

Abstract
Sulfate-reducing bacteria (SRB) belonging to the metabolically versatile Desulfobacteriaceae are abundant in marine sediments and contribute to the global carbon cycle by complete oxidation of organic compounds. Desulfobacterium autotrophicum HRM2 is the first member of this ecophysiologically important group with a now available genome sequence. With 5.6 megabasepairs (Mbp) the genome of Db. autotrophicum HRM2 is about 2 Mbp larger than the sequenced genomes of other sulfate reducers (SRB). A high number of genome plasticity elements (> 100 transposon-related genes), several regions of GC discontinuity and a high number of repetitive elements (132 paralogous genes Mbp−1) point to a different genome evolution when comparing with Desulfovibrio spp. The metabolic versatility of Db. autotrophicum HRM2 is reflected in the presence of genes for the degradation of a variety of organic compounds including long-chain fatty acids and for the Wood–Ljungdahl pathway, which enables the organism to completely oxidize acetyl-CoA to CO2 but also to grow chemolithoautotrophically. The presence of more than 250 proteins of the sensory/regulatory protein families should enable Db. autotrophicum HRM2 to efficiently adapt to changing environmental conditions. Genes encoding periplasmic or cytoplasmic hydrogenases and formate dehydrogenases have been detected as well as genes for the transmembrane TpII-c3, Hme and Rnf complexes. Genes for subunits A, B, C and D as well as for the proposed novel subunits L and F of the heterodisulfide reductases are present. This enzyme is involved in energy conservation in methanoarchaea and it is speculated that it exhibits a similar function in the process of dissimilatory sulfate reduction in Db. autotrophicum HRM2.
Introduction
Shelf sediments receive the highest input of organic carbon among marine systems. They are mostly anoxic and more than 50% of the mineralization of the organic carbon is coupled in these environments to bacterial sulfate reduction (Jørgensen, 1982; Canfield et al., 1993). Typically, sulfate-reducing bacteria (SRB) utilize small reduced molecules mostly fermentation products, e.g. acetate, lactate or ethanol, placing them together with syntrophs and methanogens at the end of the anaerobic food chain (Widdel, 1988; Rabus et al., 2000). The observed high mineralization rates coupled to bacterial sulfate reduction are indicative for complete oxidation of organic substrates to CO2 (Fenchel and Jørgensen, 1977; Jørgensen, 1982). While the frequently isolated and intensively studied Desulfovibrio spp. oxidize organic substrates only to the level of acetate, complete oxidation was first demonstrated for Desulfotomaculum acetoxidans (Widdel and Pfennig, 1977), Desulfobacter postgatei (Widdel and Pfennig, 1981) and Desulfobacterium autotrophicum HRM2 (Brysch et al., 1987). The nutritionally versatile Db. autotrophicum HRM2 oxidizes a variety of organic acids and alcohols to CO2 and, in addition, is able to grow chemolithoautotrophically with H2, CO2 and sulfate. While Desulfobacter spp. so far investigated employ modified citric acid cycles for terminal oxidation of acetyl-CoA (Brandis-Heep et al., 1983), the Wood–Ljungdahl pathway is used by Db. autotrophicum HRM2 (Schauder et al., 1986) with acetyl-CoA synthase/CO dehydrogenase (ACS/CODH) as the key enzyme complex. This pathway presumably also functions in the reductive direction for CO2 fixation under autotrophic growth conditions (Länge et al., 1989; Schauder et al., 1989). Corroborating their ecophysiological role members of the Desulfobacteriaceae were repeatedly observed to dominate the population of SRB in various anoxic habitats, while Desulfovibrio species were mostly absent (e.g. Teske et al., 1998; Llobet-Brossa et al., 2002; Dhillon et al., 2003). Recognition of the environmental significance of SRB resulted in several genome-sequencing projects (for overview see Rabus and Strittmatter, 2007). Desulfobacterium autotrophicum HRM2 is the first representative of the ecophysiologically important group of the completely oxidizing sulfate reducers. The genome sequence presented here reflects its high metabolic versatility and reveals novel insights into the bioenergetics of dissimilatory sulfate reduction.
Results and discussion
General genome features
Genome size and coding sequences
The genome of Db. autotrophicum HRM2 consists of two circular replicons, a chromosome of 5 589 073 base pairs (bp) encoding 4871 CDS (Accession No. CP001087) and a plasmid (pHRM2a) of 62 962 bp encoding 76 CDS (CP001088). Three of these encode proteins for plasmid maintenance of the Par family, the remaining CDS only share weak similarities with other proteins. Therefore, the plasmid does not carry any known physiological function. With 5.5 megabasepairs (Mbp) the chromosome of Db. autotrophicumHRM2 is about 2 Mbp larger than those of the other three δ-proteobacterial SRB with to date published genomes: Desulfotalea psychrophila LSv54 (Rabus et al., 2004), Desulfovibrio vulgaris Hildenborough (Heidelberg et al., 2004) and Desulfovibrio desulfuricansG20 (Copeland et al., 2005). The principal features of the Db. autotrophicumHRM2 genome in comparison with other sequenced sulfate- and sulfur-reducing prokaryotes are summarized in Table 1. A general overview is given in the Fig. S1.
Table 1.
General genome features of Db. autotrophicum HRM2, of four other δ-proteobacterial sulfate reducers, of G. sulfurreducens and of the archaeon A. fulgidus.
| Genome feature |
Db. autotrophicum HRM2 |
Dt. psychrophila LSv54 |
Dv. vulgaris Hildenborough |
Dv. desulfuricans G20 |
G. sulfurreducens PCA |
A. fulgidus VC-16 |
Dc. oleovorans Hxd3 |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Replicon | Chrom. | Plasmid | Chrom. | Large plasmid | Small plasmid | Chrom. | Plasmid | Chrom. | Chrom. | Chrom. | Chrom. |
| Size (bp) | 5 589 073 | 62 962 | 3 523 383 | 121 587 | 14 664 | 3 570 858 | 202 301 | 3 730 232 | 3 814 139 | 2 178 400 | 3 944 167 |
| G+C content (mol%) | 48.9 | 42.0 | 46 | 43 | 28 | 63 | 66 | 58 | 60 | 48 | 56 |
| Stable RNAs | |||||||||||
| rRNAs | 19 | – | 22 | – | – | 15 | – | 12 | 6 | 3 | 3 |
| tRNAs | 51 | – | 64 | – | – | 67 | 1 | 66 | 49 | 46 | 46 |
| Coding sequences (CDS) | 4 871 | 76 | 3 116 | 101 | 17 | 3 379 | 152 | 3 775 | 3 446 | 2 420 | 3 265 |
| Coding (%) | 89 | 79 | 85 | 77 | 72 | 86 | 85 | 90 | 90 | 91 | n.d. |
| Assigned functions (%) | 71 | 41 | 54 | 60 | 24 | 63 | 63 | 61 | 68 | 48 | n.d. |
| Hypothetical proteins (%) | 29 | 59 | 46 | 40 | 76 | 37 | 37 | 39 | 32 | 62 | n.d. |
| Paralogues genes (Mbp−1) | 132 | n.d. | 35 | n.d. | n.d. | 39 | n.d. | 94 | 77 | 87 | n.d. |
| Orthologues genes | ref | – | 1 534 | n.d. | n.d. | 1 472 | n.d. | 1 526 | 1 428 | 681 | 1 688 |
| % of Db. auto in X | ref | – | 31 | n.d. | n.d. | 30 | n.d. | 31 | 24 | 14 | 34 |
| % of X in Db. auto | ref | – | 47 | n.d. | n.d. | 42 | n.d. | 40 | 34 | 28 | 51 |
| Selenocysteine- containing proteins | 31 | 0 | 9 | 0 | 0 | 8 | 1 | n.d. | 14 | 0 | 0 |
n.d., no data; Db., Desulfobacterium, G., Geobacter, Dt., Desulfotalea, Dv., Desulfovibrio, A., Archaeoglobus, Dc., Desulfococcus; ref, reference genome for BiBaG (A. Wollherr and H. Liesegang, pers. comm.).
Paralogous proteins and repeats
The Db. autotrophicum HRM2 genome contains 2357 exact DNA repeats greater than or equal to 50 bp, giving an average of 422 repeats per Mbp (Mbp−1). The total number of genes with one or more paralogues is 1460. This corresponds to 265 genes with paralogues per Mbp, and is the highest number of repetitive DNA stretches and the highest number of paralogous genes in all compared genomes (Table 1, Tables S1 and S2). The distribution of the number of paralogous genes is strongly biased, e.g. key enzymes of the sulfate reduction pathway, CO2 fixation and central metabolism are unique or have only few copies (≤ 3). This is in clear contrast to genes for substrate utilization where for instance 11 paralogues of acyl-CoA synthetases and 17 paralogues of acyl-CoA dehydrogenases could be identified. The presence of multiple copies of metabolic genes could represent an ecological advantage allowing an expansion of functional capabilities in response to varying environmental conditions (e.g. redox states or substrate concentrations).
Comparative genomics
A bidirectional blast comparison of Db. autotrophicum HRM2 with Dt. psychrophila LSv54 (Rabus et al., 2004), Dv. desulfuricans G20 (Copeland et al., 2005), Dv. vulgaris Hildenborough (Heidelberg et al., 2004), Archaeoglobus fulgidus VC-16 (Klenk et al., 1997), Geobacter sulfurreducens PCA (Methéet al., 2003) and Desulfococcus oleovorans Hxd3 (Copeland et al., 2008) revealed that these organisms share between 681 and 1688 orthologous proteins (Table 1). However, genome alignments did not show significant synteny between Db. autotrophicum HRM2 and any other SRB. Apparently, there is no comprehensive ‘core’-genome characteristic for all sulfate-reducing prokaryotes (see also Fig. S2).
Carbon metabolism
Fatty acids
Important features of the carbon metabolism of Db. autotrophicum HRM2 are depicted in Fig. 1. One of the ecologically important features of Db. autotrophicum HRM2 is the utilization of fatty acids up to a carbon chain length of 16. These fatty acids are probably taken up by a H+-driven symporter, then activated to the respective CoA esters (FadD) and broken down to acetyl-CoA moieties by classical β-oxidation (Bcd, FabGH, FadBGA). Db. autotrophicum HRM2 contains at least 17 acyl-CoA dehydrogenase genes (acd) and 11 acetyl-CoA synthetase genes (acs) with specificities for short-, medium- and long-chain fatty acids. Three sets of genes, one short, one medium and one long chain specific, have homologues in other SRB despite the finding that complete fatty acid degradation has not been found in these organisms (Fig. 2). The remaining sets of genes have orthologues in anaerobic firmicutes (Moorella thermoacetica and Desulfitobacterium hafniense) and in proteobacteria known for their ability to utilize a broad spectrum of substrates (Ralstonia eutropha JMP134, Pseudomonas putida and ‘Aromatoleum aromaticum’ EbN1) (Fig. 2).
Fig. 1.

