

Abstract
Objective
The transcription factors, peroxisome proliferator-activated receptors (PPAR) alpha (α) and gamma (γ), which are involved in lipid and glucose homeostasis, also exert modulatory actions on vascular cells where they exhibit anti-inflammatory and anti-proliferative properties. Hence PPAR agonists potentially can affect atherogenesis both via metabolic effects and direct effects on the vessel wall. We tested whether the dual PPAR-α/γ agonist, tesaglitazar (TZ), would reduce atherosclerosis in a non-diabetic, atherosclerosis-prone mouse model, independent of effects on plasma lipids.
Methods and results
Low-density lipoprotein receptor deficient (LDLr −/−) mice were fed a Western type diet consisting of 21% butterfat and 0.15% cholesterol, with or without TZ 0.5 μmol/kg of diet, for 12 weeks. TZ reduced atherosclerosis in the female, but not male, LDLr −/− mice without affecting cholesterol and triglyceride levels, HDL binding to biglycan, or the inflammatory markers serum amyloid A (SAA) and serum amyloid P (SAP). TZ also decreased adiposity in both genders.
Conclusions
TZ reduced atherosclerosis in the female LDLr−/− mice via lipid-independent mechanisms, probably at least in part by direct actions on the vessels. The body weight changes in these mice are different from the effects of dual PPAR agonists seen in humans.
Keywords: Atherosclerosis, dual PPAR agonist, LDL receptor deficient mice, tesaglitazar
Introduction
PPARs are nuclear receptors present in several cell types that regulate multiple genes involved in lipid metabolism, glucose homeostasis and cellular differentiation. PPARs also inhibit expression of several pro-inflammatory genes, partly by interfering with signal transduction pathways like that of nuclear factor-κB (NF-κB) [1]. There are three known isotypes, PPAR alpha, beta/delta and gamma, which have different patterns of tissue expression. PPAR-α and PPAR-γ are the two best described isotypes. PPAR-α is expressed predominantly in the liver, heart and muscle and controls the expression of genes that regulate lipid metabolism [2]. PPAR-γ is expressed most abundantly in adipose tissue and is very important in regulating glucose homeostasis and adipogenesis [2]. All PPARs are expressed in endothelial cells (EC) and vascular smooth muscle cells (VSMC) where they can modulate inflammatory and atherogenic properties [1]. Both PPAR-α and PPAR-γ agonists have been shown to inhibit macrophage foam cell formation by different mechanisms [3]. Because of their direct effects on vascular cells and their beneficial effects on metabolic abnormalities that are known to promote atherogenesis, PPARs are an attractive target for anti-atherogenic therapy.
Fibrates, which are PPAR-α agonists, are the best currently available agents to lower triglycerides, while they also have a modest effect in raising high density lipoproteins (HDL) [4]. The PPAR-γ agonists, thiazolidinediones, are used in the management of diabetes by virtue of their insulin-sensitizing effect. Agents that are dual agonists for both PPAR-α and γ have been in development with the hope that they would offer the therapeutic advantages of both fibrates and thiazolidinediones, including their putative anti-inflammatory and direct vascular effects. It is conceivable that the complex interactions involved in PPAR activation may produce outcomes from dual activation that would not ordinarily be predicted from known effects of agonists of either PPAR isotypes.
In this study, we tested the effect of TZ, a dual PPAR-α and γ agonist, on atherosclerosis in an atherosclerosis-prone mouse model. We used LDLr −/− mice, a model that has been shown to develop marked atherosclerosis when fed a western-type diet. After 12 weeks of TZ administration, atherosclerosis was reduced in female, but not male, mice without any effect on the lipid profile.
Methods
Animals, diets and specimens
Six to eight weeks old male and female LDLr−/− mice, bred into a C57BL/6 background, were purchased from The Jackson Laboratory (Stock Number: 002207, Bar Harbor, ME). They were fed either a Western-type diet containing 21% butterfat and 0.15% cholesterol (diet # 88137, Harlan Teklad) or the Western-type diet supplemented with TZ (0.5 μmol/kg diet) added by Harlan Teklad (diet # TD 04155) for 12 weeks. The concentration of TZ in the diet was based on advice from scientists from AstraZeneca, the makers of TZ, who have extensive experience with the drug in their own rodent studies. The mice were maintained in a temperature-controlled (25°C), modified specific pathogen-free facility with a strict 12-hour light/dark cycle and given free access to food and water. They were housed in cages with 3 animals per cage and were weighed every week. At the end of the second and sixth weeks, a crude assessment of food intake was made by measuring the rate of food disappearance per cage over a 3-day period and dividing it by the number of mice (three) in the cage. Three mice were lost during the study and the number that completed the study were: control females 14, TZ females 15, control males 13, TZ males 15. This project was approved by the Animal Care and Use Committee of the University of Washington.
Blood samples, after a 4-hour fast, were obtained via the retro-orbital sinus at baseline, after 6 weeks of diet feeding, and just before the mice were euthanized at 12 weeks. The blood was collected into tubes containing <1% sodium EDTA and then centrifuged to obtain plasma. The mice were euthanized by cervical dislocation. The heart and the entire aorta were removed and placed in formalin. The inguinal, abdominal (perigonadal), and retroperitoneal fat depots were removed and weighed. The liver also was removed and weighed.
Glucose
Blood glucose was measured using the OneTouch® Profile Meter.
