

Abstract
Background
Candida albicans resides on epithelial surfaces as part of the physiological microflora. However, under certain conditions it may cause life-threatening infections like Candida sepsis. Human β-defensins (hBDs) are critical components of host defense at mucosal surfaces and we have recently shown that hBD-2 and hBD-3 are upregulated in Candida esophagitis. We therefore studied the role of Candidate signalling pathways in order to understand the mechanisms involved in regulation of hBD-expression by C. albicans. We used the esophageal cell line OE21 and analysed the role of paracrine signals from polymorphonuclear leukocytes (PMN) in an in vitro model of esophageal candidiasis.
Results
Supernatants of C. albicans or indirect coculture with C. albicans induces upregulation of hBD-2 and hBD-3 expression. PMNs strongly amplifies C. albicans-mediated induction of hBDs. By EMSA we demonstrate that C. albicans activates NF-κB and AP-1 in OE21 cells. Inhibition of these pathways revealed that hBD-2 expression is synergistically regulated by both NF-κB and AP-1. In contrast hBD-3 expression is independent of NF-κB and relies solely on an EGFR/MAPK/AP-1-dependent pathway.
Conclusion
Our analysis of signal transduction events demonstrate a functional interaction of epithelial cells with PMNs in response to Candida infection involving divergent signalling events that differentially govern hBD-2 and hBD-3 expression.
Background
Candida albicans colonize distinct microanatomical regions such as the oro-gastointestinal tract as a commensal, but also accounts for more than 50% of all fungal systemic infections [1]. Candida esophagitis represents a severe threat to an immunocompromised body and, especially in neutropenic patients, often is the first manifestation before Candida sepsis develops [2,3]. Host defense mechanisms preventing mucosal (e.g. esophageal) candidiasis are poorly understood, but include both innate and adaptive responses [4,5].
The gastrointestinal epithelial layer represents a barrier that is usually adequate to restrain commensal microbes, but is often insufficient to protect against microbial pathogens. Once this physical barrier is penetrated, recognition of invading microbiota is the first step in the initiation of a fast immune response and involves the activation of pattern recognition receptors by microbial pathogens and their products [6-8]. C. albicans have been shown to activate a subset of pattern recognition receptors, the Toll-like receptors (TLRs). This family of transmembrane receptors recognizes a broad variety of signature motifs on microbes and transduces signals leading to the activation of transcription factors [9], production of cytokines and antimicrobial peptides [10,11]. However there is some controversy and inconclusive data which TLR subtypes are activated by C. albicans. Some reports indicated a critical role for TLR2 and TLR4 in activating the host defense response alone or in combination with the ss-glucan receptor Dectin-1,[5,12,13] whereas other studies showed that TLR-1 and TLR-6 are responsible for the recognition of C. albicans [14,15].
In addition the downstream effectors in the immune response against C. albicans are largely unknown. There is growing evidence that human β-defensins (hBDs) are critical components of both the innate and adaptive immune responses to Candida infections with distinct antifungal efficacies and mechanisms for hBD-2 and hBD-3 [16-19]. Expression of hBDs is regulated by a plethora of proinflammatory cytokines like IL-1β, TNF-α and EGF-receptor ligands activating downstream effectors like the transcription factors NF-κB or AP-1 [20-22]. hBDs are secreted by epithelial cells and protect the gastrointestinal mucosa by antimicriobial activity as well as by chemotactic properties recruiting polymorphonuclear leukocytes (PMN) to the site of infection [21,23]. Transepithelial migration of PMNs is observed during oral Candida infection and is believed to play a crucial role in the clearance of infection and in epithelial homeostasis [5,24]. This protective phenotype is associated with production of epithelial proinflammatory cytokines, including IL-8, IL-1β and TNF-α. However, the mechanisms by which PMNs and epithelial cells interact to protect mucosal surfaces from C. albicans invasion are mostly unknown.
We have recently shown that hBD-2 and hBD-3 are highly expressed in Candida esophagitis and that the α-defensins Human Neutrophil Peptides 1–3 were also upregulated [25] indicating involvement of neutrophils in the immune response to the C. albicans infection [26]. This is in line with the observation that IL-8 is strongly expressed in the mucosa of patients suffering by Candida infection [25], since this cytokine is involved in recruitment of polymorphonuclear leukocytes (PMNs) to sites of microbial infection [27].
In the present study we explored the mechanisms involved in the induction of hBD expression in an in vitro model of Candida esophagitis. Using this co-culture model we analyzed the contribution of PMNs in the regulation of epithelial hBD expression. We found that hBD-2 and hBD-3 are strongly upregulated through high concentrations of C. albicans alone. If lower concentrations of C. albicans were used, only a moderate induction of these hBDs were observed that was strongly enhanced by coculture of the epithelial cell line with PMNs, emphasizing the important role of mesenchymal-epithelial interactions in early host defense against fungal infection. Furthermore we were able to show that the NF-κB and MAPK signalling pathways contribute to the regulation of hBD-2 and that hBD-3 is a downstream target of a distinct EGFR/MAPK/AP-1 pathway in Candida esophagitis.
Results
Supernatants of C. albicans induce hBD expression in OE21 cells
To establish an in vitro model for Candida esophagitis we first treated several oesophageal cell lines (i.e. OE21, KYSE70, KYSE7150, KYSE7180, KYSE410 and Colo680N) with established inducers of hBD-expression. Only for the OE21 cell line a reliable induction of the mRNA of hBD-2 and hBD-3 could be detected (Figure 1A). We next challengend OE21 cells with supernatants of C. albicans cell culture growing under various concentrations of C. albicans. Supernatants of C. albicans induce upregulation of hBDs in a concentration-dependent manner. For the supernatants of the density of 0.5 × 104 C. albicans cells/ml no significant induction of hBD-2 and hBD-3 was detected (Figure 1B). In contrast the supernatants of 1 × 105 C. albicans cells/ml resulted in a significant upregulation of hBD-3 and to a smaller extent of hBD-2 (Figure 1B). hBD1 expression was not altered by one of the inducers or the supernatants (Figure 1).
Figure 1.
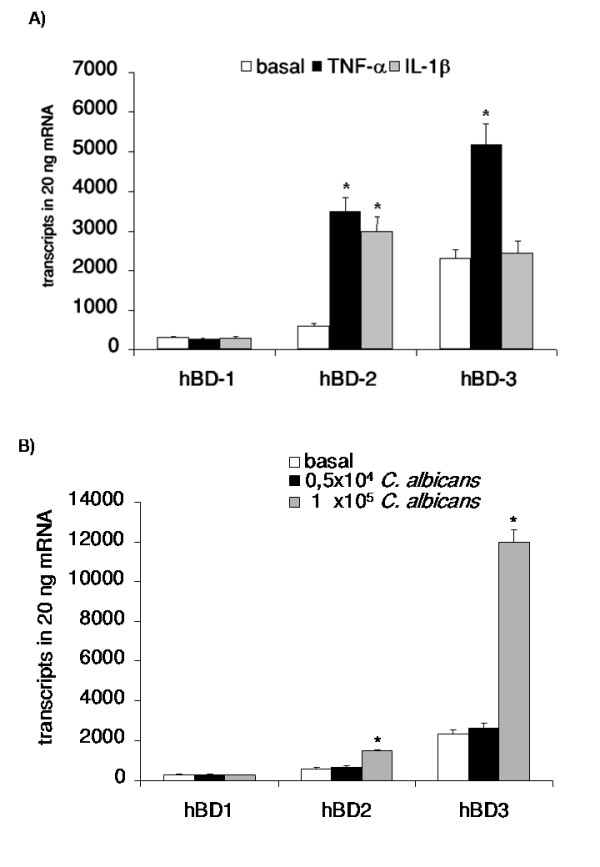
Supernatants of C. albicans induce hBD expression in OE21 cells. OE21 cells were stimulated with A) TNF-α (20 ng/ml), IL-1β (20 ng/ml) or B) supernatants of 0.5 × 104 C. albican/ml or 1 × 105 C. albican/ml for 24 h. hBD-1, hBD-2 and hBD-3 gene expression was assessed by real time RT-PCR. Means ± s.d. of three independent experiments performed in triplicate are shown, * indicates p-value <0,05.