Metabolic reconstruction of Db. autotrophicum HRM2 based on known growth substrates and metabolic capacities. Complete oxidation of organic substrates and CO2 fixation under autotrophic growth conditions proceeds via the Wood–Ljungdahl pathway (blue box). The sulfate reduction is given in a dark red box, heterodisulfide reduction is given in a dark orange box and electron transfer from Qmo to sulfate reduction is given in an orange box. All selenium-dependent proteins are marked by dark grey backgrounds and an asterisk (*). The cytoplasmic membrane is given in light grey. Arrows indicate metabolic flows; dashed lines indicate assumed or putative electron flows. For reasons of simplicity, cytochromes, multihaem proteins and the following proteins with one or more paralogues are displayed only once in the figure: Acs, CysAW, EtfAB, FdhAB, GlpAB, HdrA, HdrL, MvhD, PorABC, RnfA–E, Sat, Suc, SulP, TmcBCA. Abbreviations are: CM, cytoplasmic membrane; OM, outer membrane; Acs/CODH, acetyl-CoA synthase/CO dehydrogenase; Acs, acetyl-CoA synthetase; AcsF, CODH maturation factor; Adh, aldehyde dehydrogenase; AMP/ADP/ATP, adenosine-monophosphate/-diphosphate/-triphosphate; AprAB, adenylylsulfate reductase; AtpA–I, ATP synthase F0/F1; Cit, citrate synthase; CO2, carbon dioxide; Cdh1/2, carbon monoxide dehydrogenase; CdhA–E, bifunctional acetyl-CoA synthetase/carbon monoxide dehydrogenase; DctPQM, TRAP-type C4-dicarboxylate transporter; DsrABC, dissimilatory sulfite reductase complex; EtfABox/EtfABred, electron transfer flavoprotein, oxidized and reduced form; FabGH, 3-oxoacyl-acyl carrier synthase/reductase; FadDBA, long-chain fatty acid-CoA ligase; FdhABCD, formate dehydrogenase; Fdox/Fdred, ferredoxin, oxidized and reduced form; FdrABC, fumarate reductase; Fhs, formate-tetrahydrofolate ligase; Fdx, ferredoxin, 4Fe-4S cluster; Fhs, formate-tetrahydrofolate ligase; FolD, methylenetetrahydrofolate dehydrogenase; FumAC, fumarate hydratase; GlpAB, glycerol-3-phosphate dehydrogenase; HdrADFL, heterodisulfide reductase; HmeCDEP, Hdr-like menaquinol-oxidizing complex; HydA, [Fe]-only fusion hydrogenase; HyfBCDEFGI, hydrogenase Hyf homologue; HynAB, periplasmic [Ni/Fe] hydrogenase; HysAB, periplasmic [Ni/Fe/Se] hydrogenase; Idh, isocitrate dehydrogenase; LdhAB, lactate dehydrogenase; LldP, l-lactate permease; Mae, malic enzyme; MetF-ABC, methylenetetrahydrofolate reductase; MQ/MQ-H2, menaquinone pool, oxidized and reduced form; MvhADG, methylviologen non-reducing hydrogenase; NAD+, NADH/H+, nicotinamide-adenine dinucleotide, oxidized and reduced form; NADP+, NADPH/H+, nicotinamide-adenine dinucleotide phosphate, oxidized and reduced form; NqrA–F, Na+-translocating NADH-quinone reductase; PccB, propionyl-CoA carboxylase; Pfl, pyruvate formate lyase; PorABC, pyruvate:ferredoxin oxidoreductase; PPi, pyrophosphate; QmoABC, quinone-interacting membrane-bound oxidoreductase complex; RnfA–E, electron transport complex protein; Sat, sulfate adenylyl transferase; Sbm, methylmalonyl-CoA mutase; SdhABC, succinate dehydrogenase/fumarate reductase; SseA, thiosulfate sulfur transferase; SucCD, succinyl-CoA synthetase; SulP, high-affinity H+/SO42− symporter; TmcABC, acidic type II cytochrome complex; Tst, thiosulfate sulfur transferase; X/XH2, unknown carrier of reducing equivalent, oxidized and reduced form.
Fig. 2.
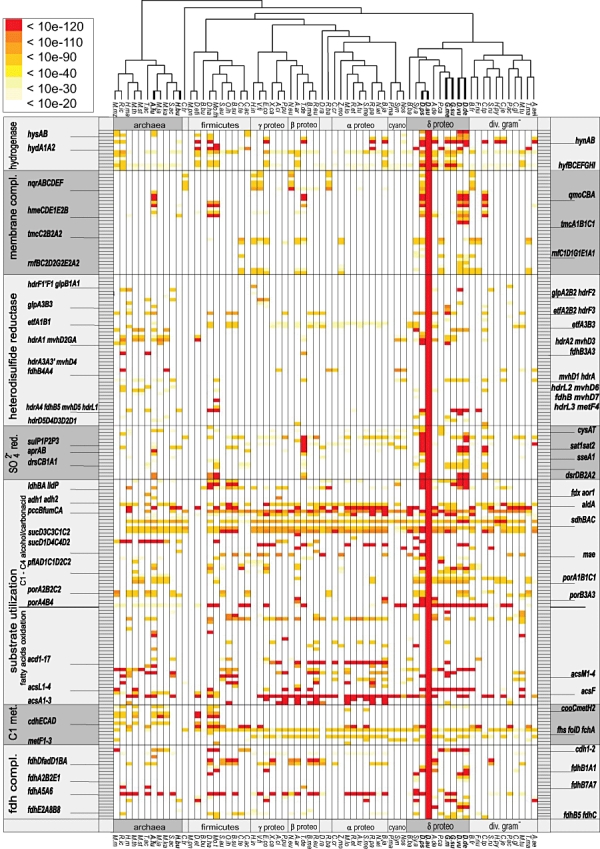
Metabolic heat map of Db. autotrophicum HRM2 (white-orange-red scale on similarity; metabolic genes versus organisms). In the red column 206 metabolic genes of Db. autotrophicum HRM2 are grouped into seven functional categories: hydrogenase, membrane complexes, hdr genes, sulfate reduction, substrate utilization, C1-metabolic pathway and formate dehydrogenases. First and best bidirectional alignments with the proteins of 67 phylogenetically important, but taxonomically diverse prokaryotes are given by colour-coded boxes. Colours correspond to similarity as indicated on the top left panel. A white background indicates no similarities and no hits, dark-red boxes indicate high similarities and best hits. The grey ladders on the left and the right side indicate the 206 genes compared. Paralogous genes in Db. autotrophicum HRM2 were not indicated by name but by corresponding boxes, e.g. 17 grey boxes for acd1–17. The complete bidirectional blast data with 700 to date sequenced prokaryotes genomes and the 4947 genes of Db. autotrophicum HRM2 are given in Table S1. Order of organism abbreviations follows the grouping used in this figure: Archaea:M.mz, Methanosarcina mazei; R.ic, Rice cluster RC1; H.ma, Haloarcula marismortui ATCC 43049; M.th, Methanothermobacter thermoautotrophicus str. ΔH; M.st, Methanosphaera stadtmanae; T.ac, Thermoplasma acidophilum; A.fu, Archaeoglobus fulgidus; M.ja, Methanococcus jannaschii; M.ka, Methanopyrus kandleri; S.ac, Sulfolobus acidocaldarius DSM 639; H.bu, Hyperthermus butylicus; Chlamydiae: C.tr, Chlamydia trachomatis; Firmicutes: M.pn, Mycoplasma pneumoniae; D.et, Dehalococcoides ethenogenes 195; B.bu, Borrelia burgdorferi; D.ha, Desulfitobacterium hafniense Y51; Mo.th, Moorella thermoacetica ATCC 39073; S.au, Staphylococcus aureus MRSA252; O.ih, Oceanobacillus iheyensis; B.su, Bacillus subtilis 168; C.te, Clostridium tetani E88; C.ac, Clostridium acetobutylicum; γ-proteobacteria: H.in, Haemophilus influenzae; V.fi, Vibrio fischeri ES114; E.co, Escherichia coli K12; X.ca, Xanthomonas campestris; A.ci, Acinetobacter sp. ADP1; P.pu, Pseudomonas putida KT2440; β-proteobacteria: N.eu, Nitrosomonas europaea; A.ar.,‘Aromatoleum aromaticum’ EbN1; T.de, Thiobacillus denitrificans ATCC 25259; B.ma, Burkholderia mallei ATCC 23344; R.eu, Ralstonia eutropha JMP134; Deinococcus-Thermus: D.ra, Deinococcus radiodurans; Planctomycetes: R.ba, Rhodopirellula baltica; α-proteobacteria: C.cr, Caulobacter crescentus; Z.mo, Zymomonas mobilis ZM4; M.lo, Mesorhizobium loti; R.et, Rhizobium etli CFN 42; A.tu, Agrobacterium tumefaciens C58; S.me, Sinorhizobium meliloti; R.pa, Rhodopseudomonas palustris CGA009; N.wi, Nitrobacter winogradskyi Nb-255; B.ja, Bradyrhizobium japonicum; Cyanobacteria: P.ma, Prochlorococcus marinus CCMP1375; Syn, Synechocystis PCC6803; Nos, Nostoc sp.; α-proteobacteria: B.ba, Bdellovibrio bacteriovorus; Sy.a, Syntrophus aciditrophicus SB; D.ps, Desulfotalea psychrophila LSv54; D.au, Desulfobacterium autotrophicum HRM2; A.de, Anaeromyxobacter dehalogenans 2CP-C; P.ca, Pelobacter carbinolicus; G.me, Geobacter metallireducens GS-15; G.su, Geobacter sulfurreducens; D.vu, Desulfovibrio vulgaris Hildenborough; D.de, Desulfovibrio desulfuricans G20; diverse Gram-negative bacteria: B.fr, Bacteroides fragilis NCTC 9434; F.nu, Fusobacterium nucleatum; C.tp, Chlorobium tepidum TLS; S.ru, Salinibacter ruber DSM 13855; H.py, Helicobacter pylori J99; C.je, Campylobacter jejuni; P.ac, Propionibacterium acnes KPA171202; C.gl, Corynebacterium glutamicum ATCC 13032; M.tu, Mycobacterium tuberculosis H37Rv; T.ma, Thermotoga maritima; A.ae, Aquifex aeolicus.
Organic acids
Db. autotrophicum HRM2 utilizes formate, lactate as well as succinate, fumarate and malate (Brysch et al., 1987) and the genes for all required enzymes were detected. The fdhAD genes encode for three periplasmic formate dehydrogenases with TAT-signal peptides. Eight genes for cytoplasmic formate dehydrogenases are present, three of which have orthologues in Archaea. Genes for enzymes involved in the utilization of the other acids mentioned correspond to the homologous genes in other SRB (see Fig. 2). This is also true for the single alcohol dehydrogenase gene present.
Degradation of propanol and uneven fatty acids
The degradation of all these carbon sources necessitates channelling through the methylmalonyl-CoA pathway to convert intermediate propionyl-CoA via succinyl-CoA to acetyl-CoA. A gene coding for a methylmalonyl-CoA mutase (sbm) could be identified on the chromosome. blast analysis showed that Sbm has the highest similarity with the α-subunit of methylmalonyl-CoA mutase from Leptospira interrogans (Ren et al., 2003). It seems that a gene encoding the small β-subunit of methylmalonyl-CoA mutase, which is known in some bacteria (Birch et al., 1993; Bott et al., 1997), is absent in Db. autotrophicum HRM2. Thus the enzyme of Db. autotrophicum HRM2 could be a homodimeric enzyme as reported for the methylmalonyl-CoA mutase from Escherichia coli (Roy and Leadly, 1992). The analysis of the sbm gene product revealed the presence of the highly conserved signature sequence RIANT at position 370–376 and a classical binding motive for the cobalamin cofactor, DxHxxG(41)SxL(26–28)GG (Charles and Aneja, 1999). In close proximity of the sbm gene all genes required for the other enzymes of the methylmalonyl-CoA pathway are present on the chromosome of Db. autotrophicum HRM2 resulting in an operon-like organization.
