prey plasmid was the C-terminal 250 amino acids of GPS1. Thus, the interaction between these molecules involves the ankyrin repeat region of Asb-4 and the C-terminal portion of GPS1. Consistent with other reports, we observed that GPS1 is localized in both the nucleus and cytosol [7]. In contrast, Asb-4 was localized solely in the cytosol, and interactions between Asb-4 and GPS1 are presumed to be cytosolic. The physiologic role of Asb-4 is currently unknown. Asb proteins are involved in numerous processes, as diverse as cellular differentiation, arteriogenesis and muscle growth [8–10]. Asb-6 has been reported to regulate components of the insulin signaling pathway in adipocytes [11].
The interaction of Asb-4 with GPS1 resulted in the reduced levels of GPS1. The SOCS box of Asb-4 was not required for the inhibitory effect of Asb-4 on GPS1. Asb-4 constructs with various ankyrin repeat truncations did not retain inhibitory activities. It has been suggested that all SOCS box proteins have two functional domains. The first is an ankyrin repeat domain upstream from the SOCS box where unique protein-protein interactions occur. The second is the SOCS box domain, which serves as a generic adapter site for the elongin BC components of the E3 ubiquitin ligase [1]. This complex mediates polyubiquitination and proteasomal degradation of targeted proteins [1]. The inhibitory effect of Asb-4 on GPS1 level did not involve polyubiquitination, suggesting that additional degradation pathways may exist.
GPS1 (CSN1) is a component of the COP9 signalosome (CSN) multiprotein complex. This complex is composed of eight distinct proteins (CSN1 to CSN8) [12]. CSN is expressed in most eukaryotic organisms ranging from fission yeast to humans. In eukaryotes, the specific degradation of regulated proteins is mediated by the 26 S proteasome, which can be dissected into a proteolytic 20 S core particle and two identical 19 S regulatory subunits. The 19S regulatory particle consists of a base and a lid [13]. The overall organization and amino acid sequences of CSN subunits resemble the lid subcomplex of the 19S regulatory particles [14–16]. CSN has been shown to interact with ubiquitin E3-ligase and to exhibit deneddylase activity [17,18].
CSN has also been shown to interact with protein kinases and to affect kinase activity [19]. Sun et al reported that inositol 1,3,4,- trisphosphate 5/6-kinase associates with CSN through an interaction with CSN1 (GPS1) and that overexpression of CSN1 inhibited 5/6 kinase activity [20]. Spain et al reported that overexpression of GPS1 in NIH 3T3 cell repressed JNK1 activity [21]. In contrast, we have demonstrated that transient expression of GPS1 in NIH 3T3 cell stimulated JNK activity. The actions of GPS1 were abolished by simultaneous transient expression of Asb-4. Asb-4 may affect JNK activity by reducing the cytosolic protein levels of GPS1.
The c-Jun NH2-terminal kinases (JNKs) are members of the mitogen-activated protein kinase (MAPK) family [22,23]. JNKs phosphorylate c-Jun at Ser63 and Ser73 as well as other transcription factors. Although initially identified by their ability to phosphorylate c-Jun in response to UV-irradiation, JNKs are now recognized to be involved in insulin resistance, type 2 diabetes and obesity [24]. In the insulin signaling cascade, binding of insulin to the insulin receptor results in insulin receptor substrate (IRS1) phosphorylation on tyrosine residues; tyrosine-phosphorylated IRS1 recruits and activates various effectors that contain phospho-tyrosine binding domains [25]. In the context of obesity and systemic insulin resistance, IRS1 may be phosphorylated at Ser307. Serine307 phosphorylation of IRS1 interferes with its association with the insulin receptor, reduces tyrosine phosphorylation of IRS1 in response to insulin and thereby suppresses downstream signaling and insulin action [26–29]. JNKs can phosphorylate IRS1 on Ser307 and contribute to the development of insulin resistance [27,28]. In accordance with its inhibitory effect on JNK activity, Asb-4 reduced the phosphosrylation of IRS-1 at serine 307.