PMNs significantly enhances hBD-2 and hBD-3 expression during C. albicans infection
To elucidate a potential functional interaction of PMNs and epithelial cells in the immune response to C. albicans infection we established a transwell coculture model. In this two-compartment model we tested the effect of a coculture of OE21 cells, PMNs from healthy donors and C. albicans in different settings: 1) direct contact of OE21 cells with PMNs and 2) direct contact of OE21 cells with C. albicans. The corresponding third component of the system (i.e. C. albicans or PMNs) was cocultured in the transwell system allowing for the interaction of auto- and paracrine factors (Figure 2). Since higher cell numbers than 0.5 × 104 C. albicans cells/ml or 0.5 × 106 PMN/ml repressed growth of OE21 cells in the direct setting (data not shown), all experiments were conducted with these concentrations of C. albicans and/or PMNs. The direct or indirect coculture of OE21 with C. albicans had only small effects on the expression of hBD-2 (Figure 3A) and hBD-3 (Figure 3B) comparable with effects observed with supernatants of the low concentration of C. albicans (Figure 1B). The direct interaction of OE21 cells with PMNs led to an increase of the expression of both hBDs (Figure 3A and Figure 3B). The interaction of OE21 cells with PMNs lead to a significant upregulation of C. albicans induced hBD-2 and hBD-3 expression compared to the induction with C. albicans or PMN alone (Figure 3A and Figure 3B). Substitution of the live C. albicans by supernatants of the density of 0.5 × 104 C. albicans cells/ml also lead to the synergistic effects. To keep the manuscript mor concise we only show the data for the live C. albicans.
Figure 2.

PMNs enhances hBD-2 and hBD-3 expression during C. albicans infection. OE21 cells were stimulated with 0.5 × 104 C. albican/ml or 0.5 × 106 PMNs/ml alone or indicated combinations in a transwell setting for 24 h. Shematic presentation of experimental setting.
Figure 3.
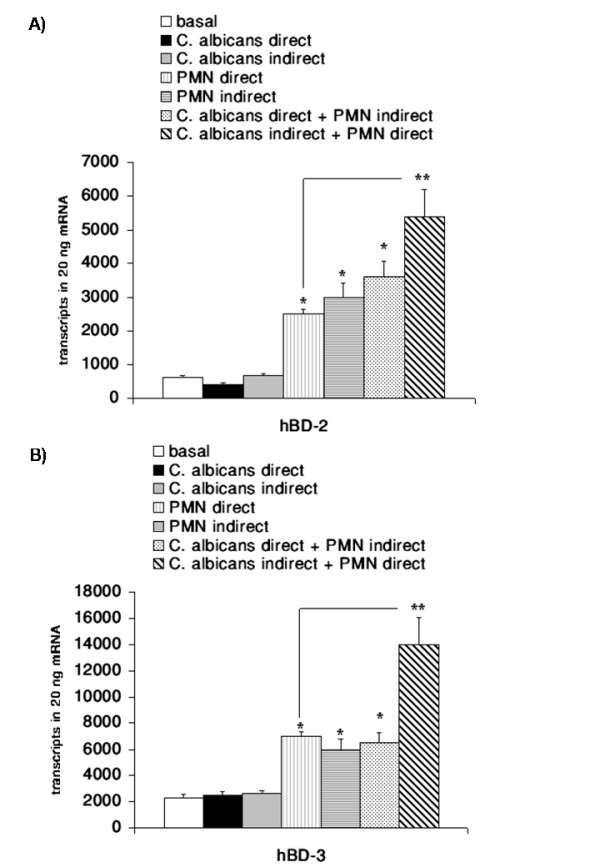
PMNs enhances hBD-2 and hBD-3 expression during C. albicans infection. OE21 cells were stimulated with 0.5 × 104 C. albican/ml or 0.5 × 106 PMNs/ml alone or indicated combinations in a transwell setting for 24 h. A) hBD-2 and B) hBD-3 gene expression was assessed by real time RT-PCR. Means ± s.d. of three independent experiments performed in triplicate are shown, * indicates p-value <0,05, ** indicates synergistic effects with a p-value <0,05.
C. albicans activate NF-κB in OE21 cells
To further analyse the mechanisms involved in the regulation of hBD expression during Candida esophagitis we determined whether the observed induction of hBD-2 and hBD-3 expression by C. albicans was associated with the activation of NF-κB. This transcription factor has been shown to be a central regulator of hBD-2 expression [22,28,29] but there is some controversy if NF-κB is also involved in the regulation of hBD-3 expression [20,30,31]. By EMSA we could show that NF-κB is activated (Figure 4) in association with C. albicans mediated induction of β-defensin expression. In parallel to the observed dependency of defensin expression on the cell number of C. albicans a concentration dependent activation of NF-κB was observed (Figure 4A). The coculture of OE21 cells with PMNs or C. albicans also led to an increased DNA-binding of NF-κB that was further enhanced in the transwell setting (direct PMN and C. albicans indirect; Figure 4B). Supershift assays revealed that the NF-κB complex consisted of the p50 and p65 subunits (Figure 4C).
Figure 4.
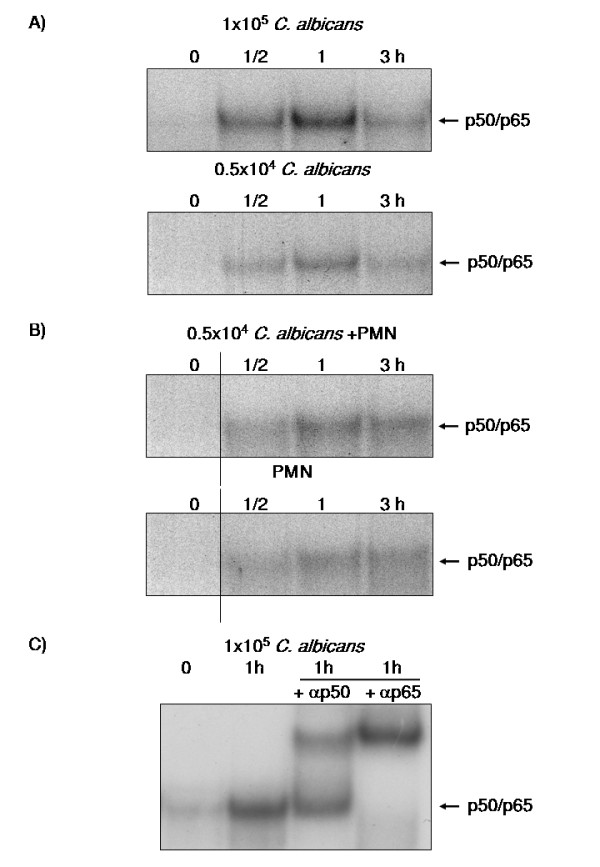
C. albicans activate NF-κB in OE21 cells. OE21 cells were stimulated with A) 0.5 × 104 C. albican/ml, 1 × 105 C. albican/ml, or B) 0.5 × 106 PMNs/ml alone or with direct contact with 0.5 × 106 PMNs/ml and coincubation with 0.5 × 104 C. albican/ml in a transwell setting for indicated time periods. Nuclear extracts of these cells were submitted to EMSA using a radiolabelled probe for NF-κB. The arrow indicates the p50/p65 heterodimer. C) Supershift experiments with antibodies directed against the p50 or p65 subunit of NF-κB on nuclear extracts of OE21 cells treated for 1 h with 1 × 105 C. albican/ml.