Plasma lipids and lipoprotein profiles
Plasma from individual mice was analyzed for total cholesterol and triglyceride concentrations with colorimetric assay kits obtained from Diagnostic Chemicals Limited (cholesterol) and Roche Diagnostics (triglycerides). The lipoprotein distribution was characterized by analyzing pooled plasma samples from each diet and gender group (3 pools of 4 or 5 from each group) by fast-phase liquid chromatography (FPLC). Briefly, 100 μL of plasma was chromatographed on a Superose 6 HR10/30 column (Amersham Biotech) and eluted with phosphate-buffered saline, 0.02% sodium azide, pH 7.4. Multiple 0.5-ml fractions were collected and fractions 11 to 40 were analyzed for cholesterol content in duplicate. VLDL and IDL were in fractions 15 to 20, LDL in fractions 21 to 27, and HDL in fractions 28 to 34.
High-density lipoprotein (HDL) and biglycan binding studies
HDL (d = 1.063 to 1.21 g/ml) was isolated by density-gradient ultracentrifugation from pooled plasma from 4 or 5 mice (3 pools from each diet and gender group) and tested for its ability to bind to purified 35SO4-labeled biglycan with an electrophoretic gel mobility shift assay, described by Hurt-Camejo et al [5]. Biglycan was prepared as described previously [6]. It is an extracellular proteoglycan and was chosen because of its putative role in trapping lipoproteins in atherosclerotic lesions [7, 8].
Measurement of serum amyloid A (SAA) and serum amyloid P (SAP) levels
SAA and SAP levels in plasma were measured by ELISA using the technique previously described [9]. Briefly, samples and standards (from Alpha Diagnostic International, Texas) in triplicates were allowed to bind to 96-well ELISA plates (Costar) for 2 hours at 37°C. Nonspecific sites were blocked with 3% (wt/vol) BSA in PBS and the primary antibody (SAP from Alpha Diagnostic International, Texas; SAA a gift from Godfrey Getz) was added and incubated for 1.5 to 2 hours at 37°C. Plates were then incubated in the presence of anti-rabbit horseradish peroxidase-conjugated IgG (Boehringer-Mannheim, diluted 1:4000 in blocking buffer) for 1 hour at room temperature. The color reaction was developed with o-phenylene diamine as the substrate (Sigma). Sulphuric acid was added to terminate the reaction and the plates were read at wavelengths 490 to 405 nm.
Quantification of atherosclerosis
Atherosclerosis was evaluated by analyzing serial sections at the aortic root and by en face analysis of the aortic arch. Lesion sizes were quantified in the aortic root essentially as described [10], with modifications. In brief, the upper sections of the hearts were fixed overnight in 10% neutral-buffered formalin and embedded in paraffin the following day. Aortic root lesion area was quantified, beginning at the termination of the aortic valve and spanning 400 μm of the ascending aorta. Every other section (5 μm thick) through the aortic root was measured for analysis. A subset of 5 sections from each animal spanning the region were stained with Movat’s stain to quantify lesion area (Image Pro Plus, Media Cybernetics). The findings were confirmed by another investigator who was blinded to the treatment groups. For the en face analysis, aortas were cut longitudinally, pinned flat on black wax, and photographed. The length of the aorta studied was 16.9 mm from the beginning of the ascending aorta, which almost spanned the entire aorta in all the animals. Total surface area and lesion area in the studied sections of aortas were quantified.
Lesion composition
Lesion characteristics were quantified using computer-assisted morphometry (ImagePro Plus, Version 4.0, Media Cybernetics) by an investigator blinded to gender/treatment group, and expressed as percent of lesion area. Glycosaminoglycan areas were determined from Movat’s-stained sections and defined as areas of blue staining. Necrotic areas were determined from Movat’s-stained sections and defined as areas of acellularity with disrupted extracellular matrix. Collagen area was determined from picrosirius red-stained sections viewed under polarized light.
For immunohistochemistry, apolipoprotein A-I (apoA-I) was detected using a goat polyclonal anti-apoA-I antiserum (titer = 1:1,500, kind gift of Dr. John Oram, University of Washington) and macrophages were detected using rat monoclonal antibody, Mac2 (titer = 1:2,500, Catalog # CL8942B-3, Cedarlane Laboratories, Ltd., Burlington, ON). Tissue sections were deparaffinized with xylene and then hydrated with graded alcohols. The slides were blocked with 3% H2O2, washed with PBS, incubated in antigen retrieval solution (DAKO Target Retrieval Solution, Catalog #: S1699, Dako, Glostrup, Denmark) for 10 min, washed with PBS, incubated with the primary antibody for 60 min, and then washed again with PBS. A biotin-labeled secondary antibody, either anti-rabbit (for ApoA-I), or anti-rat (for Mac2) was applied for 30 min, followed by an avidin-biotin-peroxidase conjugate (ABC Elite, Vector Laboratories, Burlingame, CA) for 30 min. A standard peroxidase enzyme substrate, NovaRED (Catalog #: SK-4800, Vector) was added to yield a red reaction product.
Statistical analyses
Unless otherwise stated, values shown are mean ± SEM and Student’s t test was used to assess differences between means. For data sets that were not normally-distributed (lesion size and composition), values are reported as median (interquartile range) and the Mann-Whitney U test was used to assess differences in median values. Probability values < 0.05 were considered to be statistically significant.
