

Abstract
Purpose
We describe the clinical phenotype of a Mexican family segregating Duane syndrome as an autosomal dominant trait linked to chromosome 2q31 (DURS2) and previously reported to harbor a heterozygous α2-chimaerinmissense mutation.
Methods
A five-generation Mexican family was analyzed. Ten affected subjects were available for clinical examination. Participating subjects were tested for visual acuity, ocular alignment by prism cover testing, ocular ductions and versions, and globe retraction. In children, alignment was measured with the Krimsky test in cardinal positions of gaze.
Results
Ten cases were included, 6 females and 4 males. Five cases presented with bilateral and 5 with unilateral Duane syndrome. Right side was the most commonly affected side on unilateral cases. Five cases exhibited exotropia, 4 esotropia, and 1 hypotropia. Seven patients had important limitation of abduction; two, moderate limitation. Four patients had mild adduction limitation and 4 had moderate limitation. No additional anomalies such as fourth (trochlear) nerve palsy, blepharoptosis, or dense amblyopia, reported in previous Duane syndrome families, were observed. All 3 cases that exhibited vertical dysfunction had upgaze limitation. One instance of nonpenetrance was recorded.
Conclusions
Considerable intrafamilial clinical variability was observed in this Duane syndrome pedigree carrying a α2-chimaerin mutation. The presence of bilateral involvement and associated vertical movements, commonly observed in this and others DURS2 families, could suggest the occurrence of CHN1 mutations as the source of the disease in isolated or familial DURS cases.
Introduction
Duane retraction syndrome is a congenital ocular motility disorder that accounts for up to 4% of all strabismus cases. It is considered the most common type of congenital aberrant ocular innervation.1,2 Duane syndrome can be caused by absence or hypoplasia of both the abducens nucleus and nerve with anomalous innervation of its target, the lateral rectus muscle, by a branch of the oculomotor nerve. This in turn leads to absent abduction or adduction and narrowing of the palpebral fissure, with retraction of the globe on attempted adduction.3–5 Associated ocular anomalies in subjects with Duane syndrome can include nystagmus, anisocoria, ptosis, optic nerve colobomas, epibulbar dermoids, crocodile tears, and aniridia.6–11 Systemic malformations can be found in Duane syndrome patients, particularly skeletal (limb hypoplasia, polydactyly, hypoplastic or absent radius and/or thumb), vertebral (scoliosis, spina bifida, “butterfly” vertebrae, Klippel-Feil anomaly), genitourinary (renal agenesis, vesicoureteral reflux), and cardiac (patent ductus arteriosus, auricular septal defect) defects.7,9,12–14 However, a distinction should be made between true etiologic associations and coincidental findings. Well-known syndromic entities associating Duane syndrome include Okihiro (Duane syndrome and radial ray defects), Wildervanck (Duane syndrome, Klippel-Feil anomaly, and deafness), Moebius (congenital paresis of facial and abducens cranial nerves) and Townes-Brocks (ear, limb, anal, renal and heart anomalies) syndromes, among others. Approximately 90% of nonsyndromic Duane syndrome cases occur sporadically,15,16 with predominant affectation for females and the left eye.7,17 The remaining 10% of Duane syndrome subjects have familial antecedents of the disorder and these inherited cases are commonly bilateral and have associated vertical movement abnormalities.18–20 Previously, genetic linkage studies in several of these pedigrees allowed the assignment of a locus for autosomal dominant Duane syndrome, named DURS2, at chromosome 2q31.20–22 Recently, Miyake and colleagues23 provided genetic, in vitro, and in vivo evidence that mutations in the CHN1 gene, encoding α2-chimaerin, are responsible for the 2q31-linked form of Duane syndrome. Alpha-2-chimaerin is a Rac guanosine triphosphatase-activating protein (RacGAP) signaling protein implicated in the pathfinding of corticospinal axons.
The purpose of this study is to describe the clinical phenotype resulting from a mutation in the α2-chimaerin gene in a Mexican family that segregates DURS2 as an autosomal dominant trait.23 This is the first detailed report regarding phenotypic features in Duane syndrome subjects carrying an α2-chimaerin mutation.