NF-κB is involved in the induction of hBD-2 but not hBD-3 expression
To evaluate the putative contribution of NF-κB in C. albicans induced hBD expression we transiently transfected cells with siRNA directed against RelA/p65 as the transcriptional active subunit of the p65/p50 NF-κB complex [32]. Transfection of OE21 cells led to a significant reduction of the expression of RelA/p65 (Figure 5A). Furthermore the induction of NF-κB by C. albicans was inhibited by this strategy as shown by EMSA (Figure 5B). This reduction of NF-κB activation strongly reduced the C. albicans mediated induction of hBD-2 expression but had no effect on the hBD-3 expression (Figure 5C). In the coculture settings the reduction of RelA/p65 expression also attenuated hBD-2 induction (data not shown). These results support a critical role of NF-κB in regulating hBD-2 but not hBD-3 expression.
Figure 5.
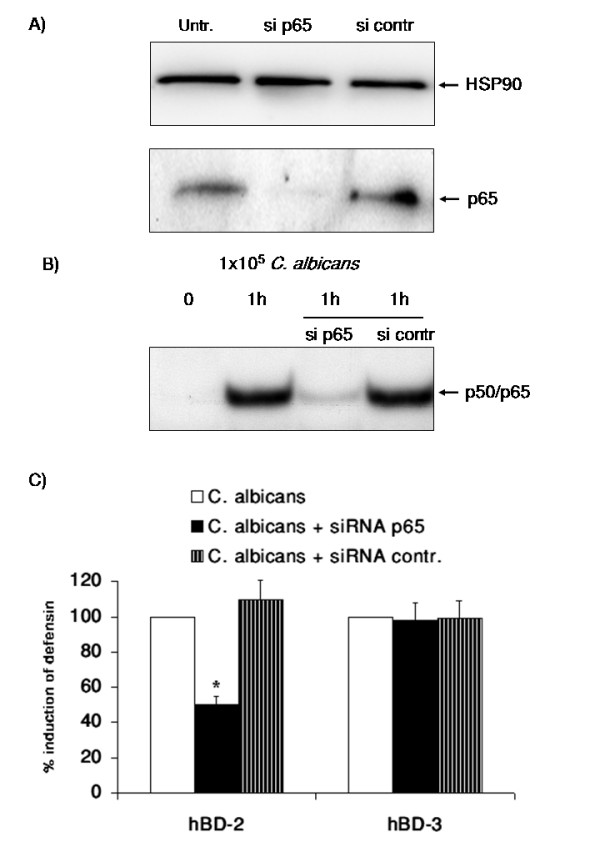
NF-κB is involved in the induction of hBD-2 but not hBD-3 expression. OE21 cells were transfected with a control siRNA or a siRNA directed against RelA/p65. A) Total protein extracts were submitted to Western Blotting for the analysis of RelA/p65 using HSP90 for normalization and B) EMSA on nuclear extracts of siRNA transfected OE21 cells treated 9 with 1 × 105 C. albican/ml for 1 h. Representative results from three independent experiments are shown. C) SiRNA transfected OE21 cells were incubated with 1 × 105 C. albican/ml for 24 h and hBD gene expression was assessed by real time RT-PCR. Means ± s.d. of three independent experiments performed in triplicate are shown, * indicates p-value <0,05.
Role of AP-1 and the MAPK Pathway in C. albicans mediated β-defensin induction
Our results indicated a critical role for NF-κB in C. albicans mediated induction of hBD-2 but not for the regulation of hBD-3 expression. Since the hBD-3 promotor contains a functional binding site for the AP-1 transcription factor [33] we conducted EMSA with a probe containing an AP-1 consensus sequence. C. albicans alone (Figure 6A) or in the coculture setting (Figure 6B) activated AP-1 in OE21 cells and supershift experiments identified c-jun as one of the central subunits involved (Figure 6C). Activation of the c-jun/AP-1 complex by the MAPK pathway is well established [34,35] and a recent report indicated a role of this pathway in the response of macrophages to the infection with C. albicans [9]. To analyse the contribution of the MAPK/AP-1 pathway in the induction of β-defensin induction by C. albicans pharmacological inhibitors selective for individual pathway were utilized.
Figure 6.
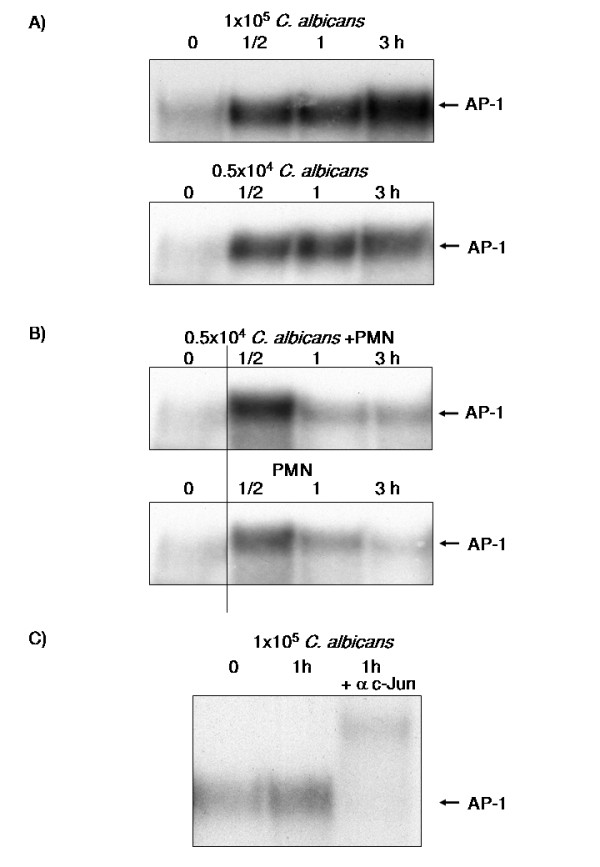
Role of AP-1 and the MAPK Pathway in C. albicans mediated hBD induction. OE21 cells were stimulated with A) 0.5 × 104 C. albican/ml, 1 × 105 C. albican/ml, or B) 0.5 × 106PMNs/ml alone or with direct contact with 0.5 × 106 PMNs/ml and coincubation with 0.5 × 104 C. albican/ml in a transwell setting for indicated time periods. Nuclear extracts of these cells were submitted to EMSA using a radiolabelled probe for AP-1. The arrow indicates the AP-1 complex. C) Supershift experiments with an antibody directed against the c-Jun subunit of AP-1 on nuclear extracts of OE21 cells treated for 1 h with 1 × 105 C. albican/ml.