Results
TZ treatment did not affect plasma lipid levels or cholesterol distribution profiles in LDLR-deficient mice
Six-week old LDLr−/− mice were fed for 12 weeks with a western type diet consisting of 21% butterfat and 0.15% cholesterol, with or without added TZ (0.5 μmol/kg diet). At baseline there were similar cholesterol and triglyceride levels among the groups (data not shown) with marked increases in all groups at 6 and 12 weeks. There were no differences in plasma lipids between control and TZ treated mice at 6 weeks (data not shown). At 12 weeks also, there was no significant difference in plasma cholesterol between the control and TZ treated mice (females, 35.02 ± 2.42 versus 35.61 ± 2.95 mmol/L, p = 0.88; males, 46.38 ± 2.67 versus 40.38 ± 2.25 mmol/L, p = 0.1) (Fig. 1A). Likewise, TZ treatment did not affect triglyceride levels at 12 weeks (females, 2.74 ± 0.38 versus 3.29 ± 0.44 mmol/L, p = 0.35; males, 5.70 ± 0.61 versus 6.67 ± 0.81 mmol/L, p = 0.36) (Fig. 1B).
Figure 1.

Plasma cholesterol (Fig 1A) and triglycerides (Fig 1B) in LDLr−/− mice fed a western-type diet, with (open bars) or without (closed bars) TZ, for 12 weeks. No significant difference between the groups. Values are means ± SEM. Female controls (n = 14), TZ (n = 15); Male controls (n = 13), TZ (n = 15). Size distribution of lipoprotein particles in female (Fig 1C) and male (Fig 1D) mice after 12 weeks (closed squares/circles, control mice; open squares/circles, TZ treated mice). Plasma was pooled from 4 or 5 mice in each treatment group (3 pools per group) and fractionated by FPLC. Cholesterol content was determined in each fraction. FPLC data shown represents the average cholesterol in each fraction, weighted for the number of mice in each pool.
To assess whether TZ caused changes in lipoprotein profiles, pooled 12-week plasma samples from each group were analyzed by FPLC. TZ treatment did not change the lipoprotein distribution profile in LDLr−/− mice (Figs. 1C and 1D).
TZ reduced average blood glucose at 12 weeks
As PPAR-γ agonists are insulin sensitizers used clinically in the treatment of diabetes mellitus, the effect of TZ on blood glucose was assessed in these mice. Compared to controls, TZ treated mice had significantly decreased blood glucose at 12 weeks, although they were all within the non-diabetic range (table 1).
Table 1.
Blood glucose, SAA and SAP levels at 12 weeks.
| Females | Males | |||
|---|---|---|---|---|
| Control (n=14) | TZ (n=15) | Control (n=13) | TZ (n=15) | |
| Glucose (mmol/l) | 6.22 ± 0.17 | 5.05 ± 0.28* | 6.77 ± 0.44 | 5.72 ± 0.17** |
| SAA (mg/l) | 51.2 ± 5.8 | 52 ± 10.1† (n=10) | 74.6 ± 7 (n=12) | 58.7 ± 4.1‡ |
| SAP (mg/l) | 17.5 ± 0.7 | 18.5 ± 0.7† | 17.3 ± 0.6 | 17 ± 0.9† |
Blood glucose, SAA and SAP levels at 12 weeks. Data is shown as means ± SEM. P values when compared with control;
= 0.003,
= 0.03,
= non-significant,
= 0.051.
TZ did not affect plasma levels of SAA and SAP
The effect of TZ on the inflammatory marker, SAA, was assessed. At 12 weeks, there was no significant difference in SAA levels between the control and TZ treated mice, although there was a trend towards a reduction in the TZ treated males (Table 1). Samples with SAA levels above 1000 mg/L (5 from the female TZ group and 1 from the male control group) were excluded from the analysis as outliers.
Another inflammatory marker, SAP, was measured. SAP is an acute phase reactant (APR) with structural similarities to the main human APR, C-reactive protein (CRP) [11]. CRP in mice is present only in trace amounts and is not an APR in this species [11]. At 12 weeks, there was no significant difference between the control and TZ treated mice (Table 1).
TZ reduced atherosclerosis in female, but not male, LDLr−/− mice at 12 weeks
Atherosclerosis was assessed in en face preparations of the aorta, using image analysis software, after 12 weeks. The sum of the surface areas of all atherosclerotic plaques that were seen in the aorta was calculated as a percentage of the total area of that aortic segment. Compared to controls, the TZ treated female, but not male, mice had significantly reduced atherosclerosis in the en face aortic preparations (median values of 10.7 versus 8.4 %, p = 0.018, females; 11.7 versus 11.9 %, p = 0.85, males) (fig. 2A and B).
Figure 2.

Atherosclerosis in LDLr−/− mice fed a western-type diet, with or without TZ, for 12 weeks. Shown are En Face Aortic Lesion Areas (A, B) and Aortic Sinus Lesion Areas (C, D) for Female (A, C) and Male (B, D) mice. Control groups are indicated by filled symbols and TZ groups by open symbols. Group median values are shown to the right of the median bars. Differences in group median values were analyzed using the non-parametric, Mann-Whitney test.
Atherosclerosis also was assessed in cross-sections through the aortic origin, using image analysis software. Five sections per mouse, at the same sampling levels, were analyzed and the average lesion areas obtained. Compared to controls, TZ treated female, but not male, mice had significantly reduced cross-sectional lesion areas at the aortic origin (median values of 3.5 versus 2.8 × 105 μm2/section, p = 0.009, F; 3.0 versus 3.4 × 105 μm2/section, p = 0.27, M) (fig. 2B).
TZ altered the composition of the atherosclerotic lesions in female, but not male mice
To further define the composition of atherosclerotic lesions, areas for macrophages, collagen, apoA-I (as a measure of lipoprotein deposition) and necrosis were measured and expressed as a percentage of total lesion area. In the male animals, there was no difference in any of these characteristics between control and TZ groups (Table 2). In the female TZ group, there were non-significant trends towards increased macrophage and necrotic areas, a significant decrease in collagen, and a significant increase in apo A-1, as compared to controls (table 2). There was no difference in glycosaminoglycan content of the lesions in either sex (table 2).
