

Abstract
The neuropeptide VIP is anti-inflammatory and protective in the immune and nervous systems, respectively. The present study demonstrated in corneal endothelial (CE) cells injured by severe oxidative stress (1.4mM H2O2) in bovine corneal organ cultures that VIP pre-treatment (0, 10−10, 10−8, and 10−6 M; 15 min), in a VIP concentration-dependent manner, switched the inflammation-causing necrosis to inflammation neutral apoptosis (showing annexin V-binding, chromatin condensation, and DNA fragmentation) and upheld ATP levels in a VIP antagonist (SN)VIPhyb-sensitive manner, while up-regulated mRNA levels of the anti-apoptotic Bcl-2 and the differentiation marker N-cadherin in a kinase A inhibitor-sensitive manner. As a result, VIP, in a concentration-dependent and VIP antagonist-sensitive manners, promoted long-term CE cell survival. ATP levels, a determining factor in the choice of apoptosis vs necrosis, measured after VIP pre-treatment and 0.5 min post- H2O2 were 39.6±3.3, 50.8±6.2, 60.1±4.8, and 53.6±5.3 pmoles/μg protein (mean±sem), respectively (p<0.05, ANOVA). VIP treatment alone concentration-dependently increased levels of N-cadherin (Koh et al., 2008), the phosphorylated cAMP-responsive-element binding protein and Bcl-2, while10−8 M VIP, in a VIP antagonist (SN)VIPhyb-sensitive manner, increased ATP level by 38% (p< 0.02) and decreased glycogen level by 32% (p< 0.02). VPAC1 (not VPAC2) receptor was expressed in CE cells. Thus, CE cell VIP/VPAC1 signaling is both anti-inflammatory and protective in the corneal endothelium.
Keywords: Apoptosis, necrosis, VIP, corneal endothelium, cell survival, oxidative stress
INTRODUCTION
VIP, a 28-amino-acid neuropeptide binding to two types of adenylyl cyclase stimulatory heterotrimeric G protein-coupled receptors VPAC1 and VPAC2, transduces signals through cAMP-dependent and -independent pathways (Langer and Robberecht, 2007). VIP is widely distributed in the central and peripheral nervous systems, where its neuroprotective role has been demonstrated in a variety of in vitro and in vivo systems (Moody et al., 2003; Rangon et al., 2005; Brenneman; 2007; Delgado et al, 2008), as well as in the immune system, where it is an anti-inflammatory agent (Delgado et al., 2004; Gonzalez-Rey et al., 2007).
Recent studies demonstrate that dying cells, particularly those in the necrotic death mode, transduce signals that trigger the immune system to display an inflammatory response (Chen et al., 2007; Kono and Rock, 2008; Rock and Kono, 2008). Thus, by preventing cell death, the protective effect of VIP is intrinsically anti-inflammatory. However, whether VIP can reduce the inflammatory potential of dying cells in injured tissues has never been studied. It is a distinct possibility that down regulation of the inflammatory potential of dying cells is an integral part of the VIP protective effects observed in the nervous systems.
Necrosis and apoptosis are two cell death pathways sharing common initiating events such as the oxidative stress. Early plasma membrane rupture is the hallmark of necrotic death, while the apoptotic cell maintains the plasma membrane integrity within the same time frame. The choice of death modes impacts the outcome of injured tissues since necrotic cells enhance the inflammatory responses while those dying by apoptosis inhibit the proinflammatory cytokine expression (Li et al., 2001; Cocco and Ucker, 2001; Cvelanovic and Ucker, 2004; Erwig and Henson, 2007 & 2008). Furthermore, cells dying of apoptosis, but not necrosis, can be rescued by neurotrophic factors (Fujita and Ueda, 2003a; Kim et al., 2004; Ueda et al., 2007; Ueda 2008). The possibility exists that, as an integral part of the protective mechanism, VIP switches the necrotic cell death to an apoptotic one and allows dying cells to be rescued by available neurotrophic factors, including VIP itself.
VIP is an autocrine of the corneal endothelium (Koh and Waschek, 2000), which is a neural crest-derived tissue ( LeLievre e a., 1975; Noden et al., 1978; Johston et al., 1979; Hayashi et al., 1986) of a single-layer of corneal endothelial (CE) cells The corneal endothelium has a very limited capacity for regeneration ( Joyce, 2005) and is where the immune-privilege enjoyed by the cornea resides (Niederkorn 2003, Cursiefen, 2007; Hori et al., 2007). How the corneal endothelium survives through out life and maintains its immune suppressive capacity is not known at the present time. In a recent report (Koh et al., 2008), VIP is shown to be a differentiation state-maintaining factor of CE cells in human corneas ex vivo in that VIP gene knockdown in CE cells results in a dramatic alteration in cell shape and size as well as a diminished level of the CE differentiation marker N-cadherin. Up-regulation of the endogenous VIP expression in CE cells by a well known injury factor, the ciliary neurotrophic factor (CNTF) supports a role of VIP in CE cell survival (koh et al., 2007). In addition, CNTF (together with its CNTF-biding α receptor subunit) is released by CE cells surviving the oxidative stress (Koh, 2002), indicating a role of VIP in CE cell surviving the oxidative stress. In corneal organ cultures, exogenous VIP protects CE cells against the acute killing effect of severe oxidative stress (Koh and Waschek, 2000). The present study examined the possibility that this protective effect of VIP has an anti-inflammatory component. Specifically, the present study examined if VIP played a role in down-regulating the inflammatory potential of dying CE cells by promoting apoptosis at the expense of necrosis. In addition, the possibility that VIP, by promoting the apoptotic cell death mode over that of necrosis allowed the recovery and long-term survival of injured CE cells in corneoscleral explants.
Apoptotic cells display several hallmarks including exposure of phosphatidylserine (PtdSer) to the outer phospholipid leaflet of the plasma membrane early in apoptosis (Vermes et al., 1995), chromatin condensation, and oligonucleosomal DNA fragmentation (Susin et al., 2000; Joselin et al, 2006). In the present study, the effects of VIP pretreatment on the display of these hallmarks in CE cells in corneoscleral explants under severe oxidative stress, which brought about a sizable necrotic cell population, were investigated.
In the choice of apoptosis vs necrosis, the intracellular ATP level of the dying cells plays a crucial role. When the ATP level is maintained, cells die of apoptosis, whereas necrosis is the death mode of injured cells with depleted ATP (Leist et al., 1997; Kim et al., 2003; Atlante et al. 2005; Miyoshi et al., 2006). Earlier studies of VIP have demonstrated that VIP exerts metabolic effect through stimulation of glycogenolysis in astrocytes, retinal cells, a sarcoma cell line (Van Valen et al. 1989; Ghazi and Osborne, 1989; Pellerin et al. 1997), and in cortical slices (Magistretti et al., 1981). Glycogen is present in CE cells (Malinin and Bernstein, 1979), the effect of VIP on glycogen and intracellular ATP levels were studied in the present study.