As shown in Figure 7A and 7B inhibition of each of the major MAPK pathways resulted in a reduction of hBD-2 and hBD-3 induction by C. albicans.
Figure 7.
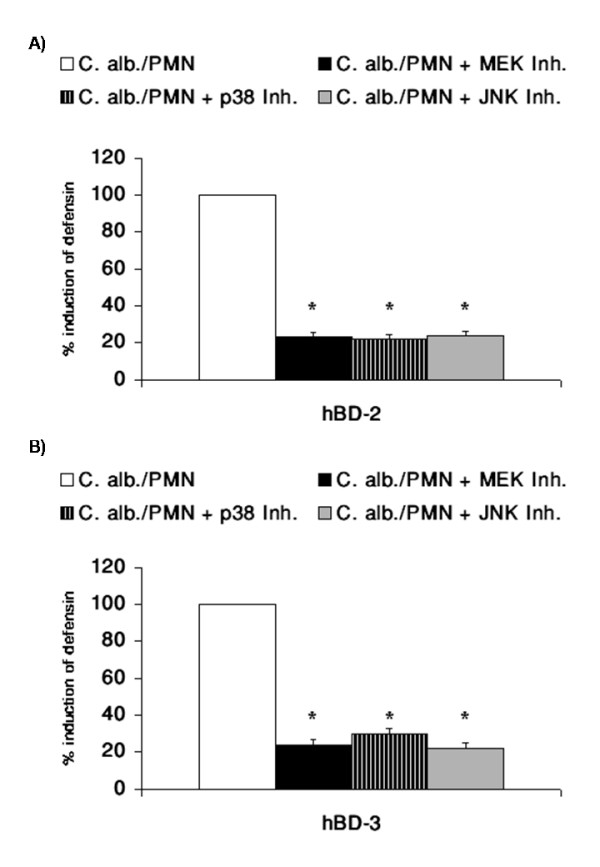
Role of AP-1 and the MAPK Pathway in C. albicans mediated hBD induction. OE21 cells were preincubated with inhibitors for MEK (PD98059, 10 μM), p38 (SB203580 10 μM) and JNK (SP600125 20 μM) kinase pathways for 1 h and then incubated with direct contact with 0.5 × 106 PMNs/ml and coincubation with 0.5 × 104 C. albican/ml in a transwell setting for additional 24 h. A) hBD-2 and B) hBD-3 gene expression was assessed by real time RT-PCR. Means ± s.d. of three independent experiments performed in triplicate are shown, * indicates p-value <0,05.
Activation of the EGFR is an upstream event in regulation of hBD-3 expression
Upstream of the MAPK pathway several growth factor receptors have been shown to be involved in the immune response to microbiota [20,36,37]. To investigate the upstream events leading to the activation of the MAPK pathway in infection with C. albicans we used blocking antibodies against several growth factor receptors. The results revealed a clear involvement of the EGF-receptor in the regulation of hBD-3 expression by C. albicans. Whereas induction of hBD-2 expression was slightly inhibited by a neutralizing antibody targeting EGFR transactivation, a significant reduction of C. albicans-induced hBD-3 expression observed (Figure 8).
Figure 8.

Activation of the EGFR is an upstream event in regulation of hBD-3 expression. OE21 cells were preincubated with a blocking EGFR antibody (10 μg/ml) for 1 h and then incubated with direct contact with 0.5 × 106 PMNs/ml and coincubation with 0.5 × 104 C. albican/ml in a transwell setting for additional 24 h. hBD gene expression was assessed by real time RT-PCR. Means ± s.d. of three independent experiments performed in triplicate are shown, * indicates p-value <0,05.
TGF-α is involved in EGFR mediated hBD-3 induction
Finally we searched for the ligand activating the EGFR. By using blocking antibodies for the established EGFR-ligands EGF, HB-EGF, amphiregulin and TGF-α we were able to show that TGF-α is involved in the EGFR mediated upregulation of hBD-3 expression (Figure 9).
Figure 9.

TGF-α is involved in EGFR mediated hBD-3 induction. OE21 cells were preincubated with the blocking EGFR antibody (10 μg/ml) or neutralizing antibody against EGF (0,5 μg/ml), HB-EGF (10 μg/ml), amphiregulin (10 μg/ml) or TGF-α (1 μg/ml) for 1 h and then incubated with direct contact with 0.5 × 106 PMNs/ml and coincubation with 0.5 × 104 C. albican/ml in a transwell setting for additional 24 h. hBD gene expression was assessed by real time RT-PCR. Means ± s.d. of three independent experiments performed in triplicate are shown, * indicates p-value <0,05.
Discussion
Candida esophagitis represents a severe threat to an immunocompromised body and the course of the infection is determined by both pathogen- and host-dependent factors [15,24,38,39]. It is well established that epithelial cells of the esophagus are the central target of an oro-esophageal invasive Candida infection but there are only very limited data on the host response preventing a Candida esophagitis. In the present study we identified the NF-κB and MAPK/AP-1 pathways as central regulators of epithelial hBD-2 and hBD-3 expression during C. albicans infection. Furthermore we were able to show a crucial role of the interaction of PMNs with the epithelial cell compartment for the induction of hBD-2 and hBD-3 expression during Candida infection of esophageal epithelial cells. Finally hBD-3 expression is dependent on transactivation of EGFR by TGF-α. This is the first report delineating molecular mechanisms leading to upregulation of hBD-2 and hBD-3 in esophageal Candida infection suggesting differential regulation of these two central epithelial antibiotic peptides in response to a C. albicans infection. We were able to show a concentration dependency of hBD-2 and hBD-3 induction upon stimulation with C. albicans supernatants (Figure 1B). This dose dependent effect could be at least in part be mediated through stronger induction of NF-κB (Figure 4) and AP-1 (Figure 6) through higher concentrations of C. albicans. A comparable concentration-dependent effect of supernatants of environmental airborne fungi on cytokine release of eosinophils [40] and of C. albicans on the activation of NF-κB [9] was recently reported.