Table 2.
Effect of TZ on Lesion Composition
| Female | Male | |||||
|---|---|---|---|---|---|---|
| Control | TZ | P value* | Control | TZ | P value* | |
| Macrophage | 19.9 (9.8, 28.9) | 24.4 (16.5, 45.0) | 0.12 | 25 (12.2, 44.9) | 18.3 (13.3, 26.7 | 0.46 |
| Collagen | 7 (3.8, 9.4) | 4.3 (2.3, 6.2) | 0.03 | 7.7 (3.9, 11.5) | 7.6 (6.1, 13.3) | 0.58 |
| Glycosaminoglycan | 24.6 (20.0, 31.4) | 25.8 (14.6, 31.5) | 0.62 | 18.9 (13.4, 22.6) | 18.5 (15.6, 23.8) | 0.89 |
| Apo A-I | 3.7 (1.6, 5.1) | 9.6 (5.5, 15.8) | 0.0016 | 8.2 (2.4, 14.6) | 5.8 (3.6, 10.9) | 0.82 |
| Necrotic Area | 11.5 (6.7, 20.8) | 17 (13.1, 22.8) | 0.12 | 12 (6.1, 18.8) | 14.4 (10.0, 18.6) | 0.58 |
Areas containing macrophage, collagen, glycosaminoglycan, apo A-I, and necrosis, expressed as a percentages of total lesion areas. All results are expressed as Median (IQR).
Mann-Whitney U test.
TZ did not affect the binding of HDL to biglycan
Our group has shown previously that HDL is retained in lesions of LDLr−/− fed either chow diet [12, 13] or high-fat diet with or without added cholesterol [9]. Biglycan is an extracellular matrix proteoglycan that binds HDL and is present in atherosclerotic lesions [6–8]. HDL was isolated from plasma by density-gradient ultracentrifugation and tested for its ability to bind to purified 35SO4-labeled biglycan with an electrophorectic gel mobility shift assay. There was no difference, between control and TZ treated animals, in the ability of their HDL to bind to biglycan (Fig. 3). Females, 26.3 ± 3.42 versus 24.4 ± 0.35 % bound, p = 0.61; Males 43.7 ± 1.97 versus 47 ± 3.09 % bound, p = 0.42.
Figure 3.
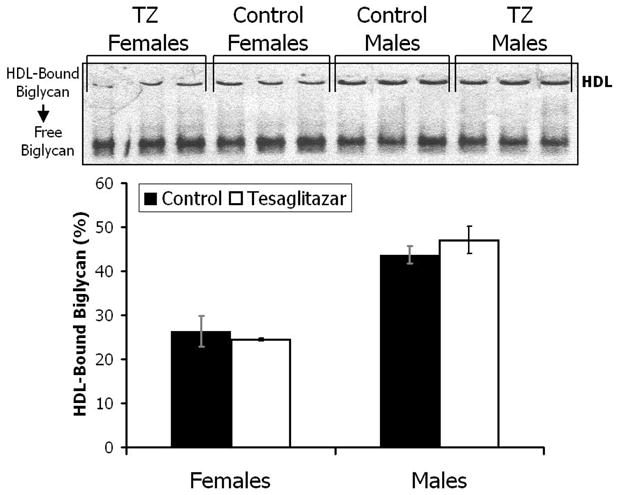
HDL was isolated from pooled plasma from 4 or 5 LDLr−/− mice (3 pools per group) fed a western-type diet, with or without TZ, for 12 weeks and tested for its ability to bind to purified 35SO4-labeled biglycan using an electrophoretic gel mobility shift assay as described in methods. The data shows the percentage of labeled biglycan that was bound to HDL from the mice. Values are means ± SEM. Closed bars, control; open bars, TZ treated.
TZ treatment resulted in reduction of white adipose tissue (WAT) mass and body weight
The effect of TZ on visceral and subcutaneous fat and body weight was assessed. The body weights were assessed in terms of percentage weight gain between baseline and the end of the 12-week study. After sacrifice of the animals, the weights of visceral (abdominal and retro-peritoneal) and subcutaneous (inguinal) WAT depots were calculated as a percentage of their total body weights. Food intake, assessed at the ends of week 2 and week 6, was not different between control and TZ treated mice (data not shown). Compared to controls and despite similar food intake, TZ treated mice gained significantly less weight, had reduced abdominal fat, reduced retro-peritoneal fat and reduced inguinal fat (figs. 4A – 4D).
Figure 4.

Body weight increase and WAT size in LDLr−/− mice fed a western-type diet, with (open bars) or without (closed bars) TZ, for 12 weeks. A) Percentage increase in body weight from baseline to 12 weeks. B, C, D) After 12 weeks, the weights of their abdominal, retro-peritoneal and inguinal white adipose tissue depots were calculated as a percentage of their total body weights. Values are means ± SEM. Female controls (n = 14), TZ (n = 15); Male controls (n = 13), TZ (n = 15).
TZ increased liver weight
The livers of the animals were weighed and calculated in terms of their percentage of total body weight (% tbw). Compared to controls, the TZ treated mice had increased liver weights (5.14 ± 0.2 versus 7.12 ± 0.3 % tbw, p < 0.001, females; 5.81 ± 0.3 versus 9.14 ± 0.3 % tbw, p < 0.001, males).