Report of Cases
A five-generation Mexican family (Figure 1) segregating Duane syndrome as an autosomal dominant trait was analyzed. All subjects were of Mexican Mestizo origin and without history of additional genetic or inherited diseases. Of a total of 15 affected subjects identified, 10 were available for clinical examination. A number of unaffected relatives were also examined. Participating subjects were tested for visual acuity, ocular alignment, ocular ductions and versions, and globe retraction. Ductions were assessed by two independent examiners (CM-C and VK-J). Ocular alignment was measured by prism cover testing with the subject viewing a distant object in the seven cardinal positions of gaze. In children, alignment was measured with the Krimsky test in cardinal positions of gaze. When possible, Titmus stereo test was performed to evaluate binocular visual function. None of the subjects exhibited biomicroscopic or fundoscopic anomalies. Significant ametropia was not observed in any case. Table 1 summarizes the clinical findings in affected subjects. The detailed results of clinical examination in each subject are listed in e-Supplement 1 (available at jaapos.org).
FIG 1.
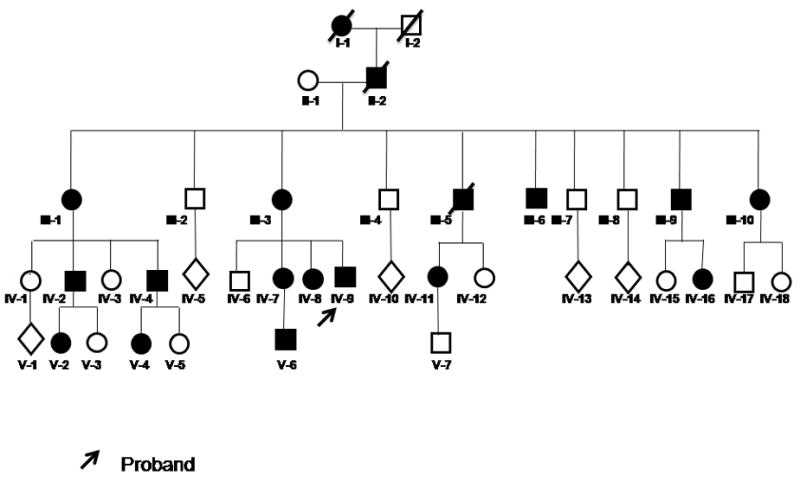
Genealogical tree of a five-generation Mexican family segregating DURS as an autosomal dominant trait. Black symbols denote affected individuals. Arrowed subject is the propositus.
Table 1.
Subjects and clinical examination
| Case | Gender | Age(years) | Deviation | Limitation | Previous surgery | Affected side | |
|---|---|---|---|---|---|---|---|
| ABD | ADD | ||||||
| 1 | M | 15 | XT | ++ (RE) | ++ (RE) | No | Bilateral |
| + (LE) | ++ (LE) | ||||||
|
| |||||||
| 2 | F | 28 | HT | ++ | Yes | Right | |
|
| |||||||
| 3 | F | 30 | XT | ++ (RE) | + (RE) | No | Bilateral |
| ++ (LE) | |||||||
|
| |||||||
| 4 | F | 26 | ET | +++ (RE) | ++ (RE) | No | Bilateral |
| +++ (LE) | + (LE) | ||||||
|
| |||||||
| 5 | F | 39 | XT | + (RE) | + (RE) | Yes | Bilateral |
| + (LE) | |||||||
|
| |||||||
| 6 | M | 45 | ET | ++ | + | Yes | Right |
|
| |||||||
| 7 | F | 59 | XT | ++ | ++ | Yes | Right |
|
| |||||||
| 8 | F | 50 | ET | ++ | No | Right | |
|
| |||||||
| 9 | M | 4 | XT | ++ (RE) | + (RE) | No | Bilateral |
| ++ (LE) | + (LE) | ||||||
|
| |||||||
| 10 | M | 5 | ET | ++ | + | Yes | Left |
ET, esotropia; XT, exotropia; HT, hypotropia; RE, right eye; LE, left eye; ABD, abduction; ADD, adduction
Previous molecular analysis in affected subjects from this family reported a 378 T>G transversion in exon 6 of the CHN1 gene, predicting an I126M (isoleucine to methionine) missense mutation at residue 126 of α2-chimaerin (Pedigree RF in reference 23).