There is some controversy about the relevance of the NF-κB and AP-1 binding sites in the promotor of hBD-2 for full induction of hBD-2 expression after treatment with IL-1β or infection with microbiota. Wehkamp and collegues showed that the parallel activation of NF-κB and AP-1 is needed for full transcriptional activation of the hBD-2-promotor after IL-1β stimulation, treatment of keratinocytes with supernatants of Pseudomonas aeruginosa [22], or E. coli mediated hBD-2 induction [41]. On the other hand another study demonstrated induction of hBD-2 expression through Fusobacterium nucleatum in human gingival epithelial cells independent of NF-κB activation [42] and there is also evidence for induction of hBD-2 through Salmonella enteritidis [43] or H. pylori [44] in the absence of functional AP-1. In the present work we were able to show that both NF-κB and AP-1 activation are required for full upregulation of the hBD-2 mRNA after treatment with either supernatants of C. albicans or the coculture of C. albicans with PMNs. Inhibiton of the NF-κB or of the MAPK/AP-1 pathway significantly reduced the induction of hBD-2 expression confirming the central role of both transcription factors. These observations are in line with recent results for H. pylori infection [20] and the effects of lactobacilli and the VSL#3 bacterial mixture on enterocytes [45]. The role of NF-κB is less clear in the regulation of hBD-3 expression. In contrast to a recently reported role of NF-κB in the regulation of the hBD-3 gene in keratinocytes [31] the majority of data failed to show a functional relevance for NF-κB in controlling hBD-3 expression [33,46]. By using siRNA targeting the RelA/p65 subunit of NF-κB we could demonstrate that C. albicans mediates hBD-3 upregulation through a MAPK/AP-1 pathway independently of the observed NF-κB acitvation. To investigate the proposed role of PMNs in the immune response the C. albicans infection [25,26] we established an in vitro model (Figure 2). PMNs alone induced NF-κB and AP-1 leading to hBD-2 and hBD-3 expression. Coincubation of PMNs with C. albicans lead to a significant upregulation of hBD-2 and hBD-3 expression compared to effects of PMNs or C. albicans alone. Inhibition of the MAPK/AP-1 pathway reduced the expression of both hBDs under this condition. The fact that PMNs alone induced hBD expression indicated that PMNs could contribute to the induction of antimicrobial peptides in epithelia during inflammation. There is some evidence that a TLR-4 mediated interplay between PMNs and epithelial cells is important for the protective response against Candida infections [5] but the exact mechanisms remained elusive. One the one hand PMNs induced expression and release of IL-6 and IL-8 in O21 cells (Additional file 1) which might in turn activate NF-κB and AP-1 leading to hBD-2 and hBD-3 expression. On the other hand the induction of a TH1 response, i.e. TNF-α and interferon-γ might directly induce or enhance the expression of hBDs as reported recently [47]. On the other hand PMNs are able to establish an anti-fungal response by upregulate the expression of TLR4 in epithelial cells [5]. The observed more pronounced effect on hBD-3 expression are in line with the theory that hBD-3 might be clinically more relevant than hBD-2 since hBD-2 and hBD-3 have potent fungicidal activity against C. albicans at micromolar concentrations, with hBD-3 being about 10 times more fungicidal than hBD-2 [16,17,19]. Finally we demonstrated that hBD-3 expression was induced by transactivation of the EGFR independent of EGF (Figure 8 and 9). Many signals besides EGF converge and result in EGFR-dependent signaling which is important for various biological processes including normal growth, development and as shown recently for inflammation or innate immune response [48,49]. Using blocking antibodies for the EGFR-ligands EGF, HB-EGF, amphiregulin and TGF-α we were able to show that most likely TGF-α is involved in the EGFR mediated upregulation of hBD-3 expression (Figure 9). Since we are also able to inhibit the induction of hBD3 by the supernatants of 1 × 105 C. albicans cells/ml by EGFR and/or TGF-α blocking antibodies we speculate that the EGFR-ligand is secreted by OE21 cells. Ligands of the EGFR are expressed as transmembrane precursors. These are released from the cell surface following shedding of the extracellular domain by a family of metalloproteinases (a disintegrin and metalloprotease (ADAM)). ADAM10, -12 and -17 are the sheddases of the EGFR ligands in response to various stimuli. Since human cathelicidin cationic antimicrobial protein (hCAP)-18 and its active peptide LL-37 have been shown to be involved in the transactivation of the EGFR at the airway epithelial surfaces [50,51] it is tempting to speculate that the activation of PMNs by C. albicans leads to secretion of proinflammatory mediators including leukotriene B4 (LTB4) and LL-37 [52,53] which in turn amplifies the inflammatory response [54,55] and leads to the shedding of EGFR-ligands.
In conclusion we were able to establish a complex in vitro model for Candida infection of esophageal cells to investigate the signalling events leading to upregulation of hBD-2 and hBD-3 expression.
Conclusion
This analysis of signal transduction events in esophageal Candida infection demonstrated a potential functional interaction of epithelial cells with PMNs and that EGFR, NF-κB and MAPK/AP-1 are involved in divergent signalling events governing hBD-2 and hBD-3 expression. Especially the observed effect of PMNs on hBD expression might explain the high incidence of Candida esophagitis and Candida related deaths in neutropenic patients.
Methods
Materials
Cell culture medium was purchased from PAA-Labs (Linz, Austria), fetal bovine serum from Seromed (Berlin, Germany), recombinant TNF-α from Sigma. Supershift and Westernblot p65, p50 and c-Jun antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA).
Organism and Cell culture
OE21 cells, a moderately differentiated oesophageal squamous carcinoma cell line were cultured in RPMI 1670 supplemented with 10% fetal calf serum and divided every 3 days. Cells for experimental purposes were cultured in 6 well cell culture plates with the same medium and used at subconfluence. C. albicans (a clinical isolate from oesophagitis [25]) was taken fom a frozen strain suspension (BHI, glycerine). 10 μl was transferred into 10 ml TSB-Bouillon and incubated overnight at 37°C. The cells were then separated from the medium by centrifugation at 2800 × g for 5 min, washed two times with PBS, and finally resuspended in RPMI for each experiment. The final concentration of C. albicans was 1 × 104/ml.
Inhibitor studies
OE21 cells were pre-treated with specific inhibitors or neutralizing antibodies for 30–60 min prior to and during Candida or cytokine stimulation. The neutralzing antibodies against the EGFR (Upstate, USA), TGF-α (R&D Systems), HB-EGF (R&D Systems), amphiregulin (R&D Systems), and EGF (Pepro Tech, Germany) were used at the indicated concentrations. Inhibitors utilized were: PD98059 (Calbiochem) at 10 μM, SB203580 (Tocris, Ellusville, USA) at 10 μM, SP600125 (Tocris, Ellusville, USA) at 20 μM. Inhibitor were disolved in DMSO and DMSO was used as a vehicel control.
Generation of Supernatants from C. albicans
To analyze the effects of secreted factors from C. albicans on OE21 cells, C. albicans were cultured with respective medium for 48 h in the indicated density (0.5 × 104 to 1 × 105 cells/ml). Before use, these conditioned media were precleared by centrifugation (10,000 rpm for 10 min) and used for additional experiments.
Isolation of polymorphonuclear leukocytes (PMN)
PMN were purified using LeucoSep tubes according to the instructions of the manufacturer (Greiner Bio-One). In brief, 3 ml of Ficoll-Paque was preloaded in a 14 ml LeucoSep tube by centrifugation for 30 s at 1,000 g. The heparinized whole-blood samples of healthy volunters were diluted with equal volumes of PBS, and 6 ml of the diluted blood was added to a LeucoSep tube. The cell separation tubes were centrifuged for 15 min at 800 g without braking at room temperature. The cell suspension was collected, and the cells were washed twice in PBS (for 10 min at 640 and 470 g, respectively, for the two successive wash steps) and resuspended in complete RPMI medium before counting.