Discussion
In our study the dual PPAR-α/γ agonist TZ reduced atherosclerosis in female, but not male, LDLr−/−mice fed a western diet without affecting the plasma lipid levels or lipoprotein distribution. It also did not affect the ability of HDL to bind to biglycan, which is a vascular matrix proteoglycan implicated in the lipoprotein retention hypothesis for the mechanism by which atherosclerosis is initiated [7, 8, 14]. Therefore the effect of TZ on atherosclerosis was independent of lipids and HDL retention properties. However, while TZ did not affect the biglycan-binding properties of lipoproteins, it could potentially have affected native biglycan (as well as other proteoglycans) in such a way as to influence its ability to bind to lipoproteins. Also, in this study, VLDL/IDL was not isolated and tested for its proteoglycan-binding affinity in vitro. It is possible that TZ could have altered the biglycan-binding affinity of these other lipoprotein fractions. It should be noted that the lipoprotein profile in these mice is very different from that of humans and so may not necessarily be directly applicable. In addition, heparan sulfate proteoglycans, such as perlecan, are the predominant proteoglycans implicated in HDL retention in mice [12]. Although this study was not performed in a diabetic model, there was a statistically significant reduction in fasting blood sugar in the TZ treated mice. However, since blood sugars in all the groups were still within the normal range, the effect on blood sugar is unlikely to have directly contributed to its effect on atherosclerosis. It is conceivable that different effect would have been observed had these animals been insulin resistant. TZ treatment also had no significant effect on the inflammatory markers SAA and SAP. Therefore inflammatory pathways involving SAA and SAP did not appear to play a direct role in the observed effect of TZ in these mice. However, since PPAR α and γ have been reported to have anti-inflammatory effects [1], it is possible that we missed these by only measuring SAA and SAP at a single time point. Also, we have found in previous studies that addition of cholesterol to diet, as was the case in this study, leads to elevated SAA levels [9]. This increased inflammatory state could conceivably have blunted the atheroprotective effects of TZ in these animals.
Considering the pleiotropic actions of PPARs, there are several potential mechanisms by which TZ might have led to the reduced atherosclerosis seen in the female mice in our study. In vitro studies show that PPAR-α activation can produce both anti-atherogenic and pro-atherogenic effects. For example, PPAR-α agonists decrease cytokine-induced expression of vascular cell adhesion molecule-1 (VCAM-1) leading to impaired adhesion of monocytes to stimulated ECs [15]. These same investigators also showed that PPAR-α activation in human ECs had no effect on gamma interferon-induced monocyte chemoattractant protein-1 (MCP-1) expression [16]. On the other hand, another group showed that oxidized phospholipids can activate PPAR-α in human ECs, resulting in the expression of the pro-inflammatory molecules MCP-1 and interleukin-8 (IL-8) [17]. Therefore, given the diverse biological effects of PPAR-α activation, only studies using in vivo models can show whether it has a net anti-atherogenic or pro-atherogenic effect. Studies of PPAR-α agonists in rodent models of atherosclerosis have been variable, depending on the model used. Apo E knockout (apoE −/−) mice lacking PPAR-α receptors had reduced atherosclerosis, suggesting that PPAR-α activation may increase atherosclerosis [18]. In another study, apoE −/− mice treated with a PPAR-α agonist showed no change in atherosclerotic lesional area and increase in plasma lipids, when compared with control [19]. However, apo E −/− mice with human apo A1 transgene showed significant reduction in atherosclerosis and no change in plasma lipids when treated with a PPAR-α agonist [19]. LDLr−/− mice showed a significant reduction in atherosclerosis when treated with a PPAR-α agonist [3]. In clinical trials such as the Bezafibrate Coronary Atherosclerosis Intervention Trial, fibrates were shown to decrease the progression of atherosclerosis [20], while in the Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial (VA-HIT), they were shown to decrease the incidence of coronary heart disease, a benefit that was only partially explained by the increase in HDL cholesterol [4]. However more recently, in the large FIELD study, therapy with the PPAR-α agonist fenofibrate led to a reduction in non-fatal myocardial infarction and revascularization procedures, but a trend towards increased cardiovascular mortality, which did not achieve statistical significance [21].
In vitro studies suggest that PPAR-γ activators inhibit VSMC proliferation and migration [1]. They also decrease EC expression of MCP-1, endothelial intracellular adhesion molecule-1 (ICAM-1) and VCAM-1 [1]. These are potentially atheroprotective effects of PPAR-γ agonists. PPAR-γ agonists were shown to inhibit angiogenesis in vitro as well as in vivo in rat cornea [22]. This could be advantageous as neovascularization may contribute to plaque progression and intraplaque hemorrhage. On the other hand this may be counterproductive by possibly inhibiting formation of collateral vessels in diabetic patients with small vessel coronary artery disease. PPAR-γ agonists increase expression of the scavenger receptor CD36 on macrophages, thereby promoting uptake of oxidized LDL [23]. This can potentially facilitate foam cell formation, an important step in the formation of atherosclerosis. PPAR-γ agonists however where shown to have a net effect of reduced atherosclerosis in both LDLr −/− and the apoE −/− mice [24–26]. In human studies, troglitazone decreased progression of carotid intimal medial thickness in premenopausal women with previous histories of gestational diabetes, suggesting a benefit in subclinical atherosclerosis [27]. Pioglitazone and rosiglitazone, other PPAR-γ agonists, both decreased CRP and resistin levels in non-diabetic patients with the metabolic syndrome [28, 29]. Rosiglitazone decreased matrix metalloproteinase 9 (MMP-9) in type 2 diabetic patients with coronary artery disease [16]. In the large prospective clinical trial, the PROactive study, therapy with pioglitazone led to a 10% reduction in the composite cardiovascular primary end point, which did not achieve statistical significance [30].