Complete oxidation of acetyl-CoA
A central element of the metabolic network of Db. autotrophicum HRM2 is the ACS/CODH pathway, since complete oxidation of acetyl-CoA to CO2 (heterotrophy) and CO2 fixation (autotrophy) proceed via this pathway. A gene cluster could be identified on the chromosome of Db. autotrophicum HRM2 encoding all proteins required for the Wood–Ljungdahl pathway to operate in oxidative as well as reductive direction (Fig. 3). The genomic locus is arranged in four operon-like structures. The genes encoding a ACS/CODH form one of these units (classification of CODHs according to Lindahl, 2002). The ACS/CODH protein exhibit the highest similarities to proteins from sulfite-reducing firmicutes, i.e. Mo. thermoacetica (Morton et al., 1991), Desulfitobacterium hafniense (Nonaka et al., 2006) and archaea; the complex lacks any orthologues to the SRB from the δ-proteobacterial SRB (Fig. 2). The genes encoding the proteins of the methyl branch of the pathway are organized in three distinct groups containing (i) methylene-tetrahydrofolate reductase (MetF), (ii) methylene-tetrahydrofolate dehydrogenase (FolD) and (iii) C1-tetrahydrofolate synthetase (THF synthetase) (Fhs). Orthologous genes of the methyl branch can be found in all taxonomic groups, but again sulfite-reducing firmicutes and archaea contain the most similar orthologues (Fig. 2).
Fig. 3.
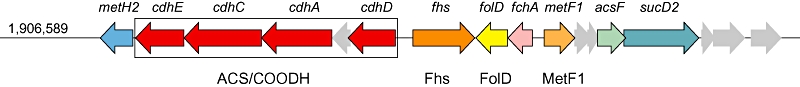
Genomic organization of the Wood–Ljungdahl pathway. In Db. autotrophicum HRM2 the genes encoding the key enzymes from the Wood–Ljungdahl pathway are organized in four operon-like structures in a single chromosomal locus. The genes encoding a bifunctional acetyl-CoA synthase/CO dehydrogenase (ACS/CODH) form one colinear group. The genes of the methyl branch of the Wood–Ljungdahl pathway are organized in three distinct groups containing (i) methylene-tetrahydrofolate reductase (MetF1), (ii) methylene-tetrahydrofolate dehydrogenase (FolD) and (iii) C1-tetrahydrofolate synthetase (THF synthetase) (Fhs).
Chemolithoautotrophy
The unique and physiologically important property of Db. autotrophicum HRM2 is the ability to grow with H2, CO2 and sulfate. It already has been mentioned that CO2 fixation may proceed via the Wood–Ljungdahl pathway. In addition, two further genes for monofunctional CODHs (cdh1 and cdh2) could be identified at different locations on the chromosome. In contrast to acs/codh, their genetic context displays no relation to any other proteins of the Wood–Ljungdahl pathway. CODH2 has highly similar orthologues in Methanosarcina mazei (Deppenmeier et al., 2002), Mo. thermoacetica (Morton et al., 1991), Ds. hafniense (Nonaka et al., 2006) and the SRB Dv. vulgaris (Heidelberg et al., 2004) and Dv. desulfuricans (Copeland et al., 2005). In Ms. mazei the orthologous gene product is the β-subunit of ACS/CODH, which catalyses the reversible oxidation of CO (Deppenmeier et al., 2002). Synthesis of pyruvate is then accomplished by one of the eight present pyruvate:ferredoxin oxidoreductases (por) and this compound can be further carboxylated to oxaloacetate by the biotin dependent pyruvate carboxylase PcB. The hydrogenases which, of course, are essential for chemolithoautotrophic growth will be discussed in the next session.
Energy conservation during sulfidogenesis
Dissimilatory sulfate reduction
Reduction of sulfate to sulfide can be divided into two steps: (i) the reduction of sulfate to sulfite, which is connected with the conversion of ATP to AMP and pyrophosphate, and (ii) the six-electron reduction of sulfite to sulfide. It was shown by Badziong and Thauer (1978) that bacterial growth yields with sulfite and hydrogen (H2) are higher than those with sulfate and hydrogen (H2). This is in agreement with an ATP-consuming first step but requires that the second step is coupled to the generation of a protonmotive force, which then can be taken advantage of for ATP synthesis. Sulfate uptake in Db. autotrophicum HRM2 is performed via three high-affinity, H+-driven symporters (sulP1–3), while homologues to the permease subunit CysP of the ABC-type transport system (cysATP) are absent. Two genes for ATP sulfurylase (sat) as well as genes for the reduction of the adenosine-5′-phosphosulfate (APS) as catalysed by a single APS reductase (aprAB) were detected (Fig. 1). Genes encoding the dissimilatory sulfite reductase (dsrABCD) converting sulfite to sulfide were shown to be present in two loci on the chromosome. In agreement with the capacity of Db. autotrophicum HRM2 to utilize thiosulfate as alternative electron acceptor, the chromosome contains a thiosulfate sulfurtransferase gene (tst).
Evolutionary aspects of sulfate reduction genes
The key enzymes of dissimilatory sulfate reduction, i.e. ATP sulfurylase (sat), APS reductase (aprAB) and dissimilatory sulfite reductase (dsrABC), are scattered around the chromosome of Db. autotrophicum HRM2 (Fig. S1), as has been observed for other sulfate reducers with known genome sequences. However, DNA fragments were recently obtained from uncultured prokaryotes that contained clustered genes of sulfate reduction (Mussmann et al., 2005). This finding supports speculations about a metabolic island for the pathway of dissimilatory sulfate reduction (Klein et al., 2001; Friedrich, 2002). Moreover, Mussmann and colleagues (2005) speculate that the obtained gene cluster could represent a conserved progenitor that was upon its lateral acquisition increasingly scattered due to genome plasticity of the recipient organism. The latter could be driven by mobile elements, which are highly abundant in the genome of Db. autotrophicum HRM2. There are two paralogues of sat, whereas the aprAB and the dsrABC genes are unique in the genome of Db. autotrophicum HRM2. This is a surprisingly low number, considering that on average each gene has five paralogues in the genome.
Electron donors
Genes for one periplasmic [Ni/Fe/Se] hydrogenase (hysAB), one periplasmic [Ni/Fe] hydrogenase (hynAB), as well as three periplasmic formate dehydrogenases (fdhAB) are present in the genome of Db. autotrophicum HRM2 (Figs 1 and 2). The [Ni/Fe/Se] hydrogenase is apparently soluble, therefore requiring a soluble c-type cytochrome to transfer the electrons to the type II cytochrome c3 complex for transmembrane passage (Pieulle et al., 2005). The putative membrane association of the [Ni/Fe] hydrogenase suggests a direct transfer of electrons derived from hydrogen oxidation onto a thus far unknown transmembrane carrier. All corresponding enzymes are capable of producing scalar protons, and the electrons can be transferred to redox-active complexes such as TpII-c3 (TmcBCA), Qmo (QmoCAB) or Hme (HmeCDE1E2P) either directly, or indirectly via the menaquinol pool (MQ/MQ-H2) (Fig. 1). H2 can be produced in the cytoplasm by a formate–hydrogen-lyase complex consisting of FdhABCD and an associated [Ni/Fe] hydrogenase HyfBCEFGI (Hedderich and Forzi, 2005; Pereira et al., 2006). Apparently, Db. autotrophicum HRM2 does not contain the membrane-bound EchABCDE [Ni/Fe]- or the CO-dependent hydrogenases (CooMLKXUHF), which are enzymatically active towards the cytoplasmic side. Both of these hydrogenases are present in Dv. vulgaris Hildenborough (Heidelberg et al., 2004) and Ech also in Desulfovibrio gigas (Rodrigues et al., 2003). The enzymes are assumed to play a prominent role in the proposed hydrogen cycling (Voordouw, 2002; Heidelberg et al., 2004). But genes encoding a [Fe]-only hydrogenase (HydA) catalysing ferredoxin reduction with H2 and a cytoplasmic selenocysteine-containing hydrogenase (MvhA2DG) are present in Db. autotrophicum HRM2. The genes mvhA2DG are clustered with the genes for the selenocysteine-containing heterodisulfide reductase HdrA1 and the [Ni/Fe/Se] hydrogenase HysAB (Figs 1 and 4).
Fig. 4.
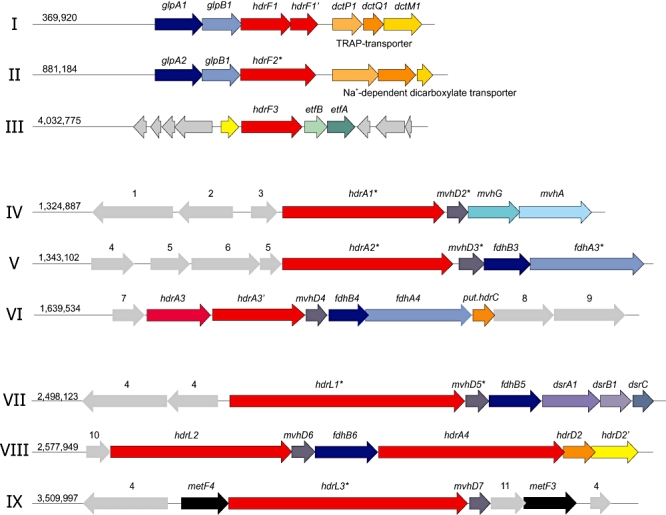
Genomic context of the hdrA, hdrF, hdrD and hdrL genes of Db. autotrophicum HRM2. Deduced heterodisulfide reductase genes are marked in red. The HdrA/HdrL proteins are encoded at nine genetic loci, one of which contains a tandem associated hdrL/hdrA copy (VIII). Each hdrA or hdrL locus is associated with a methylviologen non-reducing hydrogenase subunit D (mvhD), beside the hdrA4 locus, which is followed by a deduced hdrD2 gene activity (VIII). The loci V, VI, VII and VIII contain genes for a formate dehydrogenase subunit B, which is followed directly by formate dehydrogenase subunit A genes in loci V and VI. In locus VIII hdrA4 gene is associated with genes for deduced orthologues of subunits HdrB and HdrC, giving an hdrACB-like operon as can be found in Desulfovibrio species. The HdrF1 and HdrF2 genes are associated with genes for anaerobic glycerol-3-phosphate dehydrogenase activity (GlpAB) which probably transfers electrons from dicarboxylic acid degradation and genes for dicarboxylic acids transporters (Dct). hdrF3 is clustered with electron transfer flavoprotein subunits EtfBA, which might transfer electrons from the β-oxidation of fatty acids. Abbreviations are: hdrA, heterodisulfide reductase, subunit HdrA; hdrL, predicted heterodisulfide reductase/glutamate synthase fusion protein, subunit HdrL; hdrF, heterodisulfide reductase subunit HdrF; hdrD, heterodisulfide reductase, subunit HdrD (HdrCB homologous protein); dsrA, dissimilatory sulfite reductase, α-subunit; dsrB, dissimilatory sulfite reductase, β-subunit; dsrC, dissimilatory sulfite reductase, γ-subunit; mvhD, methylviologen non-reducing hydrogenase, iron-sulfur subunit MvhD; mvhG, methylviologen non-reducing hydrogenase, iron-sulfur subunit MvhG; mvhA, methylviologen non-reducing hydrogenase, nickel-iron subunit MvhA; glpA, anaerobic glycerol-3-phosphate dehydrogenase large subunit; glpB, anaerobic glycerol-3-phosphate dehydrogenase small subunit; etfA, electron transfer flavoprotein α-subunit; etfB, electron transfer flavoprotein β-subunit; dctP1 TRAP-type C4-dicarboxylate transporter, periplasmic solute-binding component; dctM1, TRAP-type C4-dicarboxylate permease, large subunit; dctQ1, TRAP-type C4-dicarboxylate permease, small subunit; 1, HysB, periplasmic [Ni/Fe/Se] hydrogenase, large subunit; 2, HysD, periplasmic [Ni/Fe/Se] hydrogenase, small subunit; 3, HyaD2, hydrogenase expression/formation protein; 4, hypothetical protein; 5, response regulator; 6, histidine kinase; 7, ferredoxin; 8, Fe/S cluster protein; 9, Aor5, tungsten-containing aldehyde reductase; 10, CheY-like regulator; 11, putative regulatory protein.