Discussion
Duane syndrome has an estimated prevalence of about 1 in 1000 in the general population. Earlier electrophysiologic,24 post mortem,4,25 and magnetic resonance imaging (MRI)26–28 data have established that the disorder can be caused by deficient abducens (sixth) nerve innervation of the lateral rectus muscle. In addition, however, recent studies using high-resolution, multipositional MRI showed that some 2q31-linked Duane syndrome subjects (DURS2), subsequently found to harbor CHN1 mutations, also have hypoplastic oculomotor nerves and small oculomotor-innervated muscles,19–23 indicating that, at least in some cases, Duane syndrome is a diffuse congenital cranial dysinnervation anomaly not limited to the abducens cranial nerve.19 Several genes have been associated to syndromic forms of Duane syndrome, including SALL4,29 and HOXA1.30
Recently, Miyake and colleagues23 identified heterozygous gain of function mutations in the CHN1 gene as the cause of the 2q31-linked Duane syndrome in 7 families from different ethnic backgrounds. In vitro and in vivo analysis indicated that these mutations hyperactivate α2-chimaerin RacGAP activity or enhance α2-chimaerin membrane translocation.23 Furthermore, in ovo expression of mutant CHN1 alters the development of ocular motor axons demonstrating that hyperactivated α2-chimaerin results in aberrant cranial motor neuron development.23
Here, we have described the clinical characteristics of a Duane syndrome Mexican family carrying a α2-chimaerin I126M amino acid substitution.23 In this family, five generations were known to be affected, with a total of 18 diseased individuals, of whom 15 were alive and 10 were available for clinical examination. Individual III-9 carries the I126M CHN1 mutation but detailed ocular movement examination failed to demonstrate any abnormality of ocular motility. Consistent with this observation, lack of penetrance has been reported in other DURS2 pedigrees harboring CHN1 mutations.19,23
Remarkably, of the seven original DURS2 pedigrees reported to harbor CHN1 mutations, three were of Mexican descent. The other two pedigrees, FY and IJ,23 harbor different α2-chimaerin mutations resulting in L20F and P252Q amino acid substitutions, respectively. The occurrence of distinct CHN1 mutations in three DURS2 Mexican pedigrees argues against a founder mutation in this ethnic group. In this sense, no CHN1 mutational hot-spots have been recognized in DURS2.23
A common finding in these three Mexican DURS2 pedigrees is the cosegregation of Duane syndrome types 1 and 3 but not type 2 (isolated limitation of adduction). Interestingly, we found a 50% incidence of unilateral Duane syndrome (5 of 10) in affected members of pedigree RF, and four of these involved the right side. All but one of the unilateral cases had history of previous ocular surgeries and this factor could be related to the high proportion of unilateral Duane syndrome. However, as most surgeries were performed during childhood and in other institutions, this probability cannot be addressed. In contrast to the high proportion of unilateral cases in the present family, only 1 of 4 affected subjects of pedigree FY had unilateral (left) Duane syndrome19 and only 1 of 25 affected subjects of pedigree IJ exhibited unilateral (left) Duane syndrome.18
In the present pedigree, RF, no affected individuals had fourth (trochlear) nerve palsy, blepharoptosis, or dense amblyopia. However, 2 bilateral cases (3 and 4) exhibited amblyopia (ie, difference of vision between both eyes of≥2 lines of visual acuity). In contrast, in pedigree FY, one subject with bilateral Duane syndrome had no right eye retraction, three had limitation of vertical gaze, and one had blepharoptosis. No instance of dense amblyopia was observed in this pedigree.19 In pedigree IJ, there was a high incidence of strabismus and amblyopia, which are uncommon in unilateral Duane syndrome, as well as a striking degree of variability in the amount of restriction and vertical dysfunction.18 Additional neuro-ophthalmologic anomalies were observed in a number of subjects including trochlear nerve palsy and ptosis. No instances of hypotropia were recorded in pedigrees IJ and FY.18,19 Interestingly, if compared with cases from the literature, the degree of strabismus in bilateral cases in the pedigree RF, reported here, is small.31
DURS2 interfamilial phenotypic differences could be explained by distinct effects of a given CHN1 mutation, with some mutations affecting the development of the trochlear and oculomotor nerves more than others. The marked intrafamilial phenotypic differences among subjects carrying the same α2-chimaerin mutation, however, favor genetic or environmental modifiers.
In conclusion, we present the clinical description of individuals affected with Duane syndrome in a Mexican family in which a α2-chimaerin mutation was identified as the source of the disease. Overall, the clinical features of this family were similar to those described in other familial Duane syndrome cases that have been shown recently to be caused by mutations in α2-chimaerin, including a subset of affected family members with bilateral involvement and vertical movement abnormalities.18–20 Considerable intrafamilial clinical variability and one case of non-penetrance were recorded. Although more studies are needed to establish if a genotype-phenotype correlation exists, we suggest that the presence of bilateral involvement and associated vertical movements in isolated or familial Duane syndrome cases could suggest the occurrence of CHN1 mutations as the source of the disease.