Stimulation with Supernatants, direct cell contact and transwell coculture model
2 × 105 OE21 cells were seeded into a six-well culture plate. For stimulation experiments medium was replaced after 24 h with the C. albicans supernatants and cultured for indicated times. For direct cell contact suspension cultures of C. albicans (final concentration 0.5 × 104 cells/ml) or PMNs (final concentration 0.5 × 106 cells/ml) were added to the OE21 cells after initial 24 h culture and incubated for the indicated times. In the transwell setting (for a schematic representation please refer to Figure 2) 2 × 105 OE21 cells were cultured into the bottom compartment of a six-well culture plate. After 24 h PMNs or C. albicans were added to the bottom compartment and the corresponding cell type was seeded into the top transwell compartment (Costar GmbH, Bodenheim, Germany) and cultured for additional 24 h.
RNA isolation and cDNA synthesis
Total RNA was isolated using the RNeasy Kit from Qiagen (Hilden, Germany) and reverse-transcribed into single-stranded cDNA, as described previously [56].
Primer
The oligonucleotide sequence and product size for each primer pair used were as described previously [25].
Quantification of gene expression by real time PCR
Real-time PCR analyses were performed as previously described [56]. Standard curves for each mRNA were constructed by cloning the purified PCR-products containing the target sequence into pCR-Blunt II-TOPO vector (Invitrogen). Concentration of the reference plasmid was measured spectrophotometrically and transformed into number of copies/μl by calculation. The absolute mRNA transcript number in each sample was calculated by use of calibration curves. Identical results were obtained in control experiments.
Cytokine profilling
Culture supernatants were cleared by centrifugation (6.000 rpm, 5 min, 4°C) and protein concentration was determined by the BioRad assays. For a qualitative screening for the cytokine content, samples were adjusted to equal protein concentration and submitted to cytokine antibody arrays (Cytokine Profiler kit, R&D systems) following the manufacturer's instructions.
siRNA transfection
For knock down of RelA/p65, cells were seeded into 6 well plates (2 × 105 cells/well) and grown overnight, then transfection with 12 μl/well RNAiFect reagent (Invitrogen) and 2 μg/well of either Stealth negative control siRNA (Invitrogen) or Stealth RelA/p65 siRNA (Invitrogen) was performed for 48 h.
Western Blotting
Cellular lysates were prepared and western blotting was performend as described previously [57,58].
EMSA
Nuclear extracts were prepared as described previously [58] and incubated with a γ32P-labelled oligonucleotide containing a consensus NF-κB-binding or consensus AP-1 site (Promega; Mannheim, FRG). After 30 min incubation at room temperature, samples were separated by gel electrophoresis at 100 V and 4°C. Gels were dried and exposed to X-ray Hyperfilm (Amersham; Freiburg, FRG).
Statistics
Data are presented as mean ± SD and analyzed by Student's t-test. A p-value < 0.05 (indicated as * in the figures) was considered as statistically significant.
Authors' contributions
NS, GB, RP and KK carried out the experimental studies and drafted the manuscript. KR, RP, KHH, SS, URF and PR participated in the design of the study and performed the statistical analysis. AA conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
PMNs induce IL-6 and IL-8 in OE21 cells. Supernatants of OE21 cells kept in culture alone (upper panel) or with with direct contact with 0.5 × 106 PMNs/ml (lower panel) for 24 h were submitted to a peptide arrays detecting a broad panel of chemo- and cytokines.
Acknowledgments
Acknowledgements
The authors would like to thank Silvia Voss, Christiane Sandberg, Cornelia Wilgus and Frauke Grohmann for excellent technical support. This work is part of a PhD thesis (N.S.) and a MD thesis (R.P.). This study was supported by grants of the German Research Society (DFG/SFB617) to K.K., P.R. and A.A.
Contributor Information
Nadine Steubesand, Email: nadine@steubesand.org.
Karlheinz Kiehne, Email: kkiehne@1med.uni-kiel.de.
Gabriele Brunke, Email: gbrunke@1med.uni-kiel.de.
Rene Pahl, Email: rene.pahl@gmx.de.
Karina Reiss, Email: kreiss@dermatology.uni-kiel.de.
Karl-Heinz Herzig, Email: Karl-Heinz.Herzig@oulu.fi.
Sabine Schubert, Email: schubert@infmed.uni-kiel.de.
Stefan Schreiber, Email: s.schreiber@mucosa.de.
Ulrich R Fölsch, Email: urfoelsch@1med.uni-kiel.de.
Philip Rosenstiel, Email: p.rosenstiel@mucosa.de.
Alexander Arlt, Email: aarlt@1med.uni-kiel.de.
References
- Tortorano AM, Kibbler C, Peman J, Bernhardt H, Klingspor L, Grillot R. Candidaemia in Europe: epidemiology and resistance. Int J Antimicrob Agents. 2006;27:359–366. doi: 10.1016/j.ijantimicag.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Jensen T, Obel N. Preserved neutrophil response to Candida albicans stimulation in AIDS patients with candida esophagitis. Apmis. 1996;104:591–597. doi: 10.1111/j.1699-0463.1996.tb04916.x. [DOI] [PubMed] [Google Scholar]
- Gornig M, Emmert-Buck M, Walsh TJ. [Patterns of Candida esophagitis in cancer and AIDS patients: histopathological study of 23 patients] Mycoses. 1997;40:81–85. doi: 10.1111/j.1439-0507.1997.tb00547.x. [DOI] [PubMed] [Google Scholar]
- Vilanova M, Correia A. Host defense mechanisms in invasive candidiasis originating in the GI tract. Expert Rev Anti Infect Ther. 2008;6:441–445. doi: 10.1586/14787210.6.4.441. [DOI] [PubMed] [Google Scholar]
- Weindl G, Naglik JR, Kaesler S, Biedermann T, Hube B, Korting HC, Schaller M. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J Clin Invest. 2007;117:3664–3672. doi: 10.1172/JCI28115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- Rosenstiel P, Till A, Schreiber S. NOD-like receptors and human diseases. Microbes Infect. 2007;9:648–657. doi: 10.1016/j.micinf.2007.01.015. [DOI] [PubMed] [Google Scholar]
- West TE, Ernst RK, Jansson-Hutson MJ, Skerrett SJ. Activation of Toll-like receptors by Burkholderia pseudomallei. BMC Immunol. 2008;9:46. doi: 10.1186/1471-2172-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder A, Kirschning CJ, Schaller M, Weindl G, Wagner H, Korting HC, Rupec RA. Induction of nuclear factor- kappa B and c-Jun/activator protein-1 via toll-like receptor 2 in macrophages by antimycotic-treated Candida albicans. J Infect Dis. 2004;190:1318–1326. doi: 10.1086/423854. [DOI] [PubMed] [Google Scholar]