TZ decreased body weight and white adipose tissue (WAT) mass in both male and female mice in our study. This effect has not been shown in clinical studies of dual PPAR-α/γ agonists [31]. It is most likely due to PPAR-α activation, which has been shown to decrease body weight and WAT mass in various rodent models[32–35]. The increase in liver weight is again a PPAR-α effect, may be specific to rodents and possibly relates to peroxisomal activation [36].
Our study showed that TZ significantly reduced the development of atherosclerosis in female LDLr−/−mice but not in the males. The reason for this sexual dimorphism is not clear. In contrast with our findings, Li et al found that two different PPAR-γ agonists reduced atherosclerosis in male but not female LDLr−/− mice [25]. They found gender-specific differences in their effects on lipoprotein size distribution and expression levels of TNF-α in the aorta. Gender-specific differences in the effect of PPAR- activation on weight have also been found in C57BL/6J mice and some of these differences appear to be modulated by estrogen [35].
Selectivity in the ability of different PPAR- agonists to recruit specific cofactors in the transcription process would lead to differential biological responses to different agonists of a particular PPAR-subtype in any given species [37, 38]. The pleiotropic effects of activating different PPAR- subtypes by a particular substance could potentially produce net biological responses that would be difficult to predict or explain at first. For example, ciprofibrate, a PPAR-α agonist was shown to increase atherosclerosis in apoE −/− mice [33], while fenofibrate, another PPAR-α agonist had no effect on atherosclerosis in the same mouse model [19]. Yet in this same model, a dual PPAR-α/γ agonist reduced atherosclerosis over and above what was seen with a PPAR-γ agonist alone (32.4% versus 17.9%) [26]. This suggests an added benefit of PPAR-α activation when coupled with PPAR-γ activation, which was not apparent when a PPAR-α agonist was used alone.
Zadelaar et at found an anti-atherosclerotic effect of TZ in female apoE*leiden mice fed a high cholesterol diet [39]. Male mice were not included in that study, so it was not possible for those investigators to examine whether TZ has a gender-specific effect on lesion size and composition in apoE*Leiden mice. In that study, TZ lowered cholesterol; they corrected for this effect by including a third group fed a low cholesterol diet to achieve plasma cholesterol levels comparable to those of TZ-treated mice. In our Western diet-fed LDLr−/− mice, the TZ and control groups had similar plasma cholesterol levels. Similar to our findings, Zadelaar et al also found that TZ treatment was associated with an increase in macrophage content and a decrease in collagen content, expressed as percentages of total lesion area. Our results extend these findings by demonstrating that, in female mice, TZ was associated with increased immunostaining for apo A-I area in female mice, indicating increased lipid content, but also a trend towards increased necrotic area. We also demonstrate that there are no differences in lesion composition in male mice treated with TZ or control. Our findings of increased macrophage and lipid areas in TZ-treated female mice are consistent with the possibility that these were earlier and less advanced lesions. However, these characteristics, along with the increase in necrotic area and decrease in collagen in female mice also is consistent with the possibility that these lesions are more unstable [40–43].
Since TZ had no major metabolic effects in our mouse model, it is likely that its effect on atherosclerosis is at least in part due to direct vascular effects. Some of these putative vascular effects in our mouse model may conceivably be present in the human subject. It is possible that there could be significant differences in net biological effects between earlier developed agents, due to differential potencies in the various transcription pathways. Receptor activation profile for TZ, as determined from reporter gene assays in cells transfected with mouse PPAR-α and mouse PPAR-γ showed EC50 values of 32 μmol and 0.25 μmol, respectively [44]. We did not measure plasma levels of TZ in our mice. However, in the in vivo study by Zadelaar et al, the steady state plasma concentration in female apoE Leiden mice, also after a dose of 0.5 μmol/kg diet, was 38.6 ± 11.4 nmol/L [39]. This apparent discrepancy between the effective plasma concentration in vivo (38 nmol/L) and the EC50 values in reporter gene assays (32 μmol and 250 nmol) likely reflects the fact that the in vitro study was done with isolated cells transfected only with the ligand-binding domain of the receptors [44]. Also, it is possible that the steady state TZ plasma concentrations do not reflect intracellular concentrations achieved after prolonged treatment. Unfortunately clinical development programs of two dual PPAR-α/γ agonists, muraglitazar and tesaglitazar, have recently been discontinued, following analysis and interpretation of results from phase II/III trials. However, as our understanding of the numerous transcription effects of PPAR-activation at different tissue sites continues to improve, newer agents hopefully will be developed that target the desirable actions while minimizing the undesirable ones.