The protective effect of VIP against apoptosis induced by serum withdrawal, cigarette smoke, and glutamate-induced oxidative stress has been demonstrated (Gutierrez-Canas et al., 2003; Onoue et al., 2004; Sahin et al, 2006). The VIP anti-apoptotic signaling leads to induction of the anti-apoptotic protein Bcl-2 expression (Gutierrez-Canas et al., 2003; Sahin et al., 2006) and protein kinase A-dependent phosphorylation (inactivation) of the pro-apoptotic protein BAD (Sastry et al, 2006).
VIP stimulates production of large amounts of cAMP in CE cells (Koh and Yue, 2002), whereas VIP-stimulated cAMP-dependent pathway leads to cAMP-responsive element binding protein (CREB) phosphorylation in a variety of cells (Hokari et al., 2005; Fernandez et al., 2005). CREB phosphorylation mediates neuronal survival against glutamate toxicity through induction of the anti-apoptotic Bcl-2 (Mabuchi et al., 2001). The anti-apoptotic effect of insulin-like growth factor is dependent on activated CREB-induced Bcl-2 expression (Mehrhof et al, 2001). In the promoter region of the Bcl-2, a cAMP responsive element has a positive regulatory role in Bcl-2 expression and cell survival (Wilson et al., 1996; Xiang et al., 2006), whereas insulin-like growth factor-I induces Bcl-2 promotor through CREB (Pugazhenthi et al, 1999). Here, the effects of VIP on the levels of phospho-CREB in CE cells and Bcl-2 expression in CE cells surviving the oxidative stress were studied. Furthermore, since endogenous VIP is a CE differentiated state-maintaining factor modulating the expression of the differentiation marker N-cadherin (Koh et al., 2008), the present study investigated the effect of VIP on the status of N-cdherin in CE cells injured by oxidative stress.
Finally, the type of VIP receptor in CE cells was investigated.
MATERIALS AND METHODS
Corneoscleral explants
Corneoscleral explants were dissected from bovine eyes obtained from the local abattoir within six h of sacrifice (Koh and Waschek, 2000). Those corneoscleral explants that were used in experiments that did not require a H2O2 treatment step were sterilized by immersing bovine eyes in a 0.5% iodine solution (1:4 dilution in DPBS of Providone iodine prep solution [Baxter Health Care Co., Deerfield, IL]) for five seconds and rinsed in DPBS before corneoscleral explants were dissected.
With its concave (the corneal endothelium) side up, explants were rinsed in 14 ml ice-cold DPBS in 60 mm Petri dishes and incubated in 3.5 ml of the semi-complete medium (Eagle’s minimal essential medium with Earl’s salts plus 20 mM HEPES) in 35 mm Petri dishes in a moisture box under 5% CO2/95% air at 37°C for1 h.
VIP pretreatment of corneal endothelium in corneal cups
The corneoscleral explant placed on the cap of a 50 ml conical centrifuge tube, with the corneal endothelium up, formed a corneal cup. VIP (0, 10−12-10−6 M) in semi-complete medium (0.9 ml) was immediately placed in the cups to treat and incubate the CE cells (15 min). To test the effects of blocking VIP binding to its receptor, VIP antagonist (SN)VIPhyb (Koh, 2008; Moody et al., 2001) (Bachem, Torrance, CA) was included in the medium during the VIP pretreatment. To test the effects of kinase A inhibition, H-89 (Sigma) was included in the medium during the VIP pretreatment. After the VIP pretreatment, corneoscleral explants were either treated with H2O2 as described below or incubated (25 h) in the semi-complete medium supplemented with antibiotics and fungizone (100 U/ml penicillin, 100 μg/ml streptomycin sulfate, and 250 ng/ml amphotericin; Invitrogen; Grand Island, NY).
H2O2 treatment of corneoscleral explants
Following VIP pretreatment, corneoscleral explants were transferred to 3.5 ml of 1.4 mM H2O2/DPBS or DPBS alone in 35 mm Petri dishes and incubated for the specified periods.
CE cell viability and chromatin condensation in H2O2-treated corneoscleral explants
Viable and dead CE cells in explants were revealed simultaneously using fluorescence microscopy (Nikon Diaphot-TMD) and a kit (LIVE/DEAD Viability/Cytotoxicity; Molecular Probes). The mixture of calcein acetoxymethyl ester (AM) and ethidium homodimer (0.9 ml) was added to cover and incubate the CE cells in the corneal cup (15 min, 37°C in 5%CO2-95% air) before the explant was flat-mounted on a piece of coverslip (45×50 mm), corneal endothelium side down. Neuclei including those with chromatin condensation in the dead cells, which allowed entrance of the DNA-binding fluorescent ethidium homodimer, appeared red, while live cells with intracellular esterase activities appeared green.
Annexin V/propidium iodide (PI) binding
Annexin V binding to the exposed PtdSer of apoptotic cells was demonstrated using a kit containing Alexa Fluor® 488 annexin V/PI (Vybrant® Apoptosis Assay Kit #2, Molecular Probes) at four h after H2O2 treatment. Explants were flat-mounted, corneal endothelium side down, on 45×50mm cover slips holding 200 μl of annexin V/PI mixture and incubated (room temperature, 15 min). The corneal endothelium was examined under a fluorescence microscope (X 400 magnification; Nikon Diaphot-TMD). For negative control, explants were treated with DPBS alone (no H2O2). To demonstrate annexin V binding to CE cells free of intercellular junctions, corneal cups were washed twice with Ca/Mg-free DPBS, incubated with 0.9 ml of the same containing collagenase (0.06 u/ml), BSA (0.02 %), and EDTA (0.05 mM) for 12 min (37°C). The contents of corneal cups were centrifuged (30 sec, 12,000 × g) and the pellets were washed with Ca/Mg-free DPBS, and resuspended in 100 μl of annexin V/propidium mixture.
Isolation of genomic DNA from attached and free-floating detached CE cells in corneal cups
Genomic DNA was isolated from CE cells using a kit from BioRad (Cat. No 7326340). Free-floating CE cells were collected as pellets following their transfer from the corneal cups into 1.5 ml eppendorph tubes and centrifugation (10 min, 12,000 × g). Lysis buffer (150 μl) was added to both pellets and the attached CE cells in corneal cups. Lysed cells were homogenized using micro pestles and the genomic DNA was isolated following procedures of the manufacturer. Genomic DNA content was determined by absorbance at 260 nm (GeneQuantpro, Biochrom).
Agarose gel electrophoresis of genomic DNA
DNA samples were electrophoresed in 1.8/2.0 % agarose gels, stained with ethidium bromide and examined under UV light.