Electron donors during chemoorganotrophic growth are membrane-bound lactate (LdhAB) and glycerophosphate (GlpAB) dehydrogenases as well as NADH/H+ and EtfAB from β-oxidation of organic acids (FadBGA) and various dehydrogenase reactions (Fig. 1). Electrons from the membrane-bound dehydrogenases can be channelled into the quinone pool or to the heterodisulfide reductase subunits (HdrF) either directly or indirectly via the Hme (HmeCDE1E2P) complex. The genomic proximity of two GlpAB gene clusters and an EtfAB gene cluster with the F-type heterodisulfide reductases (HdrF1–HdrF3) supports the idea of an electron transfer via the heterodisulfide reductases. NADH can be oxidized by the NADH-quinone oxidoreductase (NqrA–F), which also will provide electrons for the quinone pool, or possibly by the novel heterodisulfide reductase HdrL1 to HdrL3 (Figs 4 and 5 and Fig. S3A and B).
Fig. 5.
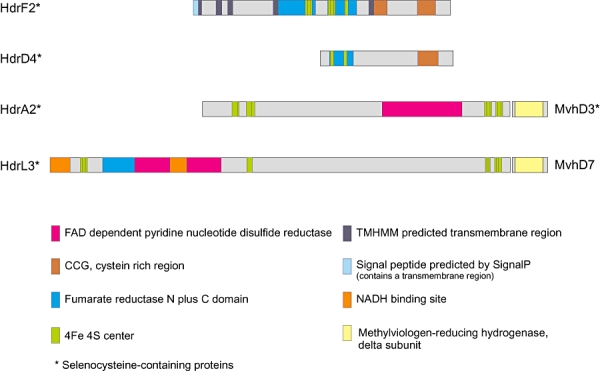
Pfam (domain scans analysis) and domain alignments of four types of heterodisulfide reductase subunits (Hdr) of Db. autotrophicum HRM2. Relevant Pfam domains are given in colour codes. All Hdr proteins depicted contain selenocysteine; the HdrA and the HdrL type of the proteins are colocated with methylviologen non-reducing hydrogenase subunit D (mvhD). The deduced HdrF protein contains a domain with several transmembrane regions indicating a possible membrane integration of the protein. In contrast, HdrD, HdrA and HdrL have domain structures typical for soluble proteins.
Membrane-bound redox complexes
The electrons, which originate from H2, NADH or ETFs, are transferred to redox complexes of which sulfate-reducing prokaryotes are very rich. In recent years, several membrane-bound complexes have been discovered in Desulfovibrio and Archaeoglobus spp., which could serve this redox function (see Matias et al., 2005). The 16-haem cytochrome Hmc of Dv. vulgaris (Rossi et al., 1993; Czjzek et al., 2002), the Hme complex of A. fulgidus (Mander et al., 2002), the 9Hc complex of Dv. desulfuricans (Saraiva et al., 2001) and the TpII-c3 complex of Dv. vulgaris (Valente et al., 2001; Pereira et al., 2007) all possess periplasmic cytochrome c3 subunits for the reception of electrons generated by periplasmic enzymes. Genes encoding several cytochrome c3 family proteins and multihaem cytochrome family proteins as well as cytochrome c oxidase and cytochrome c assembly protein CcmC are present in Db. autotrophicum HRM2. This supports the idea that electrons if necessary can be stored within the periplasm as described previously (Heidelberg et al., 2004). Genes for an Hmc complex are apparently not present in Db. autotrophicum HRM2 but two TpII-c3 (TmcBCA) and one Hme encoding gene cluster (HmeDCPE1E2) were detected (Figs 1 and 2). In addition, genes for a Qmo complex (QmoCAB) were found, which could accept electrons, transferred from the NADH-quinone oxidoreductase. The Qmo complex of Dv. desulfuricans and Dv. vulgaris does not contain a periplasmic cytochrome, but was shown to interact with a soluble menaquinone analogue and suggested to function as a menaquinol/APS reductase oxidoreductase (Pires et al., 2003; Haveman et al., 2004). Similarly, a direct electron transfer from HmeDEAB has been proposed for Dv. vulgaris (Haveman et al., 2004). Recent experimental studies with Dv. desulfuricans ATCC 27774 suggest that Hme channels electrons from the periplasm and menaquinone pool to sulfite reductase (Pires et al., 2006). In the obligatory hydrogenotrophic Archaeoglobus profundus, a hydrogenase was found to form a tight complex with a soluble heterodisulfide reductase (Mvh:Hdl), supporting the assumed role of heterodisulfide reductase in electron transfer of sulfate-reducing prokaryotes (Mander et al., 2004).
Role of heterodisulfide reductases (Hdr)
The archaeal Hdr plays a key role during methanogenesis. The enzyme reduces the heterodisulfide of coenzymes M and B which is formed in the methyl-coenzyme M reductase reaction by which methane is produced. Hdr of Ms. mazei has been shown to be a membrane protein consisting of the subunits D and E. Receiving the electrons from methanophenazine via cytochromes it functions as a proton pump (Deppenmeier et al., 2002). Methanogens growing with H2 and CO2 but not with acetate or methanol do not contain cytochromes and contain a different Hdr consisting of the subunits ABC, which is also true for Methanosphaera stadtmanae growing with methanol and H2 (Fricke et al., 2006). In silico analysis indicates that Db. autotrophicum HRM2 may express 15 paralogues of Hdr that can be divided into four protein types and that could form differently composed protein complexes (Figs 1 and 4). As shown in Fig. 4 the nine loci at which Hdr genes occur are spread over the chromosome of Db. autotrophicum HRM2. Additionally, Hdrs differ considerably not only in size but also in domain compositions (Fig. 5 and Fig. S3A and B). Four of the Hdr were designated as hdrA and share high similarities with the corresponding subunits of the archaea. In addition to the four HdrA subunits of which only HdrA3/A3′ contains a small predicted transmembrane region, a large HdrL type has been defined. Especially the selenocysteine protein HdrL3 is of interest because it contains predicted regions for a fumarate reductase domain, and additionally a NADH binding site, which is also found in the two other HdrL subunits. HdrL therefore may be essential for electron cycling during sulfidogenesis (Fig. 1). All the CDS for HdrAs and HdrLs except HdrA4 are followed by a CDS for the D-subunit of the methylviologen non-reducing hydrogenase (Mvh) (Figs 4 and 5 and Fig. S3A and B). In Methanothermobacter marburgensis MvhD and HdrA form a tight complex in which MvhD transfers reducing equivalents to Hdr (Stojanowic et al., 2003). Genomic comparisons show that MvhD homologues are colocated with hdr genes even in organisms where no complete Mvh hydrogenase is present. In some of these organisms, including the Methanosarcina species and A. fulgidus, MvhD and HdrA have been fused to a single protein (Fig. S3A). Genes of dissimilatory sulfite reductase subunits DsrA, DsrB and DsrC follow HdrL1 and MvhD (Fig. 4). Thus, the HdrL1 gene cluster encodes all proteins necessary to transfer electrons from NADH/H+ to sulfite (SO32−).
The hdrD genes as present in Ms. mazei and A. fulgidus (Hedderich et al., 2005) could be detected also in Db. autotrophicum HRM2. Two variant genetic versions are present within the chromosome. The hdrD1 to hdrD4 gene products contain HdrC and HdrB homologue domains, but are lacking transmembrane parts (Fig. 5). Three hdrD-like gene products contain both, multiple transmembrane domains as well as domains homologous to HdrC and HdrB. These genes were designated hdrF1 to hdrF3. The protein deduced from the hdrF2 gene is a selenoprotein; it contains the transmembrane region, the CCG region and the fumarate-reductase domain (Fig. 5). All three hdrF genes are clustered with genes encoding electron providing proteins, hdrF1 and hdrF2F2′ with glpAB and dicarboxylic acid transporters and hdrF3 with etfAB involved in β-oxidation (Fig. 4).
A direct orthologue to hdrE of Ms. mazei and Dt. psychrophila LSv54 is lacking in Db. autotrophicum HRM2. Instead, the three present hdrF genes (Fig. S3B) are much larger; all genes carry sequence regions for a signal peptide and up to six transmembrane helices. Taking into account the genomic context of the hdrF genes and the fact that the Hdrs of some methanoarchaea are deduced proton pumps, one could speculate that this function may also apply to the hdrF genes of Db. autotrophicum HRM2 (Fig. 1).
A concept for electron transfer and energy conservation
When Db. autotrophicum HRM2 is growing with H2, CO2 and sulfate, eight electrons are required for sulfide production. Their generation by periplasmic hydrogenases would result in a maximum of eight scalar protons. The corresponding protonmotive force could give rise to the synthesis of 22/3 ATP. However, an equivalent of two ATP is required for the reduction of sulfate to sulfite. In addition, transport of sulfate and other compounds are ATP- or ionmotive force-consuming. The conclusion is that there must be pumps present to generate additional proton or sodium ion gradients. One candidate for such a pump is the RnfA–E cluster. Genes for a hydrogenase catalysing the reduction of ferredoxin were detected. Electron transfer from reduced ferredoxin to NAD+ via Rnf would then allow the generation of a proton (or sodium ion) motive force. Proton pumping Rnf clusters have been integrated in concepts of Clostridium tetani (Brüggemann et al., 2003) and Clostridium kluyveri (Seedorf et al., 2008) energy metabolism. Further candidates are the various heterodisulfide reductases. They could receive electrons from membrane-bound redox complexes, in case of HdrL from NADH or other electron carriers, and provide the electrons for sulfite reduction (Fig. 1). In analogy to methanogenic archaea it is tempting to speculate that these processes are also coupled to generation of a protonmotive force. Under low partial pressure of H2, HydA will not be able to reduce ferredoxin (E′ of about −500 mV) and to provide Fdred for the reduction of CO2 to CO, for pyruvate synthesis from acetyl-CoA and CO2 and for the RnfA–E complex. Under these conditions, the recently discovered electron bifurcation reaction (Thauer et al., 2008) may provide the reduced ferredoxin (Fdred) required (Fig. 1). This reaction couples the reduction of crotonyl-CoA to butuyryl-CoA with Fdred generation and is the key reaction in clostridia, notably in C. kluyveri (Seedorf et al., 2008). It also plays a role in energy metabolism of methanogens not containing cytochromes (Thauer et al., 2008). In Db. autotrophicum HRM2 the MvhADG–HdrA complex (Fig. 1) could also bifurcate electrons coming from H2 under low partial pressure to reduce the disulfide and ferredoxin. A possible reaction equation could be:
Studies on the proteome of Db. autotrophicum and in vitro investigations will be required to elucidate the role of the various Hdrs for which the genes have been detected in this work.