Acknowledgments
This work was supported by a grant from AstraZeneca who also provided the tesaglitazar, and by National Institute of Health (NIH) grants HL 30186, DK 02456, HL18645, and DK35816. E. C. Chira is supported by a NIH training grant and a grant from the Endocrine Fellows Foundation. We wish to thank Renee Lebeouf for her useful suggestions, Godfrey Getz for his generous gift of the SAA antibody, and Thomas F. Johnson for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duez H, Fruchart JC, Staels B. PPARS in inflammation, atherosclerosis and thrombosis. J Cardiovasc Risk. 2001;8:187–194. doi: 10.1177/174182670100800402. [DOI] [PubMed] [Google Scholar]
- 2.Barbier O, Torra IP, Duguay Y, et al. Pleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:717–726. doi: 10.1161/01.atv.0000015598.86369.04. [DOI] [PubMed] [Google Scholar]
- 3.Li AC, Binder CJ, Gutierrez A, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004;114:1564–1576. doi: 10.1172/JCI18730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robins SJ, Collins D, Wittes JT, et al. Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. Jama. 2001;285:1585–1591. doi: 10.1001/jama.285.12.1585. [DOI] [PubMed] [Google Scholar]
- 5.Hurt-Camejo E, Camejo G, Sartipy P. Measurements of proteoglycan-lipoprotein interaction by gel mobility shift assay. Methods Mol Biol. 1998;110:267–279. doi: 10.1385/1-59259-582-0:267. [DOI] [PubMed] [Google Scholar]
- 6.Olin KL, Potter-Perigo S, Barrett PH, Wight TN, Chait A. Biglycan, a vascular proteoglycan, binds differently to HDL2 and HDL3: role of apoE. Arterioscler Thromb Vasc Biol. 2001;21:129–135. doi: 10.1161/01.atv.21.1.129. [DOI] [PubMed] [Google Scholar]
- 7.O’Brien KD, Lewis K, Fischer JW, et al. Smooth muscle cell biglycan overexpression results in increased lipoprotein retention on extracellular matrix: implications for the retention of lipoproteins in atherosclerosis. Atherosclerosis. 2004;177:29–35. doi: 10.1016/j.atherosclerosis.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 8.O’Brien KD, Olin KL, Alpers CE, et al. Comparison of apolipoprotein and proteoglycan deposits in human coronary atherosclerotic plaques: colocalization of biglycan with apolipoproteins. Circulation. 1998;98:519–527. doi: 10.1161/01.cir.98.6.519. [DOI] [PubMed] [Google Scholar]
- 9.Lewis KE, Kirk EA, McDonald TO, et al. Increase in serum amyloid a evoked by dietary cholesterol is associated with increased atherosclerosis in mice. Circulation. 2004;110:540–545. doi: 10.1161/01.CIR.0000136819.93989.E1. [DOI] [PubMed] [Google Scholar]
- 10.Kirk EA, Sutherland P, Wang SA, Chait A, LeBoeuf RC. Dietary isoflavones reduce plasma cholesterol and atherosclerosis in C57BL/6 mice but not LDL receptor-deficient mice. J Nutr. 1998;128:954–959. doi: 10.1093/jn/128.6.954. [DOI] [PubMed] [Google Scholar]
- 11.Pepys MB, Baltz M, Gomer K, Davies AJ, Doenhoff M. Serum amyloid P-component is an acute-phase reactant in the mouse. Nature. 1979;278:259–261. doi: 10.1038/278259a0. [DOI] [PubMed] [Google Scholar]
- 12.Kunjathoor VV, Chiu DS, O’Brien KD, LeBoeuf RC. Accumulation of biglycan and perlecan, but not versican, in lesions of murine models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:462–468. doi: 10.1161/hq0302.105378. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien KD, McDonald TO, Kunjathoor V, et al. Serum amyloid A and lipoprotein retention in murine models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:785–790. doi: 10.1161/01.ATV.0000158383.65277.2b. [DOI] [PubMed] [Google Scholar]
- 14.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99:3125–3131. doi: 10.1161/01.cir.99.24.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marx N, Froehlich J, Siam L, et al. Antidiabetic PPAR gamma-activator rosiglitazone reduces MMP-9 serum levels in type 2 diabetic patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2003;23:283–288. doi: 10.1161/01.atv.0000054195.35121.5e. [DOI] [PubMed] [Google Scholar]
- 17.Lee H, Shi W, Tontonoz P, et al. Role for peroxisome proliferator-activated receptor alpha in oxidized phospholipid-induced synthesis of monocyte chemotactic protein-1 and interleukin-8 by endothelial cells. Circulation research. 2000;87:516–521. doi: 10.1161/01.res.87.6.516. [DOI] [PubMed] [Google Scholar]
- 18.Tordjman K, Bernal-Mizrachi C, Zemany L, et al. PPARalpha deficiency reduces insulin resistance and atherosclerosis in apoE-null mice. J Clin Invest. 2001;107:1025–1034. doi: 10.1172/JCI11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duez H, Chao YS, Hernandez M, et al. Reduction of atherosclerosis by the peroxisome proliferator-activated receptor alpha agonist fenofibrate in mice. The Journal of biological chemistry. 2002;277:48051–48057. doi: 10.1074/jbc.M206966200. [DOI] [PubMed] [Google Scholar]
- 20.Ericsson CG, Nilsson J, Grip L, Svane B, Hamsten A. Effect of bezafibrate treatment over five years on coronary plaques causing 20% to 50% diameter narrowing (The Bezafibrate Coronary Atherosclerosis Intervention Trial [BECAIT]) Am J Cardiol. 1997;80:1125–1129. doi: 10.1016/s0002-9149(97)00626-7. [DOI] [PubMed] [Google Scholar]
- 21.Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–1861. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- 22.Xin X, Yang S, Kowalski J, Gerritsen ME. Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo. The Journal of biological chemistry. 1999;274:9116–9121. doi: 10.1074/jbc.274.13.9116. [DOI] [PubMed] [Google Scholar]