RT-PCR and semi-quantitative RT-PCR
Total RNA was isolated from CE cells scraped from corneoscleral explants (RNA-Bee;Tel-Test, Friendswood, TX) and subjected to reverse transcription (RT) using a kit (RETROscript; Cat No 1710, Ambion; Austin, TX). For demonstrating Bcl-2 mRNA expression in CE cells, PCR was conducted. RT products (derived from 0.15 μg RNA [for 25 μl reaction] or 0.3 μg RNA [for 50μl reaction]) were subjected to PCR using the primer set corresponding to the gene sequences of bovine Bcl-2 (5′-GAGATGTCCAGTCAGCTGCACC-3′ [forward] and 5′-ATAGGCACCCAGGGTGATGC-3′ [reverse]) (Pocar et al., 2005). For quantifying mRNA levels, semi-quantitative PCR was conducted in the presence of the18S primers and the 18S competomers (Classic 18S internal standards [Cat No 1716] or Universal 18S internal standards [Cat No 1718]; Ambion, Austin, TX) in the ratio of 1:9 to generate 489 (classic) and 315 (universal) bp PCR products as 18 S internal standards. Bcl-2 primer set above and that human N-cadherin (5′-CACCCAACATGTTTACAATCAACAATGAGAC-3′ [forward] and 5′-CTGCAGCAACAGTAAGGACAAACATCCTATT-3′ [reverse]) (Lee et. al., 2007; Koh et al., 2008). Between an initial 94°C (2 min) and a final 5 min (72°C) treatments, 25–35 thermo-cycles (20 sec at 94°C, 25 sec at the annealing temperature, and 40 sec at 72°C) were conducted. The numbers of cycles and the annealing temperatures (in °C) were: 35 and 56 (Bcl-2 PCR); 27 and 56 (Bcl-2 semi-quantitative PCR); 25 and 60 (N-cadherin semi-quantitative PCR). RT-PCR products were electrophoresed (2% agarose gel) and the optical densities of bands were measured using a densitometer (Nucleo Vision; Nucleo Tech, San Carlos, CA). The identities of specific bands of appropriate sizes revealed after agarose gel electrophoresis were proven by sequencing (Biopolymer Lab, University of Maryland) and demonstrated to be identical to the sequences for bovine Bcl-2 (366bp) and bovine N-cadherin (Koh et al, 2008).
ATP determination
CE cell ATP was extracted by boiling H2O (Yang et al., 2002). At the end of H2O2 treatment (0.5 min), corneoscleral explants were removed from the Petri dishes and placed on caps of 50 ml conical centrifuge tubes. 400 μl of boiling H2O was immediately placed in the corneal cups to extract ATP from the corneal endothelium. Corneal endothelium was scraped using a loop (cat. No 254437, Nunc) and contents of corneal cups were placed in eppendorph tubes, boiled (20 min) and centrifuged (5min, 1,302 × g). Aliquots were taken form the supernatants for ATP determination using a kit (INV-22066, Invitrogen). The remaining supernatants and pellets in eppendorph tubes were sonicated for protein determination by bicinchoninic acid method (Pierce, Rockford Il).
Glycogen content of CE cells
The glycogen was isolated from CE cells following a published procedure (Prothmann et al., 2001). CE cells in each of the corneal cups were washed with 2 ml DPBS, scraped from the corneal cups, and frozen on dry ice and kept at −80°C. The frozen CE cells were lysed in 0.4 ml of 0.1 N/0.5 N NaOH and incubated for 60/30 min at 80° C. Double deionized H2O2 was added to give the final lysate NaOH concentration of 0.1 N. To precipitate the glycogen, the lysates were added 2.5 volumes of 100 % alcohol and centrifuged at 13,000 × g for 10 min. After removal of the supernatant, the glycogen precipitate was resuspended in 100 μl of 50 mM sodium acetate buffer, pH 4.8. The glycogen samples were digested by adding amyloglucosidase (Sigma, St Louis, Mo) and incubated for 120 min at 37°C. The resulting glucose samples were assayed using a kit from Sigma (GAGO-20). To each glucose sample, 350 μl of the assay reagent made according to the instruction provided by Sigma was added. After incubation for 45 min at 37°C, the reaction was stopped by 350 μl 12 N H2SO4 (1/3 dilution of the full strength) and the absorbance of the sample was read at 540 nm in a spectrophotometer.
Western blot analysis
At the conclusion of VIP treatment (above), corneal endothelium was scraped off the corneas using a razor blade and homogenized in the RIPA buffer (25 mM Tris pH 7.2, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA) supplemented with protease inhibitors (one tablet of protease inhibitor cocktail [complete, mini; Roche Diagnostics, Mannheim, Germany]/10 ml). Following centrifugation at 12,000 × g for 10 min, the supernatant (cell extract) was analyzed by Western blot analysis. Samples of CE cell extracts were electrophoresed under reducing condition using preformed Tris/Glycine polyacryl amide (8–16%) gradient gels (NuPage; Novex; San Diego, CA), electrophoretically transferred to nitrocellulose membranes for Western blot analysis using a rabbit monoclonal antibody against phospho (ser 133)-CREB (87G3, Cell Signaling Tech., Danvers, MA), and mouse monoclonal antibodies against VPAC1 (20-272-191277; GenWay, San Diego, CA) and VPAC2 (MAB5470, Chemicon, Temecula CA), and a kit containing the horseradish peroxidase-linked anti-rabbit/mouse IgG secondary antibodies and horseradish peroxidase substrate (ECL kit, Amersham Pharmacia; Piscataway, NJ). To reprobe the blots with a monoclonal anti-actin antibody (Ab-1 kit; Oncogene, Cambridge, MA), the blots were stripped using Restore Western Blot Stripping buffer (Pierce, Rockford, IL) as per instruction from the manufacturer. Densities of immunoreactive bands were determined same as in semi-quantitative RT-PCR.
Statistical analysis
Effects of multiple concentrations of VIP were analyzed by one-way analysis of variance (ANOVA) followed bypost-hoc t- test of VIP-pretreated vs control groups.
RESULTS
VIP Promotion of apoptosis, at the expense of necrosis, among CE cells injured by severe oxidative stress: annexin V-binding study
In order to test the hypothesis that VIP pretreatment switches necrotic cell death to apoptotic one, the condition that brought about a sizable population of necrotic CE cells was established and used in the present study. CE cells in corneoscleral explants that were freshly dissected from bovine eyes were nearly all alive. Fluorescence microscopy following live/dead cytotoxicity test of CE cells in fresh corneoscleral explants showed a green carpet of live CE cells. Occasionally, red nuclei of dead CE cells can be found dotting the green carpet (Fig. 1A). CE cells are resistant to oxidative stress, surviving up to 0.6 mM H2O2 treatment for 30 min with no visible cell death over that found in fresh corneoscleral explants (Koh, 2002), whereas we have previously demonstrated (Koh and Waschek, 2000) that 1.4 mM H2O2 treatment for 30 min brought about numerous necrotic CE cells (Fig. 1B). Therefore acute oxidative stress was brought about by 1.4 mM H2O2 treatment.
Fig. 1.
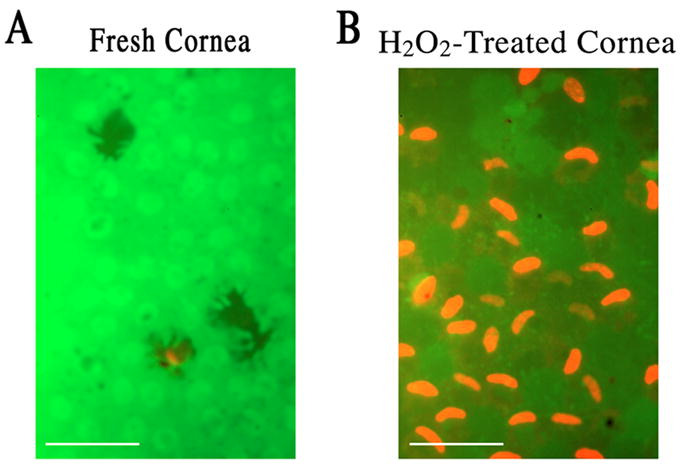
Fluorescence microscopy in a live and dead cytotoxicity test of CE cells in flat-mounted fresh (A) and severe oxidative stress-injured corneoscleral explants (B). (A) A carpet of nearly all alive CE cells in a fresh corneoscleral explant. (B) Necrotic CE cell nuclei dotting the carpet of live CE cells in a corneoscleral explant injured by severe oxidative stress (1.4 mM H2O2; 30 min). White bar length=15 μm.