Hirosumi et al reported that JNK activity is elevated in obesity and that suppression of JNK1 results in decreased adiposity and improved insulin sensitivity [24]. Prada et al reported that JNK activity was elevated in the hypothalamus of obese rats consuming a high fat diet [30]. Our studies demonstrated that insulin stimulates JNK activity in 3T3 L1 cells. This observation is consistent with studies in various of cell types and tissues [31–34]. JNK has also been reported to be a negative feedback regulator of insulin action [34]. Mediation of JNK activity by Asb-4 provides another mechanism by which cellular insulin signaling may be regulated.
5. Conclusion
The present study demonstrates that Asb-4 interacts with GPS1 and inhibits JNK activity.
Acknowledgments
Supported by NIH 5RO1 DK054032–09, 5R01 DK043225, the University of Michigan Gastrointestinal Peptide Research Center (NIH 2P30DK34933). We thank Dr. Yi Cai and Dr. Deepak Nihalani for their advice for the assay of JNK activity.
Abbreviations
- Asb-4
ankyrin repeat and SOCS box-containing protein 4
- Asb-4/Δsb
Asb-4 minus its SOCS box
- GPS1
G-protein pathway suppressor 1
- CSN1
COP9 signalosome, subunit 1
- ARC
arcuate nucleus
- POMC
pro-opiomelanocortin
- NPY
neuropeptide Y
- QDO
quadruple dropout medium
- RT-PCR
reverse transcription polymerase chain reaction
- SOCS
suppressor of cytokine signaling
- HEK293 cell
human embryonic kidney 293 cell
- Co-IP
co-immunoprecipitation
- JNK
c-Jun NH2-terminal kinase
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- Kd
kilodaltons
- EGFP
enhanced green fluorescent protein
- DAPI - 4′
6-diamidino-2-phenylindole, dihydrochloride
- IRS-1
insulin receptor substrate 1
- SOCS1
suppressor of cytokine signaling 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kile BT, Schulman BA, Alexander WS, Nicola NA, Martin HM, Hilton DJ. Trends Biochem Sci. 2002;27:235. doi: 10.1016/s0968-0004(02)02085-6. [DOI] [PubMed] [Google Scholar]
- 2.Kile BT, Viney EM, Willson TA, Brodnicki TC, Cancilla MR, Herlihy AS, Croker BA, Baca M, Nicola NA, Hilton DJ, Alexander WS. Gene. 2000;258:31. doi: 10.1016/s0378-1119(00)00402-9. [DOI] [PubMed] [Google Scholar]
- 3.Li JY, Kuick R, Thompson RC, Misek DE, Lai YM, Liu YQ, Chai BX, Hanash SM, Gantz I. J Neuroendocrinol. 2005;17:394. doi: 10.1111/j.1365-2826.2005.01317.x. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Silverstein FS, Skoff R, Barks JD. Pediatr Res. 2002;51:25. doi: 10.1203/00006450-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Cai Y, Lechner MS, Nihalani D, Prindle MJ, Holzman LB, Dressler GR. J Biol Chem. 2002;277:1217. doi: 10.1074/jbc.M109663200. [DOI] [PubMed] [Google Scholar]
- 6.Rui L, Yuan M, Frantz D, Shoelson S, White MF. J Biol Chem. 2002;277:42394. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 7.Tsuge T, Matsui M, Wei N. J Mol Biol. 2001;305:1. doi: 10.1006/jmbi.2000.4288. [DOI] [PubMed] [Google Scholar]
- 8.Kohroki J, Fujita S, Itoh N, Yamada Y, Imai H, Yumoto N, Nakanishi T, Tanaka K. FEBS Lett. 2001;505:223. doi: 10.1016/s0014-5793(01)02829-0. [DOI] [PubMed] [Google Scholar]
- 9.Boengler K, Pipp F, Fernandez B, Richter A, Schaper W, Deindl E. Biochem Biophys Res Commun. 2003;302:17. doi: 10.1016/s0006-291x(03)00095-0. [DOI] [PubMed] [Google Scholar]
- 10.McDaneld TG, Hancock DL, Moody DE. Physiol Genomics. 2003;16:275. doi: 10.1152/physiolgenomics.00127.2003. [DOI] [PubMed] [Google Scholar]
- 11.Wilcox A, Katsanakis KD, Bheda F, Pillay TS. J Biol Chem. 2004;279:38881. doi: 10.1074/jbc.M406101200. [DOI] [PubMed] [Google Scholar]