- 23.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 24.Collins AR, Meehan WP, Kintscher U, et al. Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:365–371. doi: 10.1161/01.atv.21.3.365. [DOI] [PubMed] [Google Scholar]
- 25.Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claudel T, Leibowitz MD, Fievet C, et al. Reduction of atherosclerosis in apolipoprotein E knockout mice by activation of the retinoid X receptor. Proc Natl Acad Sci U S A. 2001;98:2610–2615. doi: 10.1073/pnas.041609298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiang AH, Peters RK, Kjos SL, et al. Effect of thiazolidinedione treatment on progression of subclinical atherosclerosis in premenopausal women at high risk for type 2 diabetes. J Clin Endocrinol Metab. 2005;90:1986–1991. doi: 10.1210/jc.2004-1685. [DOI] [PubMed] [Google Scholar]
- 28.Szapary PO, Bloedon LT, Samaha FF, et al. Effects of pioglitazone on lipoproteins, inflammatory markers, and adipokines in nondiabetic patients with metabolic syndrome. Arterioscler Thromb Vasc Biol. 2006;26:182–188. doi: 10.1161/01.ATV.0000195790.24531.4f. [DOI] [PubMed] [Google Scholar]
- 29.Samaha FF, Szapary PO, Iqbal N, et al. Effects of rosiglitazone on lipids, adipokines, and inflammatory markers in nondiabetic patients with low high-density lipoprotein cholesterol and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2006;26:624–630. doi: 10.1161/01.ATV.0000200136.56716.30. [DOI] [PubMed] [Google Scholar]
- 30.Dormandy JA, Charbonnel B, Eckland DJ, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 31.Buse JB, Rubin CJ, Frederich R, et al. Muraglitazar, a dual (alpha/gamma) PPAR activator: A randomized, double-blind, placebo-controlled, 24-week monotherapy trial in adult patients with type 2 diabetes. Clin Ther. 2005;27:1181–1195. doi: 10.1016/j.clinthera.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Chaput E, Saladin R, Silvestre M, Edgar AD. Fenofibrate and rosiglitazone lower serum triglycerides with opposing effects on body weight. Biochem Biophys Res Commun. 2000;271:445–450. doi: 10.1006/bbrc.2000.2647. [DOI] [PubMed] [Google Scholar]
- 33.Fu T, Kashireddy P, Borensztajn J. The peroxisome-proliferator-activated receptor alpha agonist ciprofibrate severely aggravates hypercholesterolaemia and accelerates the development of atherosclerosis in mice lacking apolipoprotein E. The Biochemical journal. 2003;373:941–947. doi: 10.1042/BJ20030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guerre-Millo M, Gervois P, Raspe E, et al. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. The Journal of biological chemistry. 2000;275:16638–16642. doi: 10.1074/jbc.275.22.16638. [DOI] [PubMed] [Google Scholar]
- 35.Jeong S, Han M, Lee H, et al. Effects of fenofibrate on high-fat diet-induced body weight gain and adiposity in female C57BL/6J mice. Metabolism: clinical and experimental. 2004;53:1284–1289. doi: 10.1016/j.metabol.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 36.Meyer K, Volkl A, Endele R, Kuhnle HF, Pill J. Species differences in induction of hepatic enzymes by BM 17.0744, an activator of peroxisome proliferator-activated receptor alpha (PPARalpha) Arch Toxicol. 1999;73:440–450. doi: 10.1007/s002040050633. [DOI] [PubMed] [Google Scholar]
- 37.Duez H, Lefebvre B, Poulain P, et al. Regulation of human apoA-I by gemfibrozil and fenofibrate through selective peroxisome proliferator-activated receptor alpha modulation. Arterioscler Thromb Vasc Biol. 2005;25:585–591. doi: 10.1161/01.ATV.0000154140.73570.00. [DOI] [PubMed] [Google Scholar]
- 38.Camp HS, Li O, Wise SC, et al. Differential activation of peroxisome proliferator-activated receptor-gamma by troglitazone and rosiglitazone. Diabetes. 2000;49:539–547. doi: 10.2337/diabetes.49.4.539. [DOI] [PubMed] [Google Scholar]
- 39.Zadelaar AS, Boesten LS, Jukema JW, et al. Dual PPARalpha/gamma agonist tesaglitazar reduces atherosclerosis in insulin-resistant and hypercholesterolemic ApoE*3Leiden mice. Arterioscler Thromb Vasc Biol. 2006;26:2560–2566. doi: 10.1161/01.ATV.0000242904.34700.66. [DOI] [PubMed] [Google Scholar]
- 40.Richardson PD, Davies MJ, Born GV. Influence of plaque configuration and stress distribution on fissuring of coronary atherosclerotic plaques. Lancet. 1989;2:941–944. doi: 10.1016/s0140-6736(89)90953-7. [DOI] [PubMed] [Google Scholar]
- 41.Davies MJ. Stability and instability: two faces of coronary atherosclerosis. The Paul Dudley White Lecture 1995. Circulation. 1996;94:2013–2020. doi: 10.1161/01.cir.94.8.2013. [DOI] [PubMed] [Google Scholar]
- 42.Davies MJ, Richardson PD, Woolf N, Katz DR, Mann J. Risk of thrombosis in human atherosclerotic plaques: role of extracellular lipid, macrophage, and smooth muscle cell content. British heart journal. 1993;69:377–381. doi: 10.1136/hrt.69.5.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lendon CL, Davies MJ, Born GV, Richardson PD. Atherosclerotic plaque caps are locally weakened when macrophages density is increased. Atherosclerosis. 1991;87:87–90. doi: 10.1016/0021-9150(91)90235-u. [DOI] [PubMed] [Google Scholar]
- 44.Ljung B, Bamberg K, Dahllof B, et al. AZ 242, a novel PPARalpha/gamma agonist with beneficial effects on insulin resistance and carbohydrate and lipid metabolism in ob/ob mice and obese Zucker rats. Journal of lipid research. 2002;43:1855–1863. doi: 10.1194/jlr.m200127-jlr200. [DOI] [PubMed] [Google Scholar]