Following H2O2 treatment, early signs of CE cell apoptosis were examined. Phosphatidylserine (PtdSer), which normally is located to the inner leaflet of the plasma membrane, is exposed early during apoptosis allowing binding by the probe-conjugated annexin V to be used as a marker of apoptotic cells (Vermes et al., 1995). PI-staining of the neuclei, with or without annexin V-binding, was observed in necrotic cells, while apoptotic cells displayed only annexin V-binding.
After 2.5 h of H2O2 exposure, CE cells in the control corneal cups showed necrotic death and no sign of apoptosis (Fig. 2A). After 4 h of H2O2 treatment, some of the PI-stained CE cell nuclei sloughed off while others remained attached (Fig. 2B), and still no apoptotic cell was observed. In both 10−8 and 10−6 M VIP-pretreated corneal cups, numerous CE cells showed apoptotic cell death (Fig. 2C & 2D). As a negative control, 10 −8 M VIP-pretreated corneal cups that were not exposed to H2O2 were examined, and no apoptotic CE cell was detected (Fig. 2E). In order to eliminate the possibility that the Ca-dependent annexin V binding occurred at the Ca-enriched cell-cell junctions and not the exposed PtdSer per se, annexin V-binding was shown in 10−8 and 10−6 M VIP-pretreated apoptotic CE cells whose membranes were not in contact with neighboring cells (Fig. 2F & 2G).
Fig. 2.

Fluorescence microscopy of flat-mounted corneoscleral explants showing VIP pretreatment promoted apoptosis (annexin V-binding) at the expense of necrosis (PI-staining) among CE cells injured by severe oxidative stress. (A & B) Necrotic CE cells in a control cornel cup 2.5 h (A) and 4 h (B) after H2O2 treatment. With compromised cell membrane, necrotic CE cells showed both PI-staining nuclei and annexin V-binding cell membrane. (C & D) Apoptotic CE cells appeared in VIP-pretreated corneal cups following 4 h of H2O2 treatment. 10−8 (C) and 10−6 (D) M VIP-pretreated corneal cups showed numerous annexin V-binding only (no PI-staining) apoptotic CE cells. (E) Negative control for apoptosis. No annexin V-binding only apoptotic CE cell was observed in 10−8 M VIP-pretreated corneal cup that was not subjected to subsequent H2O2 treatment. (F &G) Annexin V-binding to apoptotic CE cell membranes free of cell-cell junctions (arrows). CE cells were isolated from corneal cups that have been pretreated with 10−8 M (F) and 10−6 M (G) VIP followed by 4 h (F) and 17 h (G) of H2O2treatment, respectively. White bar length=15 μm.
VIP-induced display of nuclear markers of apoptosis in oxidative stress-injured CE cells
Seventeen h after H2O2 treatment, examination of the CE cells that remained attached in corneal cups showed that VIP-pretreatment promoted the appearance of nuclear apoptosis. Distinctive light microscopic features of the apoptotic nucleus going through chromatin condensation appear in two stages. Stage I, in which chromatin condensation occurs at the periphery of the nucleus, is followed by complete chromatin degradation in stage II (Susin et al., 2000; Joselin et al, 2006). In VIP-pretreated corneal cups (but not the control ones), dead CE cells that still remained attached to the corneal cups demonstrated initial, peripheral chromatin condensation, consistent with the feature of stage I nuclear apoptosis (Fig. 3A). Supportive evidence showing that apoptosis was the mode of death of oxidative stress-injured CE cells in VIP-pretreated corneal cups was also provided by oligonucleosomal fragmentation of genomic DNA, the hallmark of stage II nuclear apoptosis. Genomic DNA of attached CE cells in VIP-pretreated corneal cups, but not that in control corneal cups, formed a ladder in electrophoresed agarose gel (Fig. 3B). Genomic DNA of detached (mostly dead) CE cells free floating in either control or VIP-pretreated corneal cups did not display a ladder (Fig. 3B). Furthermore, advanced chromatin condensation was observed in CE cells in VIP-pretreated corneal cups that were recovering in the semi-complete medium (27 h) following a 26 h H2O2 treatment (Fig. 3C).
Fig. 3.

Features of nuclear apoptosis in VIP (10−6 M)-pretreated CE cells dying of severe oxidative stress: (A) initial peripheral chromatin condensation, (B) genomic DNA fragmentation,(C) advanced chromatin condensation. White bar length=15 μm. For A: Fluorescence microscopy of flat-mounted corneoscleral explants in a live and dead cytotoxicity test of CE cells after a 17 h-exposure to H2O2. In the control corneal cups, CE cells dying of necrosis displayed spindle-shaped nuclei, while those in VIP-pretreated corneal cups displayed chromatin condensation at the periphery of the nuclei. For B: Agarose gel electrophoresis showing ladder formation by fragmented genomic DNA isolated from CE cells that remained attached (a) to VIP-pretreated, but not control corneal cups. No DNA fragmentation was observed in CE cells that detached from the corneas and became free-floating (f) in either the control or VIP-pretreated corneal cups. m: marker (100-bp DNA ladder). For C: Fluorescence microscopy of a flat-mounted corneoscleral explant in a live and dead cytotoxicity test showing advanced chromatin condensation in CE cells in VIP-pretreated corneal cups after a 27 h-exposure to H2O2, followed by a 26 h-recovery in the semi-complete medium.
CE cell ATP level increased by VIP
Since the ATP level in cells dying of oxidative stress is a determining factor in the choice of necrosis vs. apoptosis, the possibility that VIP-pretreatment made ATP more available to CE cells was examined. As shown in Table 1, ATP levels in CE cells that were exposed to H2O2/DPBS (0.5 min) following a 15 min pretreatment with 0, 10−10, 10−8, 10− 6 M VIP were (mean±sem [p value in VIP-pretreated vs control]) 39.6 ± 3.3, 50.8 ± 6.2 (p<0.05), 60.1 ± 4.8 (p<0.001), and 53.6 ± 5.3 (p<0.02) pmoles/μg protein, in 20, 9, 20, and 9 corneal cups, respectively. The difference among different treatment groups was significant (P<0.05, ANOVA). ATP levels in CE cells that were not subjected to oxidative stress (treated with DPBS alone) was also examined and were (mean±sem) 85.1 ± 10.1 and 117.2 ± 10.5 (p<0.02) pmoles/μg protein in eight control and eight 10−8 M VIP-pretreated corneal cups, respectively (Table 1).
Table 1.