- 12.Wei N, Deng XW. Plant cell. 1992;4:1507. doi: 10.1105/tpc.4.12.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumeister W, Walz J, Zühl F, Seemüller E. Cell. 1998;92:367. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 14.Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, Finley D. Cell. 1998;94:615. doi: 10.1016/s0092-8674(00)81603-7. [DOI] [PubMed] [Google Scholar]
- 15.Seeger M, Kraft R, Ferrell K, Bech-Otschir D, Dumdey R, Schade R, Gordon C, Naumann M, Dubiel W. FASB J. 1998;12:469. [PubMed] [Google Scholar]
- 16.Wei N, Tsuge T, Serino G, Dohmae N, Takio K, Matsui M, Deng XW. Curr Biol. 1998;8:919. doi: 10.1016/s0960-9822(07)00372-7. [DOI] [PubMed] [Google Scholar]
- 17.Schwechheimer C, Serino G, Callis J, Crosby WL, Lyapina S, Deshaies RJ, Gray WM, Estelle M, Deng XW. Science. 2001;292:1379. doi: 10.1126/science.1059776. [DOI] [PubMed] [Google Scholar]
- 18.Kawakami T, Chiba T, Suzuki T, Iwai K, Yamanaka K, Minato N, Suzuki H, Shimbara N, Hidaka Y, Osaka F, Omata M, Tanaka K. EMBO J. 2001;20:4003. doi: 10.1093/emboj/20.15.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uhle S, Medalia O, Waldron R, Dumdey R, Henklein P, Bech-Otschir D, Huang X, Berse M, Sperling J, Schade R, Dubiel W. EMBO J. 2003;22:1302. doi: 10.1093/emboj/cdg127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun Y, Wilson MP, Majerus PW. J Biol Chem. 2002;277:45759. doi: 10.1074/jbc.M208709200. [DOI] [PubMed] [Google Scholar]
- 21.Spain BH, Bowdish KS, Pacal AR, Staub SF, Koo D, Chang CY, Xie W, Colicelli J. Mol Cell Biol. 1996;16:6698. doi: 10.1128/mcb.16.12.6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. Cell. 1994;76:1025. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 23.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. Nature. 1994;369:156. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 24.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. Nature. 2002;420:333. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 25.White MF. Am J Physiol Endocrinol Metab. 2002;283:E413–422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 26.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, Whiteand MF, Spiegelman BM. Science. 1996;271:665. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 27.Aguirre V, Uchida T, Yenush L, Davis R, White MF. J Biol Chem. 2000;275:9047. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 28.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. J Biol Chem. 2002;277:1531. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 29.Paz K, Hemi R, LeRoith D, Karasik A, Elhanany E, Kanety H, Zick Y. J Biol Chem. 1997;272:2991. doi: 10.1074/jbc.272.47.29911. [DOI] [PubMed] [Google Scholar]
- 30.Prada PO, Zecchin HG, Gasparetti AL, Torsoni MA, Ueno M, Hirata AE, Corezola do Amaral ME, Hoer NF, Boschero AC, Abdalla Saad MJ. Endocrinology. 2005;146:1576. doi: 10.1210/en.2004-0767. [DOI] [PubMed] [Google Scholar]
- 31.Miller BS, Shankavaram UT, Horney MJ, Gore ACS, Kurtz DT, Rosenzweig SA. Biochemistry. 1996;35:8769. doi: 10.1021/bi952651r. [DOI] [PubMed] [Google Scholar]
- 32.Desbois-Mouthon C, Blivet-Van Eggelpoel MJ, Auclair M, Cherqui G, Capeau J, Caron M. Biochem Biophys Res Commun. 1998;243:765. doi: 10.1006/bbrc.1998.8181. [DOI] [PubMed] [Google Scholar]
- 33.Standaert ML, Bandyopadhyay G, Antwi EK, Farese RV. Endocrinology. 1999;140:2145. doi: 10.1210/endo.140.5.6699. [DOI] [PubMed] [Google Scholar]
- 34.Lee YH, Giraud J, Davis RJ, White MF. J Biol Chem. 2003;278:2896. doi: 10.1074/jbc.M208359200. [DOI] [PubMed] [Google Scholar]