- Froy O. Regulation of mammalian defensin expression by Toll-like receptor-dependent and independent signalling pathways. Cell Microbiol. 2005;7:1387–1397. doi: 10.1111/j.1462-5822.2005.00590.x. [DOI] [PubMed] [Google Scholar]
- Karlsson J, Putsep K, Chu H, Kays RJ, Bevins CL, Andersson M. Regional variations in Paneth cell antimicrobial peptide expression along the mouse intestinal tract. BMC Immunol. 2008;9:37. doi: 10.1186/1471-2172-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Graaf CA Van Der, Vonk AG, Verschueren I, Meer JW Van Der, Kullberg BJ. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J Infect Dis. 2002;185:1483–1489. doi: 10.1086/340511. [DOI] [PubMed] [Google Scholar]
- Ferwerda G, Meyer-Wentrup F, Kullberg BJ, Netea MG, Adema GJ. Dectin-1 synergizes with TLR2 and TLR4 for cytokine production in human primary monocytes and macrophages. Cell Microbiol. 2008;10:2058–2066. doi: 10.1111/j.1462-5822.2008.01188.x. [DOI] [PubMed] [Google Scholar]
- Saegusa S, Totsuka M, Kaminogawa S, Hosoi T. Candida albicans and Saccharomyces cerevisiae induce interleukin-8 production from intestinal epithelial-like Caco-2 cells in the presence of butyric acid. FEMS Immunol Med Microbiol. 2004;41:227–235. doi: 10.1016/j.femsim.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Netea MG, Veerdonk F van de, Verschueren I, Meer JW van der, Kullberg BJ. Role of TLR1 and TLR6 in the host defense against disseminated candidiasis. FEMS Immunol Med Microbiol. 2008;52:118–123. doi: 10.1111/j.1574-695X.2007.00353.x. [DOI] [PubMed] [Google Scholar]
- Vylkova S, Nayyar N, Li W, Edgerton M. Human beta-defensins kill Candida albicans in an energy-dependent and salt-sensitive manner without causing membrane disruption. Antimicrob Agents Chemother. 2007;51:154–161. doi: 10.1128/AAC.00478-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vylkova S, Li XS, Berner JC, Edgerton M. Distinct antifungal mechanisms: beta-defensins require Candida albicans Ssa1 protein, while Trk1p mediates activity of cysteine-free cationic peptides. Antimicrob Agents Chemother. 2006;50:324–331. doi: 10.1128/AAC.50.1.324-331.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Jiang B, Chandra J, Ghannoum M, Nelson S, Weinberg A. Human beta-defensins: differential activity against candidal species and regulation by Candida albicans. J Dent Res. 2005;84:445–450. doi: 10.1177/154405910508400509. [DOI] [PubMed] [Google Scholar]
- Joly S, Maze C, McCray PB, Jr, Guthmiller JM. Human beta-defensins 2 and 3 demonstrate strain-selective activity against oral microorganisms. J Clin Microbiol. 2004;42:1024–1029. doi: 10.1128/JCM.42.3.1024-1029.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boughan PK, Argent RH, Body-Malapel M, Park JH, Ewings KE, Bowie AG, Ong SJ, Cook SJ, Sorensen OE, Manzo BA, et al. Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection. J Biol Chem. 2006;281:11637–11648. doi: 10.1074/jbc.M510275200. [DOI] [PubMed] [Google Scholar]
- Wehkamp J, Schauber J, Stange EF. Defensins and cathelicidins in gastrointestinal infections. Curr Opin Gastroenterol. 2007;23:32–38. doi: 10.1097/MOG.0b013e32801182c2. [DOI] [PubMed] [Google Scholar]
- Wehkamp K, Schwichtenberg L, Schroder JM, Harder J. Pseudomonas aeruginosa- and IL-1beta-mediated induction of human beta-defensin-2 in keratinocytes is controlled by NF-kappaB and AP-1. J Invest Dermatol. 2006;126:121–127. doi: 10.1038/sj.jid.5700020. [DOI] [PubMed] [Google Scholar]
- Bowdish DM, Davidson DJ, Hancock RE. Immunomodulatory properties of defensins and cathelicidins. Curr Top Microbiol Immunol. 2006;306:27–66. doi: 10.1007/3-540-29916-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M, Zakikhany K, Naglik JR, Weindl G, Hube B. Models of oral and vaginal candidiasis based on in vitro reconstituted human epithelia. Nat Protoc. 2006;1:2767–2773. doi: 10.1038/nprot.2006.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehne K, Brunke G, Meyer D, Harder J, Herzig KH. Oesophageal defensin expression during Candida infection and reflux disease. Scand J Gastroenterol. 2005;40:501–507. doi: 10.1080/00365520510015386. [DOI] [PubMed] [Google Scholar]
- Cunliffe RN. Alpha-defensins in the gastrointestinal tract. Mol Immunol. 2003;40:463–467. doi: 10.1016/s0161-5890(03)00157-3. [DOI] [PubMed] [Google Scholar]
- Godaly G, Bergsten G, Hang L, Fischer H, Frendeus B, Lundstedt AC, Samuelsson M, Samuelsson P, Svanborg C. Neutrophil recruitment, chemokine receptors, and resistance to mucosal infection. J Leukoc Biol. 2001;69:899–906. [PubMed] [Google Scholar]
- Moon SK, Lee HY, Pan H, Takeshita T, Park R, Cha K, Andalibi A, Lim DJ. Synergistic effect of interleukin 1 alpha on nontypeable Haemophilus influenzae-induced up-regulation of human beta-defensin 2 in middle ear epithelial cells. BMC Infect Dis. 2006;6:12. doi: 10.1186/1471-2334-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang Z, Louboutin JP, Moser C, Weiner DJ, Wilson JM. Airway epithelia regulate expression of human beta-defensin 2 through Toll-like receptor 2. Faseb J. 2003;17:1727–1729. doi: 10.1096/fj.02-0616fje. [DOI] [PubMed] [Google Scholar]
- Sayama K, Komatsuzawa H, Yamasaki K, Shirakata Y, Hanakawa Y, Ouhara K, Tokumaru S, Dai X, Tohyama M, Ten Dijke P, et al. New mechanisms of skin innate immunity: ASK1-mediated keratinocyte differentiation regulates the expression of beta-defensins, LL37, and TLR2. Eur J Immunol. 2005;35:1886–1895. doi: 10.1002/eji.200425827. [DOI] [PubMed] [Google Scholar]
- Albanesi C, Fairchild HR, Madonna S, Scarponi C, De Pita O, Leung DY, Howell MD. IL-4 and IL-13 negatively regulate TNF-alpha- and IFN-gamma-induced beta-defensin expression through STAT-6, suppressor of cytokine signaling (SOCS)-1, and SOCS-3. J Immunol. 2007;179:984–992. doi: 10.4049/jimmunol.179.2.984. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- Menzies BE, Kenoyer A. Signal transduction and nuclear responses in Staphylococcus aureus-induced expression of human beta-defensin 3 in skin keratinocytes. Infect Immun. 2006;74:6847–6854. doi: 10.1128/IAI.00389-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim Biophys Acta. 2007;1773:1285–1298. doi: 10.1016/j.bbamcr.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Turjanski AG, Vaque JP, Gutkind JS. MAP kinases and the control of nuclear events. Oncogene. 2007;26:3240–3253. doi: 10.1038/sj.onc.1210415. [DOI] [PubMed] [Google Scholar]
- Miller LS, Sorensen OE, Liu PT, Jalian HR, Eshtiaghpour D, Behmanesh BE, Chung W, Starner TD, Kim J, Sieling PA, et al. TGF-alpha regulates TLR expression and function on epidermal keratinocytes. J Immunol. 2005;174:6137–6143. doi: 10.4049/jimmunol.174.10.6137. [DOI] [PubMed] [Google Scholar]