Intracellular ATP levels in CE cells increased by VIP.
| [ATP] (pmoles/μg protein) | ||
|---|---|---|
| mean ± SEM (number of corneas; p value) | ||
| VIP (M) | *H2O2/DPBS | **DPBS |
| 0 | 39.6 ± 3.3 (20;-----) | 85.1 ± 10.1 (13; -----) |
| 10−10 | 50.8 ± 6.2 (9; <0.05) | |
| 10−8 | 60.1 ± 4.8 (20; <0.001) | 117.2 ± 10.5 (13; <0.02) |
| 10−6 | 53.6 ± 5.3 (9; <0.02) | |
ATP was extracted from CE cells in corneal cups that were pretreated with VIP (15 min) and exposed to either 1.4 mM H2O2 in DPBS (*) or DPBS alone (**) for 0.5 min. The results were from eight (*) and four (**) separate experiments.
Statistical analysis indicated that the difference among different treatment groups was significant (P<0.05, ANOVA) (*).
CE cell glycogen level decreased by VIP
Since earlier studies have demonstrated the effect of VIP on glycogenolysis in glycogen-storing cells and CE cells also store glycogen (Introduction), VIP-stimulated glycogenolysis in CE cells may have resulted in increased ATP levels demonstrated in Table 1 above. The level of glycogen in VIP-treated CE cells was therefore measured. Eleven pairs of corneoscleral explants, with corneal cups of each pair used as control vs 1 × 10−8 M VIP-pretreated, demonstrated the effect of VIP in lowering the glycogen content in CE cells. Fifteen min after VIP pretreatment, the glycogen levels in CE cells of 11 corneal cups decreased to 0.683±0.104-fold (mean±SEM,; P=0.006 ) of those found in the paired control corneal cups (Fig. 4A). In pooled corneoscleral explants, the effect of VIP antagonist (SN)VIPhyb on attenuating the VIP effect was demonstrated (Fig. 4B). The relative glycogen levels in CE cells from the control, 10−8 M-treated, and 10−8 M VIP plus 5 × 10−7 M (SN)VIPhyb- treated corneal cups were 1±0.14 (N=7), 0.59±0.07 (N=6), and 0.79±0.03 (N=4), respectively(Fig. 4B). The significance levels for control vs VIP-treated and VIP-treated vs VIP± (SN)VIPhyb-treated were p=0.01 and p=0.05, respectively.
Fig. 4.
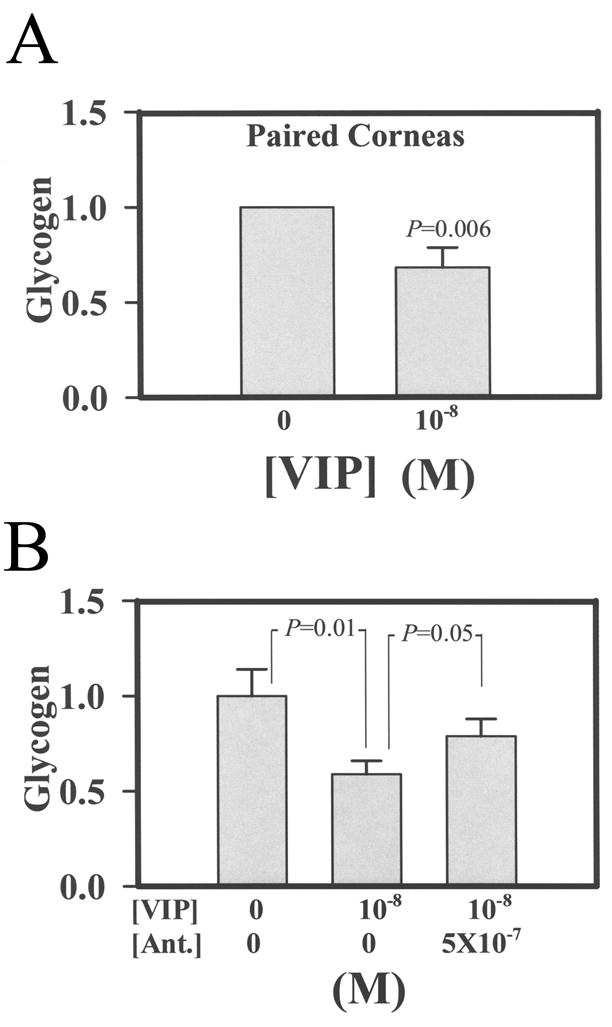
VIP decreased the glycogen content in CE cells (A) and the VIP antagonist (SN)VIPhyb (5 × 10−7 M) attenuated the VIP effect (B). For A: 11 pairs of corneoscleral explants were used. Corneal cups from each pair were used as control vs VIP (10−8 M)-treated. After a 15 min treatment, the glycogen level in CE cells from the VIP-pretreated corneal cups decreased to 0.683±0.104–fold of the control. The results were combined from four separate experiments. For B: Pooled corneosceral explants were used. The relative glycogen levels in CE cells from the control, 10−8 M-treated, and 10−8 M VIP plus 5 × 10−7 M (SN)VIPhyb-treated corneal cups were 1±0.14 (N=7), 0.59±0.07 (N=6), and 0.79±0.03 (N=4), respectively. The significance levels for control vs VIP-treated and VIP-treated vs VIP plus (SN)VIPhyb-treated corneal cups were p=0.01 and p=0.05, respectively.
VIP-promoted long-term survival of oxidative stress-injured CE cells: quantification by genomic DNA
To demonstrate that VIP promotion of apoptosis at the expense of necrosis resulted in, or gave an impression of VIP-increased survival, CE cells were examined microscopically and quantified (by genomic DNA quantification ) after 17 h of H2O2 treatment and also at 64 h into recovery from a 30 min H2O2 (1.4 mM) injury.
Seventeen h after the H2O2 treatment, fluorescence microscopy showed that dead CE cells sloughed off the cornea and became free-floating in the corneal cup, that those remained attached to the cornea were mostly viable, and that there were more attached CE cells in VIP-pretreated corneal cups than in control corneal cups (data not shown). Quantification of attached and detached CE cells was by determination of genomic DNA contents. As shown in Fig. 5, VIP, in concentration-dependent manners increased the numbers of attached CE cell while decreased those of free-floating ones. Relative amounts of genomic DNA derived from attached CE cells in corneal cups that have been pretreated with 0 (control), 10−12, 10−10, 10−8, and 10−6 M VIP were (mean±sem; [p value in VIP-pretreated vs control]) 100± 7 %, 119± 11 % (p <0.1), 152±15% (p <0.005), 140±9 % (p<0.002), and 130±16% (p<0.05) in 8, 3, 9, 8, and 7 corneal cups, respectively (Fig. 5), while those of free-floating CE cells were 100± 15%, 53± 22 %( p>0.05), 62±13% (p <0.05), 30± 7 % (p <0.001), and 28±11% (p <0.002), in 8, 3, 8, 9, and 6 corneal cups, respectively (Fig. 5). The difference among various treatment groups was significant in attached (P=0.045, ANOVA) and detached (P=0.001, ANOVA) CE cells. Sixty four h after recovering in the growth medium from injury induced by exposure to 1.4 mM H2O2 (30 min), corneal cups that have been pre-treated with VIP retained more CE cells, which were mostly alive (data not shown). Genomic DNA was extracted from attached CE cells and as shown in Fig. 6, VIP, in a concentration-dependent manner increased the numbers of attached CE cell. Relative amounts of genomic DNA derived from attached CE cells in corneal cups that have been pretreated with 0 (control), 10−10, 10−8, and 10−6 M VIP were (mean±sem; [p value in VIP-pretreated vs control]) 100± 9 %, 144± 26 % (p=0.09), 190±23% (p =0.002), and 166±18 % (p=0.004) in 7, 9, 8, and 7 corneal cups, respectively. The difference among various treatment groups was significant (P=0.045, ANOVA).