Metabolic heat map
A phylogenetic comparison was performed based on bidirectional blast comparisons of Db. autotrophicum HRM2 genes of carbon and energy metabolism with 700 to date available genome data sets (the complete data of the comparisons are given in Table S1). Among the compared were the genomes of 52 archaea including A. fulgidus VC-16, of spore formers, chemolithotrophs, phototrophs, enterobacteria and of seven sulfate reducers. Out of the 700, 67 phylogenetically diverse genomes were selected to produce a metabolic heat map (Fig. 2). None of the prokaryotes compared possesses a comparable wealth of genes encoding hydrogenases, heterodisulfide reductases, formate dehydrogenases and enzymes for fatty acid oxidation as Db. autotrophicum HRM2. Most of these genes have orthologues in other proteobacteria and firmicutes, but not in the closely related SRB of the δ-proteobacteria. This finding is especially apparent for the heterodisulfide reductases, located at nine genetic loci and present in 15 copies (Fig. 4). The closest phylogenetic relations of the four different Hdrs point to δ-proteobacteria for the HdrF type, to firmicutes for the HdrD type and to sulfate reducers in case of the HdrA genes. The HdrL type has only weak sequence similarities and seems to be unique for Db. autotrophicum HRM2. Even the most recently submitted genome of a Desulfobacteraceae member, Dc. oleovorans Hxd3, (Copeland et al., 2008) does not contain an HdrL-type heterodisulfide reductase.
Among the membrane bound redox-active complexes are two rnf and one closely related nqr system. None of these systems is found in any other SRB (Fig. 2) except Dc. oleovorans Hxd3 (Copeland et al., 2008). For each of the hydrogenases of Db. autotrophicum HRM2 we found orthologous genes in other SRB genomes, but not a single genome contains orthologous genes for all the hydrogenases of Db. autotrophicum HRM2. In contrast, the genes for the reduction of sulfite to H2S (aprAB and dsrABC) do not have paralogues in Db. autotrophicum HRM2 and have orthologous genes exclusively in other SRB.
Selenocysteine-containing proteins
Selenocysteine-containing proteins substantially increase the activity of redox-active enzymes as compared with cysteine-containing paralogues. Therefore, selenocysteine-containing proteins are found in many energy-limited anaerobic organisms (Andreesen et al., 1999; Gröbe et al., 2007). The genome of Db. autotrophicum HRM2 encodes the complete selenocysteine translation machinery consisting of selA, selB, selC and selD and the tRNASec. Genes for at least 31 selenocysteine-containing proteins were detected, which is more than has been reported for any other SRB genome sequenced (Table 1). Seventeen selenocysteine-containing proteins play a role in energy metabolism of the organism. They encode two heterodisulfide reductases (hdrA1, hdrA2), two heterodisulfide reductase fusion proteins (hdrL1, hdrL3) (see Fig. 5), one heterodisulfide reductase subunit (hdrD4), four cytoplasmic formate dehydrogenases (fdhA1, fdhA3, fdhA5, fdhA6), three periplasmic formate dehydrogenases (fdhA2, fdhA7, fdhA8), one periplasmic [Ni/Fe/Se] hydrogenase, four F420 non-reducing D-subunit hydrogenases (mvhD1, mvhD2, mvhD3, mvhD5) and others (Table S3). The extensive use of selenocysteine-containing proteins is one of the most obvious differences between Db. autotrophicum HRM2 and the other SRB with completely sequenced genomes. This may indicate that the Db. autotrophicum HRM2 genome encodes proteins that could serve as a functional replacement under selenium limitation as it has been described for the Mvh hydrogenase homologues Vhu (selenocysteine-containing) and Vhc (non-selenocysteine-containing) in Methanoccous voltae (Pfeiffer et al., 1998).
Relation to oxygen
While sulfate reducers have long been considered as strict anaerobes, they encounter dynamic changes of oxic/anoxic interfaces in their natural habitat (Jørgensen, 1987). Particularly in the photic zones of microbial mats oxygen-saturated conditions can occur during daytime (Jonkers et al., 2005). Early studies indicated that particularly Desulfovibrio spp. survive prolonged exposure to oxygen (Cypionka et al., 1985). More recent studies also indicated that SRB could in fact respire oxygen as a protective measure (Cypionka, 2000) and migrate into areas of preferential oxygen concentration (Fischer and Cypionka, 2006). The presence of cydAB genes coding for a cytochrome d ubiquinol oxidase could also enable Db. autotrophicum HRM2 to respire oxygen at low concentrations (Green et al., 1988; Lemos et al., 2001). Additionally, Db. autotrophicum HRM2 possesses catalase (Cat), a selenocysteine-containing peroxidase, rubrerythrin (Rbr), and rubredoxin oxidoreductases (Rbo) for oxygen detoxification. The latter two have recently been described as oxygen protection systems in Dv. vulgaris (Lumppio et al., 2001). However, it should be noted, that subsequent studies revealed no obvious oxidative stress phenotype for an rbr mutant (Fournier et al., 2003) and downregulation of rubrerythrins in oxygen-exposed cultures of Dv. vulgaris (Fournier et al., 2006). Db. autotrophicum HRM2 possesses several copies of F390 synthetase genes, which has previously been suggested in Methanothermobacter thermoautotrophicusΔH to react to oxygen tension or H2 limitation (Morgan et al., 1997; Vermeij et al., 1997).
Transcriptional regulation and signal transduction
When looking at its nutritional versatility, Db. autotrophicum HRM2 should possess a broad spectrum of adaption capacities, which implicate variable regulations. A search for characteristic protein domains among the group of sensory proteins in the Db. autotrophicum HRM2 genome yielded 253 proteins with putative transmembrane sensors (Table S4). Among these are 109 histidine kinases containing the characteristic Pfam domain HATPaseC (Perego and Hoch, 1996). Fifty histidine kinases contain one or more copies of the redox-sensing PAS domain (Zhulin et al., 1997; Taylor and Zhulin, 1999), reflecting also the strict dependence of Db. autotrophicum HRM2 to react to changes in the oxygen pressure.
ECF-σ-factors (σECF) have been identified by the presence of characteristic DNA-binding domains (Sigma70_rc2, Sigma70_rc4 and Sigma70_rc4.2) and the absence of a σ70-specific domain (Sigma70_rc3). We have identified five alternative σ-factor proteins in Db. autotrophicum HRM2, including σECF proteins of the RpoE and the SigM type. Additionally, six anti-σ-factors and at least two anti-anti-σ-factors could be identified.
Beside the two-component system respectively ECF-σ-factor-associated proteins 120 proteins were identified which contain sensory protein domains and therefore might represent components of additionally sensory systems.
Conclusions
Several genome features like larger genome size, higher number of repeated sequence elements and paralogous proteins, extensive use of selenocysteine-containing proteins, and partially closer relatedness to archaea and firmicutes than to other SRB, seem to underline the distinctiveness of Db. autotrophicum HRM2 among the SRB with completely sequenced genomes. The genome of Db. autotrophicum HRM2 appears to be shaped by many genetic rearrangements, gene duplications and genetic adaptation events, as well as some horizontal gene transfer (HGT). These dynamic processes might have provided the organism with a significantly broader spectrum of orthologous genes for substrate utilization and energy conversion. Oxidizing detritus and the employment of efficient electron transfer systems based on selenocysteine-containing proteins enable this slow-growing species to out-perform the fast-growing and nutritionally limited species of the genus Desulfovibrio in the natural habitat.
Experimental procedures
Organism
Desulfobacterium autotrophicum HRM2 (DSMZ 3382) was obtained from the German Collection of Microorganisms and Cell Cultures (Deutsche Sammlung von Mikroorganismen und Zellkulturen, DSMZ), Braunschweig, Germany.
Media and cultivation
Desulfobacterium autotrophicum HRM2 was cultivated as originally described by Brysch and colleagues (1987), with mineral media corresponding to saltwater medium (Widdel and Bak, 1992). Mineral media were sulfide-reduced (1 mM) and bicarbonate-buffered. Cultivation was carried out at 28°C. For maintenance, cells were grown with 10 mM sodium lactate as sole source of organic carbon and energy and sulfate as electron acceptor. Methods for anaerobic cultivation were performed as described by Widdel and Bak (1992).
Genome sequencing, assembly and gap closure
Isolation of genomic DNA from Db. autotrophicum HRM2 was performed with the Bio-Rad Aqua Pure DNA isolation kit (Bio-Rad, München, Germany). The genomic DNA sequence was determined using three conventional types of whole-genome shotgun libraries of three different insert sizes, as well as two large insert libraries derived from cosmid and fosmid cloning as previously described (Hradecna et al., 1998; Wild et al., 2002; Schwartz et al., 2003). For the whole-genome shotgun libraries DNA fragments of 1.5–2.5, 1.5–3.5 and 2.5–4.0 kb, respectively, were separated by gel electrophoresis after mechanical shearing (Nebulizer; Invitrogen, Carlsbad, USA), end-repaired and cloned using vectors pTZ19R (cmR) (Amersham, Essex, UK), pWE15R (kanR), or pCR2.1-TOPO (TOPO TA Cloning Kit for Sequencing; Invitrogen). PCR fragments were cloned using vector pCRXL-TOPO or pCR4-TOPO (TOPO XL PCR Cloning Kit; Invitrogen). For construction of the cosmid and fosmid libraries, DNA fragments of about 40 kb were isolated using a pulse field electrophoresis system (Chef Mapper; Bio-Rad), and cloned into the SuperCos cosmid vector (SuperCos I Vector Kit; Stratagene, La Jolla, CA, USA) or the pCC1FOS fosmid vector (CopyControl Fosmid Library Production Kit; Epicentre, Madison, USA). Plasmid DNAs were isolated using two BioRobots8000 (QIAGEN GmbH, Hilden, Germany). All plasmids were end-sequenced using the following primers: Forward and Reverse on ABI sequencer ABI3730XL or ABI377 (Applied Biosystems, Foster City, CA, USA), NewForward and NewReverse on MegaBace sequencer MB4000 (GE Healthcare, Munich, Germany), cos_for and cos_rev for cosmid vectors, and pCC1_Forward and pCC1_Reverse for fosmid vectors (costumn primers see Table S5). Sequences were produced using either ABI BigDye Terminator 3.1 chemistry (Applied Biosystems) or ET-Terminator chemistry (GE Healthcare). About 104 000 generated sequences were assembled into contigs using the Phrap assembly tool (http://www.phrap.org). Primer walking on plasmids, cosmid clones and fosmid clones as well as PCR-based techniques were used to close remaining gaps and to solve misassembled regions caused by the high number of repetitive sequences. Mainly the sequences derived from the over 2000 fosmid clones served for verification of orientation, linkage and consistency of all contigs. All manual editing steps were performed using the STADEN software packages and the Gap4 versions therein (Staden, 1996; Staden et al., 2000). After sequence polishing and finishing, the genome sequence had a 11.8-fold sequence redundancy. The final chromosome sequence was assembled from 110 511 reads and the plasmid from 3572 reads with a minimum PHRED score of 15 and an average used read length of 615 bp.