- Sorensen OE, Thapa DR, Rosenthal A, Liu L, Roberts AA, Ganz T. Differential regulation of beta-defensin expression in human skin by microbial stimuli. J Immunol. 2005;174:4870–4879. doi: 10.4049/jimmunol.174.8.4870. [DOI] [PubMed] [Google Scholar]
- Barker KS, Park H, Phan QT, Xu L, Homayouni R, Rogers PD, Filler SG. Transcriptome profile of the vascular endothelial cell response to Candida albicans. J Infect Dis. 2008;198:193–202. doi: 10.1086/589516. [DOI] [PubMed] [Google Scholar]
- Muller V, Viemann D, Schmidt M, Endres N, Ludwig S, Leverkus M, Roth J, Goebeler M. Candida albicans triggers activation of distinct signaling pathways to establish a proinflammatory gene expression program in primary human endothelial cells. J Immunol. 2007;179:8435–8445. doi: 10.4049/jimmunol.179.12.8435. [DOI] [PubMed] [Google Scholar]
- Inoue Y, Matsuwaki Y, Shin SH, Ponikau JU, Kita H. Nonpathogenic, environmental fungi induce activation and degranulation of human eosinophils. J Immunol. 2005;175:5439–5447. doi: 10.4049/jimmunol.175.8.5439. [DOI] [PubMed] [Google Scholar]
- Wehkamp J, Harder J, Wehkamp K, Wehkamp-von Meissner B, Schlee M, Enders C, Sonnenborn U, Nuding S, Bengmark S, Fellermann K, et al. NF-kappaB- and AP-1-mediated induction of human beta defensin-2 in intestinal epithelial cells by Escherichia coli Nissle 1917: a novel effect of a probiotic bacterium. Infect Immun. 2004;72:5750–5758. doi: 10.1128/IAI.72.10.5750-5758.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krisanaprakornkit S, Kimball JR, Dale BA. Regulation of human beta-defensin-2 in gingival epithelial cells: the involvement of mitogen-activated protein kinase pathways, but not the NF-kappaB transcription factor family. J Immunol. 2002;168:316–324. doi: 10.4049/jimmunol.168.1.316. [DOI] [PubMed] [Google Scholar]
- Ogushi K, Wada A, Niidome T, Mori N, Oishi K, Nagatake T, Takahashi A, Asakura H, Makino S, Hojo H, et al. Salmonella enteritidis FliC (flagella filament protein) induces human beta-defensin-2 mRNA production by Caco-2 cells. J Biol Chem. 2001;276:30521–30526. doi: 10.1074/jbc.M011618200. [DOI] [PubMed] [Google Scholar]
- Wada A, Ogushi K, Kimura T, Hojo H, Mori N, Suzuki S, Kumatori A, Se M, Nakahara Y, Nakamura M, et al. Helicobacter pylori-mediated transcriptional regulation of the human beta-defensin 2 gene requires NF-kappaB. Cell Microbiol. 2001;3:115–123. doi: 10.1046/j.1462-5822.2001.00096.x. [DOI] [PubMed] [Google Scholar]
- Schlee M, Harder J, Koten B, Stange EF, Wehkamp J, Fellermann K. Probiotic lactobacilli and VSL#3 induce enterocyte beta-defensin 2. Clin Exp Immunol. 2008;151:528–535. doi: 10.1111/j.1365-2249.2007.03587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WO, Dale BA. Innate immune response of oral and foreskin keratinocytes: utilization of different signaling pathways by various bacterial species. Infect Immun. 2004;72:352–358. doi: 10.1128/IAI.72.1.352-358.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly S, Organ CC, Johnson GK, McCray PB, Jr, Guthmiller JM. Correlation between beta-defensin expression and induction profiles in gingival keratinocytes. Mol Immunol. 2005;42:1073–1084. doi: 10.1016/j.molimm.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Koff JL, Shao MX, Ueki IF, Nadel JA. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1068–1075. doi: 10.1152/ajplung.00025.2008. [DOI] [PubMed] [Google Scholar]
- Pastore S, Mascia F, Mariani V, Girolomoni G. The epidermal growth factor receptor system in skin repair and inflammation. J Invest Dermatol. 2008;128:1365–1374. doi: 10.1038/sj.jid.5701184. [DOI] [PubMed] [Google Scholar]
- Zuyderduyn S, Ninaber DK, Hiemstra PS, Rabe KF. The antimicrobial peptide LL-37 enhances IL-8 release by human airway smooth muscle cells. J Allergy Clin Immunol. 2006;117:1328–1335. doi: 10.1016/j.jaci.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Tjabringa GS, Aarbiou J, Ninaber DK, Drijfhout JW, Sorensen OE, Borregaard N, Rabe KF, Hiemstra PS. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J Immunol. 2003;171:6690–6696. doi: 10.4049/jimmunol.171.12.6690. [DOI] [PubMed] [Google Scholar]
- Flamand L, Tremblay MJ, Borgeat P. Leukotriene B4 triggers the in vitro and in vivo release of potent antimicrobial agents. J Immunol. 2007;178:8036–8045. doi: 10.4049/jimmunol.178.12.8036. [DOI] [PubMed] [Google Scholar]
- Wan M, Sabirsh A, Wetterholm A, Agerberth B, Haeggstrom JZ. Leukotriene B4 triggers release of the cathelicidin LL-37 from human neutrophils: novel lipid-peptide interactions in innate immune responses. Faseb J. 2007;21:2897–2905. doi: 10.1096/fj.06-7974com. [DOI] [PubMed] [Google Scholar]
- Soehnlein O, Zernecke A, Eriksson EE, Rothfuchs AG, Pham CT, Herwald H, Bidzhekov K, Rottenberg ME, Weber C, Lindbom L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood. 2008;112:1461–1471. doi: 10.1182/blood-2008-02-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haar SF, Hiemstra PS, van Steenbergen MT, Everts V, Beertsen W. Role of polymorphonuclear leukocyte-derived serine proteinases in defense against Actinobacillus actinomycetemcomitans. Infect Immun. 2006;74:5284–5291. doi: 10.1128/IAI.02016-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehne K, Fincke A, Brunke G, Lange T, Folsch UR, Herzig KH. Antimicrobial peptides in chronic anal fistula epithelium. Scand J Gastroenterol. 2007;42:1063–1069. doi: 10.1080/00365520701320489. [DOI] [PubMed] [Google Scholar]
- Arlt A, Rosenstiel P, Kruse ML, Grohmann F, Minkenberg J, Perkins ND, Folsch UR, Schreiber S, Schafer H. IEX-1 directly interferes with RelA/p65 dependent transactivation and regulation of apoptosis. Biochim Biophys Acta. 2008;1783:941–952. doi: 10.1016/j.bbamcr.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Sebens Muerkoster S, Rausch AV, Isberner A, Minkenberg J, Blaszczuk E, Witt M, Folsch UR, Schmitz F, Schafer H, Arlt A. The apoptosis-inducing effect of gastrin on colorectal cancer cells relates to an increased IEX-1 expression mediating NF-kappa B inhibition. Oncogene. 2008;27:1122–1134. doi: 10.1038/sj.onc.1210728. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PMNs induce IL-6 and IL-8 in OE21 cells. Supernatants of OE21 cells kept in culture alone (upper panel) or with with direct contact with 0.5 × 106 PMNs/ml (lower panel) for 24 h were submitted to a peptide arrays detecting a broad panel of chemo- and cytokines.