Fig. 5.

VIP pretreatment-promoted long-term CE cell survival against the killing effect of severe oxidative stress (17 h, 1.4 mM H2O2). Amounts of genomic DNA derived from attached CE cells (0) increased and those from detached, free-floating CE cells (●) decreased in VIP-dependent manners. Relative amounts of genomic DNA derived from attached CE cells (0) in corneal cups that were pretreated with 0 (control), 10−12, 10−10, 10−8 and 10−6 M VIP, were, in %, (mean±sem [number of corneoscleral explants]) 100± 7 (8), 119± 11 (3), 152±15 (9), 140±9 (8), and 130±16 (7), respectively, and those of free- floating CE cells (●) were 100± 15 (8), 53± 22 (3), 62±13 (8), 30± 7 (9), and 28±11 (6), respectively. VIP-treated vs control significantly differed at p levels of < 0.05 (*), <0.005 (**), <0.002 (***), and <0.001 (****). Difference among treatments with varying VIP concentrations was significant (P<0.05, ANOVA) for both attached and detached CE cells. The amount of CE cell DNA in each corneal cup was normalized against the average of those found in the control corneal cups in each of the three separate experiments and the values were given on Y-axis.
Fig. 6.

VIP pretreatment-promoted long-term survival of CE cells in corneal cups that have been briefly exposed to severe oxidative stress. Sixty four h after recovering in the growth medium in corneal cups from injury induced by exposure to 1.4 mM H2O2 (30 min), CE cells that have remained attached in the corneal cups were quantified by genomic DNA. VIP, in a concentration-dependent manner increased the numbers of attached CE cell. Relative amounts of genomic DNA derived from attached CE cells in corneal cups that have been pretreated with 0 (control), 10−10, 10−8, and 10−6 M VIP were (mean±sem; [p value in VIP-pretreated vs control]) 100± 9 %, 144± 26 % (p=0.09), 190±23% (p =0.002), and 166±18 % (p=0.004) in 7, 9, 8, and 7 corneal cups, respectively. The data were combined from three separate experiments. Difference among treatments with varying VIP concentrations was significant (P=0.045, ANOVA). The amount of CE cell DNA in each corneal cup was normalized against the average value of those found in control corneal cups in each of the three separate experiments and the values were given on Y-axis.
VIP up-regulation of Bcl-2 mRNA levels in CE cells in corneoscleral explants
The expression of Bcl-2 mRNA in CE cells was demonstrated by RT-PCR (Fig. 7A). The expression of Bcl-2 mRNA in CE cells 25 h after they have been pre-treated withVIP (0, 10−12-10−6 M) was examined bysemi-quantitative RT-PCR and the results showed that the Bcl-2 levels increased in a VIP concentration-dependent manner (Fig. 7B & 7C). The normalized (against the internal standard 18S) relative Bcl-2 levels in 0. 10−12, 10−10, 10−8, and 10−6 M VIP-pretreated CE cells in corneoscleral explants were (in mean±sem[p value in VIP-pretreated vs control]) 1.00±0.11, 1.59±0.22 (p = 0.01), 2.50±0.23 (p = 0.001), 3.18±0.72 (p = 0.005), and 1.56±0.28 (p = 0.03) in 6, 3, 3, 5, and 4 corneoscleral explants, respectively. The difference among various treatment groups was significant at the level of p=0.0096.
Fig. 7.

Bcl-2 mRNA (A) and VIP up-regulated Bcl-2 mRNA in CE cells in corneoscleral explants (B and C). Agarose gel (2%) electrophoresis of RT-PCR product of Bcl-2 mRNA isolated from fresh bovine corneas (A) and that of semi-quantitative RT-PCR products of Bcl-2 and 18S (as the internal standard) mRNA isolated from VIP-pre-treated CE cells (B and C). For (A), -RT: PCR negative control (RNA not subjected to RT); H2O: reagent negative control (No RT products, H2O instead); M: DNA size markers; bp: base pair. For B and C: CE cell Bcl-2 mRNA level increased in a VIP-concentration dependent manner. Bcl-2 and 18 S mRNA levels reflected in the electrophoresed (2% agarose) semi-quantitative RT-PCR products (B). The ratio of normalized CE cell Bcl-2 cDNA (against the18S internal standard) levels over that averaged from control CE cells of the same experiment (Y-axis) was shown as a function of VIP concentration (C). Data were combined from three separate experiments. The difference between CE cells in the control (N=6) vs VIP-treated corneal cups was significant at the levels of p =0.01 (10−12 M; N=3), p=0.0001 (10−10 M; N=3), p=0.005 (10−8 M; N=5), and p=0.03 (10−6 M; N=4). The difference among various treatment groups was significant (P = 0.01, ANOVA):
VIP up-regulation of Bcl-2 and N-cadherin mRNA levels in CE cells in H2O2-treated corneoscleral explants
The VIP-up-regulated anti-apoptotic Bcl-2 mRNA levels in CE cells shown above (Fig. 7) may have increased the survivability of CE cells following the subsequent H2O2 treatment. To demonstrate that the pathway leading to VIP up-regulation of Bcl-2 mRNA survived the oxidative stress, VIP-pre-treated and H2O2 treated CE cells were subjected to semi- quantitative RT-PCR. As is shown in Fig. 8, pre-treatment with VIP (0, 10−10-10−6 M) of the CE cells prior to the H2O2 treatment of the corneoscleral explants resulted in increased CE cell Bcl-2 mRNA levels in a VIP concentration-dependent manner (Fig. 8A & 8B). The relative normalized Bcl-2 levels (against the 18S internal standard) in 0 (control), 10−10, 10−8, and 10−6 M VIP-pre-treated CE cells were (in mean±sem [Number of corneoscleral explants]) 1.00±0.13 (N=12), 1.17±0.15 (N=10), 1.47±0.18 (N=10), and 4.46±1.82 (N=9), respectively(Fig. 8B). The levels of VIP-treated vs control were significant at p=0.02 for both 10−8 and 10−6 M VIP-pre-treated CE cells in corneoscleral explants, while the difference among various groups was significant at p=0.018 (ANOVA).
Fig 8.