Prediction and annotation of CDS, gene family identification
Prediction of open reading frames (ORFs) or coding sequences (CDS) was accomplished with YACOP (Tech and Merkl, 2003) using the ORF-finding programs Glimmer (Delcher et al., 1999a,b), Critica (Badger and Olsen, 1999) and Z curve (Guo et al., 2003). Prediction of coding sequences within GenDB 2.0 to 2.4 (Meyer et al., 2003; CeBiTec, University of Bielefeld, Germany) was accomplished using the ORF-finding programs Critica (Badger and Olsen, 1999), Gismo (Krause et al., 2007), Glimmer (Delcher et al., 1999a,b) and reganor (McHardy et al., 2004). The ERGO software package (Overbeek et al., 2003) licensed by Integrated Genomics (Chicago, IL, USA) was used for manual curations of all CDS by comparing the predicted protein sequences with the publicly available data sets of SWISSPROT, GenBank, ProDom, COG and Prosite (Falquet et al., 2002). The CD-Search software (Marchler-Bauer and Bryant, 2004) was used to screen all CDS for similarities to known protein families and domains. The TMpred software was used for the prediction of transmembrane helices within the CDS (Hofmann and Stoffel, 1993). Prediction of regulatory proteins was performed by screening the whole genome protein database of Db. autotrophicum HRM2 with appropriate HMM models. The models for prediction of histidine kinases have been extracted from Pfam (Bateman et al., 2004; Galperin et al., 2001; Finn et al., 2006). Appropriate models of ECF σ-factors have been developed on the basis of a compilation of all known ECF σ-factors (Staron et al., 2007). Putative genomic islands, referred to as alien genes and/or highly expressed CDS, were searched with a score-based FSIGI-HMM program (Merkl, 2004; Waack et al., 2006).
Comparative genomics and repeat analysis
Repeated elements within the genomic sequence were calculated and analysed using the programs repfind and repvis, which both are part of the reputer software package (Kurtz et al., 2001). Pairwise graphical alignments of whole genome assemblies (e.g. synteny plots) were generated using the MUMmer system (Delcher et al., 1999a,b). The protein sequences encoded by Db. autotrophicum HRM2 were used for bidirectional blast comparisons among a selected representative set of 700 whole genome protein data sets, thus identifying putative orthologous genes. Bidirectional blast for bacterial genomes (BiBaG) has been performed using a variant method of blast developed by Antje Wollherr and Heiko Liesegang (pers. comm.). The similarities of putative orthologous proteins from 67 taxonomically diverse genomes were displayed by a colour code (see also Fig. 4). Paralogous genes have been determined by a ‘all against all’blast of all proteins within a genome. A pair of genes has been considered as paralogous if the determined protein sequences share at least 50% sequence identity in an alignment of 80% length of the shorter sequence (Table 1). Detailed results of all blast comparisons are available in the Table S1.
Acknowledgments
We would like to thank Hans-Joachim Fritz for general support and Thomas Hartsch for the initial set-up of the project. We also thank Birgit Veith, Elzbieta Brzuszkiewicz and Antje Wollherr (all Göttingen) for their help in sequence editing and polishing, plasmid annotation and comparative calculations, Jörg Wulf (Bremen) for preparation of genomic DNA and Daniela Lange (Bremen) for cultivation of Db. autotrophicum HRM2. This work was supported by the Federal Ministry of Education and Research within the REGX program, the Max-Planck-Society and the Ministry for Science and Culture of Lower-Saxony.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Genome circle. Circles (from outside to inside): 1, the outer circle gives the location of all genes of the C1 pathway (red), the heterodisulfide reductase activities (blue) and the methylviologen non-reducing hydrogenase activities (green). Arrows above indicate the orientation of the marked genes with respect to the deduced origin of replication; 2, scale circle in Mbp; 3, the third ring gives the location of all ORFs on the leading and the lagging strand, classified and colour-stained as given by the Clusters of Orthologous Groups of proteins (COG) (http://www.ncbi.nlm.nih.gov/COG/grace/fiew.cgi); 4, all genes that differ significantly from the standard codon usage of Db. autotrophicum HRM2 and therefore represent putative foreign genes are marked in green; 5, on the fifth ring all transposase/putative transposonrelated genes (dark blue), or recombinase/invertase genes (orange) are marked; arrows above indicate the orientation of the given genes with respect to the origin of replication; 6, the GC content (red) is given with marked average value (black); 7, translational components are given: rDNA clusters (green), incomplete rDNAs (red) and all tRNAs (blue); arrows above indicate the orientation of the marked genes with respect to the origin of replication.
Fig. S2. Mummer plot. Comparison of the Db. autotrophicum HRM2 chromosome with the chromosomes of the closely related δ-proteobacteria Dt. psychrophila LSv54, Dv. vulgaris Hildenborough, Dv. desulfuricans G20, G. sulfurreducens PCA and the sulfate-reducing archaeon A. fulgidus VC-16. Red dots indicate colinear similarities, green dots indicate inverted similarities. The chromosome of Db. autotrophicum HRM2 completely lacks synteny with the compared chromosomes. In contrast, a mummer plot of a comparison of Dv. vulgaris Hildenborough and Dv. desulfuricans G20 reveals genome synteny. All comparisons were performed with the programs from the MUMmer software package as described in the documentation for distantly related sequences (http://mummer.sourceforge.net/).
Fig. S3. Protein domain alignment of heterodisulfide reductases (Hdrs) from Db. autotrophicum HRM2 and experimentally characterized Hdr reference proteins. A multiple protein domain alignment of all Hdrs from Db. autotrophicum HRM2 and experimentally characterized reference proteins revealed the four distinct Hdr groups A, L, D and F. The alignment is based on colour-coded Pfam domain scans. Each protein group contains at least one selenocysteine-containing and one non-selenocysteine-containing paralogue of the enzyme. In many cases Db. autotrophicum HRM2 encodes the protein domains necessary to form an Hdr by two or more colocalized genes. It is remarkable that in each case the order of the Hdr domains is conserved in the order of the encoding genes and is colinear to the order of domains within the reference proteins. Group A contains four Hdrs of Db. autotrophicum HRM2 with similarities to HdrA of the archaea M. mazei and A. fulgidus. HdrA1 to HdrA3 are encoded by two and three genes, respectively, which contain all Pfam domains found in the single polypeptides of the reference proteins. For HdrA4 no protein encoding the MvhD domain could be identified. Group L contains three members, HdrL1 to HdrL3, each colocalized with a MvhD protein. The group L proteins are newly found in Db. autotrophicum HRM2. Group D contains HdrD1 to HdrD5 from Db. autotrophicum HRM2. The members of this group align with the domains of HdrD1 from A. fulgidus. HdrD is a paralogue of HdrD from HdrDE from M. mazei and A. fulgidus, which are encoded by one protein in the case of A. fulgidus and two proteins in the case of M. mazei respectively. An orthologue of HdrE could not be identified within the Db. autotrophicum HRM2 genome. Group F consists of three members HdrF1 to HdrF3. HdrF1/F1′ is encoded by two genes whereas HdrF2 and HdrF3 are encoded by one gene. Group F represents, as group L, a newly found type of Hdrs from Db. autotrophicum HRM2.
Table S1. Bidirectional blast comparisons of 4947 protein sequences from Db. autotrophicum HRM2 and 700 whole genome protein data sets.
Table S2. Repeat analysis of sulfate- or sulfur-reducing prokaryotes.
Table S3. Selenocysteine-containing proteins.
Table S4. Pfam domain occurrence in two-component systems from Db. autotrophicum HRM2.
Table S5. Sequence of used primers.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Andreesen JR, Wagner M, Sonntag D, Kohlstock M, Harms C, Gursinsky T, et al. Various functions of selenols and thiols in anaerobic gram-positive, amino acids-utilizing bacteria. Biofactors. 1999;10:263–270. doi: 10.1002/biof.5520100226. [DOI] [PubMed] [Google Scholar]
- Badger JH, Olsen GJ. CRITICA: coding region identification tool invoking comparative analysis. Mol Biol Evol. 1999;16:512–524. doi: 10.1093/oxfordjournals.molbev.a026133. [DOI] [PubMed] [Google Scholar]
- Badziong W, Thauer RK. Growth yields and growth rates of Desulfovibrio vulgaris (Marburg) growing on hydrogen plus sulphate and hydrogen plus thiosulphate as sole energy sources. Arch Microbiol. 1978;117:209–214. doi: 10.1007/BF00402310. [DOI] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch A, Leiser A, Robinson JA. Cloning, sequencing and expression of the gene encoding methylmalonyl-Coenzyme A mutase from Streptomyces cinnamonensis. J Bacteriol. 1993;175:3511–3519. doi: 10.1128/jb.175.11.3511-3519.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott M, Pfister K, Burda P, Kalbermatter O, Woehlke G, Dimroth P. Methylmalonyl-CoA decarboxylase from Propionum modestum: cloning and sequencing of the structural genes and purification of the enzyme complex. Eur J Biochem. 1997;250:590–599. doi: 10.1111/j.1432-1033.1997.0590a.x. [DOI] [PubMed] [Google Scholar]
- Brandis-Heep A, Gebhardt NA, Thauer RK, Widdel F, Pfennig N. Anaerobic acetate oxidation to CO2 by Desulfobacter postgatei. 1. Demonstration of all enzymes required for the operation of the citric acid cycle. Arch Microbiol. 1983;136:222–229. [Google Scholar]
- Brüggemann H, Baumer S, Fricke WF, Wiezer A, Liesegang H, Decker I, et al. The genome sequence of Clostridium tetani, the causative agent of tetanus disease. Proc Natl Acad Sci USA. 2003;100:1316–1321. doi: 10.1073/pnas.0335853100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brysch K, Schneider C, Fuchs G, Widdel F. Lithoautotrophic growth of sulfate-reducing bacteria, and description of Desulfobacterium autotrophicum gen. nov., sp. nov. Arch Microbiol. 1987;148:264–274. [Google Scholar]
- Canfield DE, Jørgensen BB, Fossing H, Glud R, Gundersen J, Ramsing NB, et al. Pathways of organic carbon oxidation in three continental margin sediments. Mar Geol. 1993;113:27–40. doi: 10.1016/0025-3227(93)90147-n. [DOI] [PubMed] [Google Scholar]
- Charles TC, Aneja P. Methylmalonyl-CoA mutase encoding gene of Sinorhizobium meliloti. Gene. 1999;226:121–127. doi: 10.1016/s0378-1119(98)00555-1. [DOI] [PubMed] [Google Scholar]
- Copeland A, Lucas S, Lapidus A, Barry K, Detter JC, Glavina T, et al. Direct sequence submission Accession No.: NC_007519.
- Copeland A, Lucas S, Lapidus A, Barry K, Glavina del Rio T, Dalin E, et al. Direct sequence submission Accession No.: NC_009943.