Supplementary Material
FIG 2.
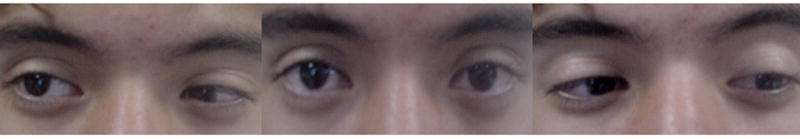
Bilateral Duane’s syndrome. Index case (IV-9 in Figure 1): left panel: right version; center panel: primary position of gaze; right panel: left version. Note palpebral narrowing, eye retraction, and mild limitation during abduction in both eyes.
Acknowledgments
This work was supported, in part, by NIH grant EY015298 to ECE.
Footnotes
The authors have no financial conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Freedman HL, Kushner BJ. Congenital ocular aberrant innervation—new concepts. J Pediatr Ophthalmol Strabismus. 1997;34:10–16. doi: 10.3928/0191-3913-19970101-04. [DOI] [PubMed] [Google Scholar]
- 2.DeRespinis PA, Caputo AR, Wagner RS, Guo S. Duane’s retraction syndrome. Surv Ophthalmol. 1993;38:257–88. doi: 10.1016/0039-6257(93)90077-k. [DOI] [PubMed] [Google Scholar]
- 3.Duane A. Congenital deficiency of abduction, associated with impairment of adduction, retraction movements, contraction of the palpebral fissure and oblique movements of the eye. Arch Ophthalmol. 1905;34:133–59. doi: 10.1001/archopht.1996.01100140455017. [DOI] [PubMed] [Google Scholar]
- 4.Hotchkiss MG, Miller NR, Clark AW, Green WR. Bilateral Duane’s retraction syndrome. A clinical-pathologic case report. Arch Ophthalmol. 1980;98:870–04. doi: 10.1001/archopht.1980.01020030864013. [DOI] [PubMed] [Google Scholar]
- 5.Raab EL. Clinical features of Duane’s syndrome. J Pediatr Ophthalmol Strabismus. 1986;23:64–8. doi: 10.3928/0191-3913-19860301-05. [DOI] [PubMed] [Google Scholar]
- 6.Denslow GT, Sims M. Duane’s retraction syndrome associated with optic nerve hypoplasia. J Pediatr Ophthalmol Strabismus. 1980;17:26–8. doi: 10.3928/0191-3913-19800101-07. [DOI] [PubMed] [Google Scholar]
- 7.Engle EC. The Genetics of Strabismus: Duane, Mobius, and Fibrosis syndromes. In: Traboulsi EI, editor. Genetic Diseases of the Eye. New York (NY): Oxford University Press; 1998. pp. 477–512. [Google Scholar]
- 8.Kansal S, Miller M. Bilateral Duane syndrome with bilateral congenital glaucoma. J AAPOS. 2001;5:325–6. doi: 10.1067/mpa.2001.117569. [DOI] [PubMed] [Google Scholar]
- 9.Pfaffenbach DD, Cross HE, Kearns TP. Congenital anomalies in Duane’s retraction syndrome. Arch Ophthalmol. 1972;88:635–9. doi: 10.1001/archopht.1972.01000030637013. [DOI] [PubMed] [Google Scholar]
- 10.Zhang F. Clinical features of 201 cases with Duane’s retraction syndrome. Chin Med J (Engl) 1997;110:789–91. [PubMed] [Google Scholar]
- 11.Khan AO, Aldahmesh M. Bilateral Duane syndrome and bilateral aniridia. J AAPOS. 2006;10:273–4. doi: 10.1016/j.jaapos.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Cross HE, Pfaffenbach DD. Duane’s retraction syndrome and associated congenital malformations. Am J Ophthalmol. 1972;73:442–50. doi: 10.1016/0002-9394(72)90074-8. [DOI] [PubMed] [Google Scholar]
- 13.Kadayifcilar S, Aydin P, Oto S. A case of Duane’s retraction syndrome with multiple congenital malformations. Eur J Ophthalmol. 1997;7:193–5. doi: 10.1177/112067219700700213. [DOI] [PubMed] [Google Scholar]
- 14.Stoll C, Alembik Y, Dott B. Association of Duane anomaly with mental retardation, cardiac and urinary tract abnormalities: A new autosomal recessive condition? Ann Genet. 1994;37:207–9. [PubMed] [Google Scholar]
- 15.Kirkham TH. Inheritance of Duane’s syndrome. Br J Ophthalmol. 1970;54:323–9. doi: 10.1136/bjo.54.5.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh P, Patnaik B. Heredity in Duane’s syndrome. Acta Ophthalmol (Copenh) 1971;49:103–10. [PubMed] [Google Scholar]