Bcl-2 and N-cadherin mRNA levels in oxidative stress-injured CE cells increased by VIP pre-treatment in VIP concentration-dependent and protein kinase A inhibitor (H89)-sensitive manners. (A & C) Electrophoresed (2% agarose) semi-quantitative RT-PCR products showing the VIP concentration-dependency of Bcl-2 mRNA (A) and N-cadherin mRNA (C). (B& D) The ratio of normalized CE cell Bcl-2 (B) and N-cadherin (D) cDNA (against the 18S internal standard) levels over that averaged from the control CE cells of the same experiment (Y-axis) was shown as a function of VIP concentration. Data were combined from four separate experiments. (E & F) The protein kinase A inhibitor H89 attenuated the effect of 10−8 M VIP pre-treatment on up-regulation of Bcl-2 (E) and N-cadherin mRNA (F) levels. (G) The relative levels of Bcl-2 and N-cadherin in 10−8 M VIP-treated CE cells decreased by H89. The data were averaged from three experiments. For B: The difference of the control vs VIP-pre-treated was significant at P =0.02 for both 10−8 and 10−6 M-VIP-pre-treated CE cells in corneoscleral explants. The difference among various groups of corneoscleral explants with CE cells pre-treated with 0 (N=12), 10−10 (N=10), 10−8 (N=10), and 10−6 (N=9) M VIP was significant (P = 0.018, ANOVA). For D: The difference of the control vs VIP-pre-treated was significant at P =0.03 for both 10−8 and 10−6 M VIP-pre-treated CE cells in corneoscleral explants. The difference among various groups of corneoscleral explants treated with 0 (N=9), 10−10 (N=8), 10−8 (N=8), and 10−6 (N=8) M VIP was significant (P = 0.049, ANOVA). For G: The difference of the control vs H89-pre-treated was significant at P =0.001 (*), p=0.0001 (**), p<0.0001 (***). The difference in Bcl-2 levels among various groups of corneoscleral explants treated with 0 (N=4), 6.6 (N=2), 9.9 (N=4), and 13.2 (N=3) μM H89 was significant (P <0.0001, ANOVA). The difference in N-Cadherin levels among various groups of corneoscleral explants treated with 0 (N=4), 6.6 (N=2), 9.9 (N=3), and 13.2 (N=4) μM H89 was significant (P =0.06, ANOVA).
Whereas we have previously demonstrated that VIP up-regulates the expression of N-cadherin and that the endogenous VIP knockdown results in a decreased level of N-cadherin in CE cells (Koh et al., 2008), the present study demonstrated that the pathway leading to VIP up-regulation of N-cadherin survived the severe oxidative stress. Pre-treatment with VIP (0, 10−10-10−6 M) ofthe CE cells prior to the H2O2 treatment of the corneoscleral explants resulted in increased CE cell N-cadherin mRNA levels in a VIP concentration-dependent manner (Fig. 8C & 8D). Semi-quantitative RT-PCR of N-cadherin and 18S (internal standard) mRNAs showed that the relative N-cadherin levels in 0 (control), 10−10, 10−8, and 10−6 M VIP-pretreated CE cells in corneoscleral explants were (in mean±sem [N=number of corneoscleral explants]) 1.00±0.09 (N=9), 1.08±0.15 (N=8), 1.45±0.21 (N=8), and 2.29±0.65 (N=8), respectively. The levels of VIP-treated vs control were significant at P=0.03 for both 10−8 and 10−6 M VIP pre-treated CE cells in corneoscleral explants, while the difference among various groups was significant at p=0.049 (ANOVA). VIP up-regulation of Bcl-2 and N-cadherin was protein kinase A-mediated, as the protein kinase A inhibitor H89 concentration-dependently attenuated the effect of 10−8 M VIP (Fig. 8E & 8F).
The phospho-CREB level increased in VIP-pretreated CE cells in corneoscleral explants
Following pretreatment of corneal cups with 0, 10−10, 10−8, 10−6 M VIP, CE cells were analyzed by Western blots for N-cadherin and actin (as internal standard). As shown in Fig. 9, the phospho-CREB level in CE cells increased in a concentration (Fig. A& B) and time (Fig. 9C) manner. The relative phospho-CREB levels in 0 (control), 10−10, 10−8, and 10−6 M VIP-pretreated CE cells in corneoscleral explants were (in mean±sem [N=number of corneoscleral explants; p in control vs VIP-pretreated]) 1.00±0.09 (N=10), 1.43±0.08 (N=2; p=0.03), 1.97±0.18 (N=7; p=0.00004), and 1.46±0.28 (N=6; p=0.039), respectively. The difference among various groups was significant at p=0.006 (ANOVA).
Fig. 9.

VIP concentration-dependency of phospho-CREB levels in CE cells. (A) Western blot analysis, (B & C) The ratio of normalized CE cell phospho-CREB (against the actin internal standard) levels over that averaged from the control CE cells of the same experiment (Y-axis) was shown as a function of VIP concentration (B) and time of VIP-pretreatment (C). For B: The ratios were (mean±sem [p values in control vs VIP-pretreted]): 1±0.085, 1.43±0.08 (p=0.03), 1.97±0.18 9 (p<0.0001), and 1.46±0.28 (p=0.04) in 0, 10−10, 10−8, and 10−6 M VIP-treated CE cells in 10, 2, 7, and 6 corneoscleral explants, respectively. Data were combined from three separate experiments. The difference among various groups of corneoscleral explants was significant (P = 0.006, ANOVA).
The VIP receptor in CE cells
Western blot analysis demonstrated that CE cells isolated from fresh bovine eyes expressed a 64 kD-VPAC1-immunoreactive molecule, while no VPAC2-immunoreactive molecule was detected (Fig. 10).
Fig. 10.
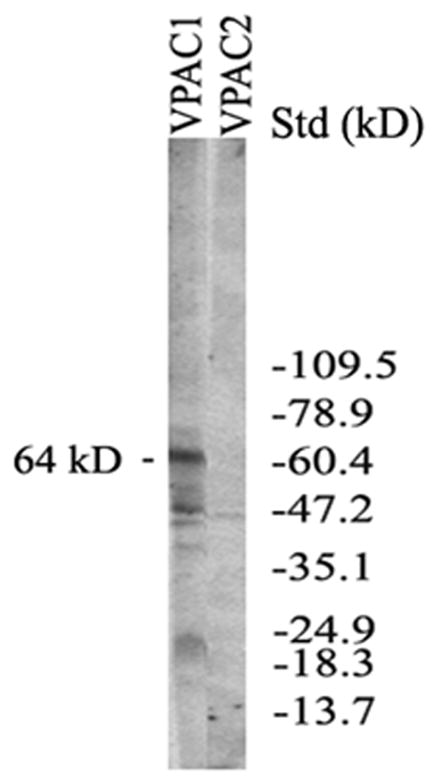
Western blot analysis showing VPAC1 (not VPAC2) expressed in CE cells from fresh bovine eyes. A 64 kDVPAC1 immunoreactive molecule
VIP antagonist (SN)VIPhyb attenuated the effects of VIP on ATP level, promoting apoptosis at the expanse of necrosis, and cell attachment in oxidative stress-injured corneal endothelium
Whereas Fig. 4 demonstrated the VIP antagonist (SN)VIPhyb attenuated the effect of VIP on decreasing the glycogen level, (Fig. 4), (SN)VIPhyb effects on the VIP-induced sustenance of ATP level, necrosis to apoptosis switch, and increased number of attached surviving CE cells in corneas under oxidative stress were examined (Table 2). The results showed that the VIP effects demonstrated throughout the present study were specific effects of VIP/VIP receptor signaling, since (SN)VIPhyb significantly (p= 0.01–0.06) attenuated these VIP effects (Table 2), although (SN)VIPhyb only marginally reversed the VIP effect on the ATP level (p=0.06). Since ATP is known to be released from the CE cells (Grome et al., 2005), it is likely that the release of ATP is also stimulated by VIP, a known secretagogue, and attenuated by (SN)VIPhyb.