- Cypionka H. Oxygen respiration by Desulfovibrio species. Annu Rev Microbiol. 2000;54:827–848. doi: 10.1146/annurev.micro.54.1.827. [DOI] [PubMed] [Google Scholar]
- Cypionka H, Widdel F, Pfennig N. Survival of sulfate-reducing bacteria after oxygen stress, and growth in sulfate-free oxygen-sulfide gradients. FEMS Microbiol Ecol. 1985;31:39–45. [Google Scholar]
- Czjzek MEI, Antak L, Zamboni V, Morelli X, Dolla A, Guerlesquin F, Bruschi M. The crystal structure of the hexadeca-heme cytochrome Hmc and a structural model of its complex with cytochrome c3. Structure. 2002;10:1677–1686. doi: 10.1016/s0969-2126(02)00909-7. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999a;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL. Alignment of whole genomes. Nucleic Acids Res. 1999b;27:2369–2376. doi: 10.1093/nar/27.11.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppenmeier U, Johann A, Hartsch T, Merkl R, Schmitz RA, Martinez-Arias R, et al. The genome of Methanosarcina mazei: evidence for lateral gene transfer between bacteria and archaea. J Mol Microbiol Biotechnol. 2002;4:453–461. [PubMed] [Google Scholar]
- Dhillon A, Teske A, Dillon J, Stahl DA, Sogin ML. Molecular characterization of sulfate-reducing bacteria in the Guaymas Basin. Appl Environ Microbiol. 2003;69:2765–2772. doi: 10.1128/AEM.69.5.2765-2772.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falquet L, Pagni M, Bucher P, Hulo N, Sigrist CJA, Hofmann K, Bairoch A. The PROSITE database, its status in 2002. Nucleic Acids Res. 2002;30:235–238. doi: 10.1093/nar/30.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenchel TM, Jørgensen BB. Detritus food chains of aquatic ecosystems: the role of bacteria. In: Alexander M, editor. Advances in Microbial Ecology. I. New York, USA: Plenum Press; 1977. pp. 1–58. [Google Scholar]
- Finn RD, Mistry J, Schuster-Böckler B, Griffiths-Jones S, Hollich V, Lassmann T, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer JP, Cypionka H. Analysis of aerotactic band formation by Desulfovibrio desulfuricans in a stopped-flow diffusion chamber. FEMS Microbiol Ecol. 2006;55:186–194. doi: 10.1111/j.1574-695X.2005.00024.x. [DOI] [PubMed] [Google Scholar]
- Fournier M, Zhang Y, Wildschut JD, Dolla A, Vourdouw JK, Schriemer DC, Vourdouw G. Function of oxygen resistance proteins in the anaerobic sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. J Bacteriol. 2003;185:71–79. doi: 10.1128/JB.185.1.71-79.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier M, Aubert C, Dermoun Z, Durand M-C, Moinier D, Dolla A. Response of the anaerobe Desulfovibrio vulgaris Hildenborough to oxidative conditions: proteome and transcript analysis. Biochimie. 2006;88:85–94. doi: 10.1016/j.biochi.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Fricke WF, Seedorf H, Henne A, Krüer M, Liesegang H, Hedderich R, et al. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J Bacteriol. 2006;188:642–658. doi: 10.1128/JB.188.2.642-658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich MW. Phylogenetic analysis reveals multiple lateral transfers for adenosine-5′-phosphosulfate reductase genes among sulfate-reducing microorganisms. J Bacteriol. 2002;184:278–289. doi: 10.1128/JB.184.1.278-289.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY, Nikolskaya AN, Koonin EV. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol Lett. 2001;203:11–21. doi: 10.1111/j.1574-6968.2001.tb10814.x. [DOI] [PubMed] [Google Scholar]
- Green GN, Fang H, Lin R-J, Newton G, Mather M, Georgiou CD, Gennis RB. The nucleotide sequence of the cyd locus encoding the two subunits of the cytochrome d terminal oxidase complex of Escherichia coli. J Biol Chem. 1988;263:13138–13143. [PubMed] [Google Scholar]
- Gröbe T, Reuter M, Gursinsky T, Söhling B, Andreesen JR. Peroxidase activity of selenoprotein GrdB of glycine reductase and stabilisation of its integrity by components of proprotein GrdE from Eubacterium acidaminophilum. Arch Microbiol. 2007;187:29–43. doi: 10.1007/s00203-006-0169-6. [DOI] [PubMed] [Google Scholar]
- Guo F-B, Ou H-Y, Zhang C-T. ZCURVE: a new system for recognizing protein-coding genes in bacterial and archaeal genomes. Nucleic Acids Res. 2003;31:1780–1789. doi: 10.1093/nar/gkg254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haveman SA, Greene EA, Stilwell CP, Voordouw JK, Voordouw G. Physiological and gene expression analysis of inhibition of Desulfovibrio vulgaris Hildenborough by nitrite. J Bacteriol. 2004;186:7944–7950. doi: 10.1128/JB.186.23.7944-7950.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedderich R, Forzi L. Energy-converting [Ni/Fe] hydrogenases: more than just H2 activation. J Mol Microbiol Biotechnol. 2005;10:92–104. doi: 10.1159/000091557. [DOI] [PubMed] [Google Scholar]
- Hedderich R, Hamann N, Bennati M. Heterodisulfide reductase from methanogenic archaea: a new catalytic role for an iron-sulfur cluster. Biol Chem. 2005;386:961–970. doi: 10.1515/BC.2005.112. [DOI] [PubMed] [Google Scholar]
- Heidelberg JF, Seshadri R, Havemann SA, Hemme CL, Paulson IT, Kolonay JF, et al. The genome sequence of the anaerobic sulfate-reducing bacterium Desulfovibrio vulgaris strain Hildenborough. Nat Biotechnol. 2004;22:554–559. doi: 10.1038/nbt959. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Stoffel W. Tmbase – a database of membrane spanning proteins segments. Biol Chem Hoppe-Seyler. 1993;347:166. [Google Scholar]
- Hradecna Z, Wild J, Szybalski W. Conditionally amplifiable inserts in pBAC vectors. Microbiol Comp Genom. 1998;3:58. [Google Scholar]
- Jonkers HM, Koh I-O, Behrend P, Muyzer G, de Beer D. Aerobic organic carbon mineralization by sulfate-reducing bacteria in the oxygen-saturated photic zone of a hypersaline microbial mat. Microbial Ecol. 2005;49:291–300. doi: 10.1007/s00248-004-0260-y. [DOI] [PubMed] [Google Scholar]
- Jørgensen BB. Mineralization of organic matter in the sea bed – the role of sulphate reduction. Nature. 1982;296:643–645. [Google Scholar]
- Jørgensen BB. Ecology of the sulphur cycle: oxidative pathways in sediments. In: Cole JA, Ferguson S, editors. The Nitrogen and Sulphur Cycles. Vol. 42. Cambridge, UK: Cambridge University Press; 1987. pp. 31–63. [Google Scholar]
- Klein M, Friedrich M, Roger AJ, Hugenholtz P, Fishbain S, Abicht H, et al. Multiple lateral transfers of dissimilatory sulfite reductase genes between major lineages of sulfate-reducing prokaryotes. J Bacteriol. 2001;183:6028–6035. doi: 10.1128/JB.183.20.6028-6035.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenk HP, Clayton RA, Tomb JF, White O, Nelson KE, Ketchum KA, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- Krause L, McHardy AC, Nattkemper TW, Pühler A, Stoye J, Meyer F. GISMO – gene identification using a support vector machine for ORF classification. Nucleic Acids Res. 2007;35:540–549. doi: 10.1093/nar/gkl1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Länge S, Scholtz R, Fuchs G. Oxidative and reductive acetyl-CoA/carbon monoxide dehydrogenase pathway in Desulfobacterium autotrophicum. 1. Characterization and metabolic function of the cellular tetrahydropterin. Arch Microbiol. 1989;151:77–83. [Google Scholar]
- Lemos RS, Gomes CM, Santana M, LeGall J, Xavier AV, Teixeira M. The ‘strict’ anaerobe Desulfovibrio gigas contains a membrane-bound oxygen-reducing respiratory chain. FEBS Lett. 2001;496:40–43. doi: 10.1016/s0014-5793(01)02399-7. [DOI] [PubMed] [Google Scholar]
- Lindahl PA. The Ni-containing carbon monoxide dehydrogenase family: light at the end of the tunnel? Biochemistry. 2002;47:2097–2105. doi: 10.1021/bi015932+. [DOI] [PubMed] [Google Scholar]
- Llobet-Brossa E, Rabus R, Böttcher ME, Könneke M, Finke N, Schramm A, et al. Community structure and activity of sulfate-reducing bacteria in an intertidal surface sediment: a multi-method approach. Aquat Microb Ecol. 2002;29:221–226. [Google Scholar]
- Lumppio HL, Shenvi NV, Summers AO, Voordouw G, Kurtz DM., Jr Rubrerythrin and rubredoxin oxidoreductase in Desulfovibrio vulgaris: a novel oxidative stress protection system. J Bacteriol. 2001;183:101–108. doi: 10.1128/JB.183.1.101-108.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHardy AC, Goesmann A, Pühler A, Meyer F. Development of joint application strategies for two microbial gene finders. Bioinformatics. 2004;20:1622–1631. doi: 10.1093/bioinformatics/bth137. [DOI] [PubMed] [Google Scholar]
- Mander GJ, Duin EC, Linder D, Stetter KO, Hedderich R. Purification and characterization of a membrane-bound enzyme complex from the sulfate-reducing archaeon Archaeoglobus fulgidus related to heterodisulfide reductase from methanogenic archaea. Eur J Biochem. 2002;269:1895–1904. doi: 10.1046/j.1432-1033.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- Mander GJ, Pierik AJ, Huber H, Hedderich R. Two distinct heterodisulfide reductase-like enzymes in the sulfate-reducing archaeon Archaeoglobus profundus. Eur J Biochem. 2004;271:1106–1116. doi: 10.1111/j.1432-1033.2004.04013.x. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer R, Bryant SH. GD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:327–332. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias PM, Pereira IAC, Soares CM, Carrondo MA. Sulphate respiration from hydrogen in Desulfovibrio bacteria: a structural biology overview. Prog Biophys Mol Biol. 2005;89:292–329. doi: 10.1016/j.pbiomolbio.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Merkl R. SIGI: score-based identification of genomic islands. BMC Bioinformatics. 2004;5:22. doi: 10.1186/1471-2105-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methé BA, Nelson KE, Eisen JA, Paulsen IT, Nelson W, Heidelberg JF, et al. Genome of Geobacter sulfurreducens: metal reduction in subsurface environments. Science. 2003;302:1967–1969. doi: 10.1126/science.1088727. [DOI] [PubMed] [Google Scholar]
- Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, et al. GenDB – an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003;31:2187–2195. doi: 10.1093/nar/gkg312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RM, Pihl TD, Nölling J, Reeve JN. Hydrogen regulation of growth, growth yields, and methane gene transcription in Methanobacterium thermoautotrophicumΔH. J Bacteriol. 1997;179:889–898. doi: 10.1128/jb.179.3.889-898.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton TA, Runquist JA, Ragsdale SW, Shanmugasundaram T, Wood HG, Ljungdahl LG. The primary structure of the subunits of carbon monoxide dehydrogenase/acetyl-CoA synthase from Clostridium thermoaceticum. J Biol Chem. 1991;266:23824–23828. [PubMed] [Google Scholar]