- 17.Khan AO, Oystreck DT, Wilken K, Akbar F. Duane retraction syndrome on the Arabian Peninsula. Strabismus. 2007;15:205–8. doi: 10.1080/09273970701632023. [DOI] [PubMed] [Google Scholar]
- 18.Chung M, Stout JT, Borchert MS. Clinical diversity of hereditary Duane’s retraction syndrome. Ophthalmology. 2000;107:500–503. doi: 10.1016/s0161-6420(99)00090-1. [DOI] [PubMed] [Google Scholar]
- 19.Demer JL, Clark RA, Lim KH, Engle EC. Magnetic resonance imaging evidence for widespread orbital dysinnervation in dominant Duane’s retraction syndrome linked to the DURS2 locus. Invest Ophthalmol Vis Sci. 2007;48:194–202. doi: 10.1167/iovs.06-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engle EC, Andrews C, Law K, Demer JL. Two pedigrees segregating Duane’s retraction syndrome as a dominant trait map to the DURS2 genetic locus. Invest Ophthalmol Vis Sci. 2007;48:189–93. doi: 10.1167/iovs.06-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Appukuttan B, Gillanders E, Juo SH, Freas-Lutz D, Ott S, Sood R, et al. Localization of a gene for Duane retraction syndrome to chromosome 2q31. Am J Hum Genet. 1999;65:1639–46. doi: 10.1086/302656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans JC, Frayling TM, Ellard S, Gutowski NJ. Confirmation of linkage of Duane’s syndrome and refinement of the disease locus to an 8.8-cM interval on chromosome 2q31. Hum Genet. 2000;106:636–8. doi: 10.1007/s004390000311. [DOI] [PubMed] [Google Scholar]
- 23.Miyake N, Chilton J, Psatha M, Cheng L, Andrews C, Chan WM, et al. Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane’s retraction syndrome. Science. 2008;321:839–43. doi: 10.1126/science.1156121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber A. Electrophysiology of the retraction syndromes. Br J Ophthalmol. 1974;58:293–300. doi: 10.1136/bjo.58.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller NR, Kiel SM, Green WR, Clark AW. Unilateral Duane’s retraction syndrome (type 1) Arch Ophthalmol. 1982;100:1468–72. doi: 10.1001/archopht.1982.01030040446016. [DOI] [PubMed] [Google Scholar]
- 26.Parsa CF, Grant E, Dillon WP, Jr, du Lac S, Hoyt WF. Absence of the abducens nerve in Duane syndrome verified by magnetic resonance imaging. Am J Ophthalmol. 1998;125:399–401. doi: 10.1016/s0002-9394(99)80158-5. [DOI] [PubMed] [Google Scholar]
- 27.Ozkurt H, Basak M, Oral Y, Ozkurt Y. Magnetic resonance imaging in Duane’s retraction syndrome. J Pediatr Ophthalmol Strabismus. 2003;40:19–22. doi: 10.3928/0191-3913-20030101-07. [DOI] [PubMed] [Google Scholar]
- 28.Kim JH, Hwang JM. Presence of the abducens nerve according to the type of Duane’s retraction syndrome. Ophthalmology. 2005;112:109–111. doi: 10.1016/j.ophtha.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 29.Al-Baradie R, Yamada K, St Hilaire C, Chan W-M, Andrews C, McIntosh N, et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am J Hum Genet. 2002;71:1195–9. doi: 10.1086/343821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tischfield MA, Bosley TM, Salih MAM, Alorainy IA, Sener EC, Nester MJ, et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nature Genet. 2005;37:1035–7. doi: 10.1038/ng1636. [DOI] [PubMed] [Google Scholar]
- 31.Khan AO, Oystreck D. Clinical characteristics of bilateral Duane syndrome. J AAPOS. 2006;10:198–201. doi: 10.1016/j.jaapos.2006.02.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.