- Mussmann M, Richter M, Lombardot T, Meyerdierks A, Kuever J, Kube M, et al. Clustered genes related to sulfate respiration in uncultured prokaryotes support the theory of their concomitant horizontal transfer. J Bacteriol. 2005;187:7126–7137. doi: 10.1128/JB.187.20.7126-7137.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka H, Keresztes G, Shinoda Y, Ikenaga Y, Abe M, Naito K, et al. Complete genome sequence of the dehalorespiring bacterium Desulfitobacterium hafniense Y51 and comparison with Dehalococcoides ethenogenes 195. J Bacteriol. 2006;188:2262–2274. doi: 10.1128/JB.188.6.2262-2274.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Larsen N, Walunas T, D'Souza M, Pusch G, Selkov E, Jr, et al. The ERGOTM genome analysis and discovery system. Nucleic Acids Res. 2003;31:164–171. doi: 10.1093/nar/gkg148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego M, Hoch JA. Protein aspartate phosphatases control the output of two-component signal transduction systems. Trends Genet. 1996;12:97–101. doi: 10.1016/0168-9525(96)81420-x. [DOI] [PubMed] [Google Scholar]
- Pereira IAC, Haveman SA, Voordouw G. Biochemical, genetic and genomic characterization of anaerobic electron transport pathways in sulphate-reducing delta-proteobacteria. In: Barton LL, Hamilton WA, editors. Sulphate-reducing Bacteria: Environmental and Engineered Systems. Cambridge, UK: Cambridge University Press; 2007. pp. 215–240. [Google Scholar]
- Pereira PM, Teixeira M, Xavier AV, Luoro RO, Pereira IAC. The Tmc complex from Desulfovibrio vulgaris Hildenborough is involved in transmembrane electron transfer from periplasmic hydrogen oxidation. Biochemistry. 2006;45:10359–10367. doi: 10.1021/bi0610294. [DOI] [PubMed] [Google Scholar]
- Pfeiffer M, Bestgen H, Burger A, Klein A. The vhuU gene encoding a small subunit of a selenium-containing [Ni/Fe]-hydrogenase in Methanococcus voltae appears to be essential for the cell. Arch Microbiol. 1998;170:418–426. doi: 10.1007/s002030050662. [DOI] [PubMed] [Google Scholar]
- Pieulle L, Morelli X, Gallice P, Lojou E, Barbier P, Czjzek M, et al. The type I/type II cytochrome c3 complex: an electron transfer link in the hydrogen-sulfate reduction pathway. J Mol Biol. 2005;354:73–90. doi: 10.1016/j.jmb.2005.09.036. [DOI] [PubMed] [Google Scholar]
- Pires RH, Lourenço AI, Morais F, Teixeira M, Xavier AV, Saraiva LM, Pereira IAC. A novel membrane-bound respiratory complex from Desulfovibrio desulfuricans ATCC 27774. Biochim Biophys Acta. 2003;1605:67–82. doi: 10.1016/s0005-2728(03)00065-3. [DOI] [PubMed] [Google Scholar]
- Pires RH, Venceslau SS, Morais F, Teixeira M, Xavier AV, Pereira IAC. Characterization of the Desulfovibrio desulfuricans ATCC 27774 DaysrMKJOP complex – a membrane-bound redox complex involved in the sulfate respiratory pathway. Biochemistry. 2006;45:249–262. doi: 10.1021/bi0515265. [DOI] [PubMed] [Google Scholar]
- Rabus R, Strittmatter A. Functional genomics of sulphate-reducing prokaryotes. In: Barton LL, Hamilton WA, editors. Sulphate-reducing Bacteria: Environmental and Engineered Systems. Cambridge, UK: Cambridge University Press; 2007. pp. 117–140. [Google Scholar]
- Rabus R, Hansen TA, Widdel F. Dissimilatory sulfate- and sulfur-reducing prokaryotes. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, editors. The Prokaryotes: An Evolving Electronic Resource for the Microbiological Community. Heidelberg, Germany: Springer Science Online; 2000. [Google Scholar]
- Rabus R, Ruepp A, Frickey T, Rattei T, Fartmann B, Stark M, et al. The genome of Desulfotalea psychrophila, a sulfate-reducing bacterium from permanently cold arctic sediments. Environ Microbiol. 2004;6:887–902. doi: 10.1111/j.1462-2920.2004.00665.x. [DOI] [PubMed] [Google Scholar]
- Ren SX, Fu G, Jiang XG, Zeng R, Miao YG, Xu H, et al. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature. 2003;422:888–893. doi: 10.1038/nature01597. [DOI] [PubMed] [Google Scholar]
- Rodrigues R, Valente FMA, Pereira IAC, Oliveira S, Rodrigues-Pousada C. A novel membrane-bound Ech [Ni/Fe] hydrogenase in Desulfovibrio gigas. Biochem Biophys Res Commun. 2003;306:366–375. doi: 10.1016/s0006-291x(03)00975-6. [DOI] [PubMed] [Google Scholar]
- Rossi M, Pollock WBR, Reij MW, Keon RG, Fu R, Voordouw G. The hmc operon of Desulfovibrio vulgaris subsp. vulgaris Hildenborough encodes a potential transmembrane redox protein complex. J Bacteriol. 1993;175:4699–4711. doi: 10.1128/jb.175.15.4699-4711.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy I, Leadlay PF. Physical map location of the new Escherichia coli gene sbm. J Bacteriol. 1992;174:5763–5764. doi: 10.1128/jb.174.17.5763-5764.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva LM, da Costa PN, Conte C, Xavier AV, LeGall J. In the facultative sulphate/nitrate reducer Desulfovibrio desulfuricans ATCC 27774, the nine-haem cytochrome c is part of a membrane-bound redox complex mainly expressed in sulphate-grown cells. Biochim Biophys Acta. 2001;1520:63–70. doi: 10.1016/s0167-4781(01)00250-0. [DOI] [PubMed] [Google Scholar]
- Schauder R, Eikmanns B, Thauer RK, Widdel F, Fuchs G. Acetate oxidation to CO2 in anaerobic bacteria via a novel pathway not involving reactions of the citric acid cycle. Arch Microbiol. 1986;145:162–172. [Google Scholar]
- Schauder R, Preuß A, Jetten M, Fuchs G. Oxidative and reductive acetyl-CoA/carbon monoxide dehydrogenase pathway in Desulfobacterium autotrophicum. 2. Demonstration of the enzymes of the pathway and comparison of CO dehydrogenase. Arch Microbiol. 1989;151:84–89. [Google Scholar]
- Schwartz E, Henne A, Cramm R, Eitinger T, Friedrich B, Gottschalk G. Complete nucleotide sequence of pHG1: a Ralstonia eutropha H16 megaplasmid encoding key enzymes of H2-based lithoautotrophy and anaerobiosis. J Mol Biol. 2003;332:369–383. doi: 10.1016/s0022-2836(03)00894-5. [DOI] [PubMed] [Google Scholar]
- Seedorf H, Fricke WF, Veith B, Brüggemann H, Liesegang H, Strittmatter A, et al. The genome of Clostridium kluyveri, a strict anaerobe with unique metabolic features. Proc Natl Acad Sci USA. 2008;105:2128–2133. doi: 10.1073/pnas.0711093105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staden R. The Staden sequence analysis package. Mol Biotechnol. 1996;5:233–241. doi: 10.1007/BF02900361. [DOI] [PubMed] [Google Scholar]
- Staden R, Beal KF, Bonfield JK. The Staden package, 1998. Methods Mol Biol. 2000;132:115–130. doi: 10.1385/1-59259-192-2:115. [DOI] [PubMed] [Google Scholar]
- Staron A, Sofia HJ, Liesegang H, Mascher T. A comparative genomics perspective on the ECF σ factor protein family: classification and functional predictions. American Society for Microbiology, 107th General Meeting Toronto, Canada.
- Stojanowic A, Mander GJ, Duin EC, Hedderich R. Physiological role of the F420-non-reducing hydrogenase (Mvh) from Methanothermobacter marburgensis. Arch Microbiol. 2003;180:194–203. doi: 10.1007/s00203-003-0577-9. [DOI] [PubMed] [Google Scholar]
- Taylor BL, Zhulin IB. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev. 1999;63:479–506. doi: 10.1128/mmbr.63.2.479-506.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tech M, Merkl R. YACOP: enhanced gene prediction obtained by a combination of existing methods. In Silico Biol. 2003;3:441–451. [PubMed] [Google Scholar]
- Teske A, Ramsing NB, Habicht K, Fukui M, Küver J, Jørgensen BB, Cohen Y. Sulfate-reducing bacteria and their activities in cyanobacterial mats of Solar Lake (Sinai, Egypt) Appl Environ Microbiol. 1998;64:2943–2951. doi: 10.1128/aem.64.8.2943-2951.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008;6:579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- Valente FMA, Saraiva LM, LeGall J, Xavier AV, Teixeira M, Pereira IAC. A membrane-bound cytochrome c3: a type II cytochrome c3 from Desulfovibrio vulgaris Hildenborough. Chembiochem. 2001;2:895–905. doi: 10.1002/1439-7633(20011203)2:12<895::AID-CBIC895>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Vermeij P, Pennings JLA, Massen SA, Keltjens JT, Vogels GD. Cellular levels of factor 390 and methanogenic enzymes during growth of Methanobacterium thermoautotrophicumΔH. J Bacteriol. 1997;179:6640–6648. doi: 10.1128/jb.179.21.6640-6648.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voordouw G. Carbon monoxide cycling by Desulfovibrio vulgaris Hildenborough. J Bacteriol. 2002;184:5903–5911. doi: 10.1128/JB.184.21.5903-5911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waack S, Keller O, Asper R, Brodag T, Damm C, Fricke WF, et al. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics. 2006;7:142. doi: 10.1186/1471-2105-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widdel F. Microbiology and ecology of sulfate-reducing bacteria. In: Zehnder AJB, editor. Biology of Anaerobic Microorganisms. Vol. 3. Munich, Germany: Carl Hanser Verlag; 1988. pp. 469–585. [Google Scholar]
- Widdel F, Bak F. Gram-negative mesophilic sulfate-reducing bacteria. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H, editors. The Prokaryotes. 2nd edn. IV. New York, NY, USA: Springer-Verlag; 1992. pp. 3352–3378. [Google Scholar]
- Widdel F, Pfennig N. A new anaerobic, sporing, acetate-oxidizing, sulfate-reducing bacterium, Desulfotomaculum (emend.) acetoxidans. Arch Microbiol. 1977;112:119–122. doi: 10.1007/BF00446665. [DOI] [PubMed] [Google Scholar]
- Widdel F, Pfennig N. Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids. I. Isolation of new sulfate-reducing bacteria enriched with acetate from saline environments. Description of Desulfobacter postgatei. Arch Microbiol. 1981;129:395–400. doi: 10.1007/BF00406470. [DOI] [PubMed] [Google Scholar]
- Wild J, Hradecna Z, Szybalski W. Conditionally amplifiable BACs: switching from single-copy to high-copy vectors and genomic clones. Genome Res. 2002;12:1434–1444. doi: 10.1101/gr.130502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhulin IB, Taylor BL, Dixon R. PAS domain S-boxes in Archaea, Bacteria and sensors for oxygen and redox. Trends Biochem Sci. 1997;9:331–333. doi: 10.1016/s0968-0004(97)01110-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.