Table 2.
Effects of VIP antagonist (SN)VIPhyb on VIP-promoted increase in ATP level, switch from necrosis to apoptosis, and cell attachment in oxidative stress-injured corneal endothelium
| VIP | VIP+VIP Antagonist | P value | |
|---|---|---|---|
| ATP | 1.00±0.04 (N=7) | 0.81±0.11 (N=7) | 0.06 |
| Apoptosis/Necrosis (%/%) | (81±4)/(19±4) (N=3/N=3) | (57±5)/(43±5) (N=3/N=3) | 0.01/0.01 |
| Cell Survival | 1.00±0.08 (N=7) | 0.78±0.05 (N=9) | 0.03 |
ATP level was determined following the same procedure as described in Table 1. Apoptotic cells (showing peripheral chromatin condensation) and necrotic cells (showing spindle-shaped nuclei) as those depicted in Fig. 3A were counted after a 17h-exposure to H2O2 under the condition described in Fig. 3. Cell survival (attachment) was determined after 17h-exposure to H2O2 following the procedure described in Fig. 5. VIP concentration was 10−8 M in all experiments and (SN)VIPhyb was used at 5×10−7 M in experiments for ATP determination and necrosis to apoptosis switch, and at 10−6 M in the experiment for cell attachment.
DISCUSSION
The present study demonstrated for the first time that VIP has the capacity of modulating the death mode, switching necrosis to apoptosis in oxidative stress-injured cells. This was also the first time that a neuropeptide was shown to switch the death mode of injured cell from necrosis to apoptosis.
Apoptotic, but not necrotic, cells can be rescued by neurotrophins, whereas necrosis, not apoptosis, causes inflammation (Introduction). Thus, creating conditions that promote apoptosis at the expense of necrosis among dying cells is therefore of therapeutic value during episodes of massive cell death in stroke, cardiac arrest, and surgery. In the brain, focal ischemia/reperfusion brings cell death via both necrosis and apoptosis. Injured cells in the infarct core and the surrounding penumbra die predominantly by necrosis and apoptosis, respectively (Ueda and Fujita, 2004). Endogenous VIP may restrict the volume of the core by promoting apoptosis at the expense of necrosis and thereby reduce the necrotic cell-induced inflammatory response.
A variety of agents have been shown to switch apoptosis to necrosis, few exerts the opposite effect, i.e. in favor of apoptosis. Glucose (Fujita and Ueda, 2003a), fructose as a substrate of glycolytic ATP generation (Kim et al., 2003), inhibitors of poly (ADP-ribose) polymerase (Virag and Szabo, 2001), whose activity consumes intracellular ATP, and factors in the conditioned medium of cortical neurons (Fujita and Ueda, 2003b) are the only known factors that can switch necrosis to apoptosis among dying cells. Recently, Ueda et al. (2007) reported identification of prothymosin-α1 as a factor present in the conditioned medium of cortical neuron that has the ability to switch necrosis to apoptosis in serum-deprived neuronal cultures. It is also very probable that VIP was one of the factors present in the conditioned medium of cortical neurons since cortical slices have been shown to release VIP in vitro (Lorezo et al., 1989; Martin and Magistretti. 1989).
Although the intracellular ATP level is a determining factor, the precise role of ATP in the choice of apoptosis vs. necrosis is not known. Formation of apoptosome and other downstream events require ATP, which must be maintained at a threshold level for the oxidative stress-injured cells to die of apoptosis (Leist et al., 1997; Kim et al., 2003; Atlante et al. 2005; Miyoshi et al., 2006). ATP production by glycolysis and oxidative phosphorylation and its consumption by the apoptosis machinery likely exist in a constant shifting balance. In the present study, the effect of VIP treatment on upholding the level of ATP was observed at 30 sec after CE cells experienced oxidative stress (Table 1). VIP also increased ATP levels in CE cells without the subsequent oxidative stress (Table 1). The reduction in the glycogen content in VIP-treated CE cells (Fig. 4), in agreement with that observed in earlier studies (Magistretti et al., 1981; Van Valen et l. 1989; Ghazi and Osborne, 1989; Pellerin et al. 1997), may have supported the higher ATP level in VIP-treated CE cells. Thus, ATP level increase in VIP-treated CE cells has made ATP more available, and afforded CE cells to die of apoptosis when injured by severe oxidative stress.
Although neurons do not store glycogen, lactate generated by glycolysis in astrocytes can fuel the energy demand of neurons (Brown et al., 2004; Tekkok, et al., 2005; Magistretti, 2006). A connection between the metabolic effect and the neuroprotective effect of VIP has not been investigated. The present study demonstrated that the metabolic effect of VIP may have afforded the dying cells to choose a non-inflammatory death mode.
Through unknown mechanism, prothymosin-α1 isolated from the cortical neuron-conditioned medium can uphold the ATP level in cultured cortical neurons stressed by serum-deprivation (Ueda et al., 2007; Ueda 2008). Since VIP is a well known secretogogue (Conconi et al., 2006) stimulating polarized protein secretion (Koh, 1991), the possibility exists that the effects of VIP observed in the present study were mediated by trophic factors released by VIP. Activity-dependent neuroprotective protein (ADNP) and activity-dependent neurotrophic factor (ADNF) released by VIP-stimulated astrocytes demonstrate neuroprotective effects in a variety of systems (Brenneman and Goes, 1996; Furman et al., 2004). Whether ADNP and ADNF are expressed by CE cells is not known at the present time, also not known is if they can switch necrosis to apoptosis death mode.
VIP is an anti-inflammatory agent down-regulating the immune-cell-mediated responses (Delgado et al., 2004; Gonzalez-Rey et al., 2007). CE cells form the front boundary of the anterior chamber, an immune-privileged site. While present in the anterior chamber (Koh et al., 2005; Niederkorn, 2006), VIP-immunoreactivity is believed to contribute to the immune-privilege of the anterior chamber (Niederkorn, 2006). Since CE cells dying of necrosis have the potential of inducing an inflammatory response in the anterior chamber and those of apoptosis do not, the present study thus demonstrated VIP down-regulating the inflammatory potential of a resident (CE) cell of an immune-privileged site. CE cells express VIP mRNA and protein (Koh and Wascheck, 2000). Thus, through VIP synthesis and release, CE cells play an active role in down-regulating their own inflammatory potential in addition to that of immune cells in the anterior chamber. It is therefore not surprising that among the three layers of the cornea, the corneal epithelium, the stroma, and the corneal endothelium, only the corneal endothelium enjoys the immune-privilege (Hori, et. al., 2007).
Although the mechanism remains to be elucidated, Bcl-2 up-regulation has been shown to protect cardiomyocytes and cardiac allograft from ischemia/reperfusion injury (Grunenfelder, 2001; Champattanachai et al., 2008). The present study demonstrated that the effect of VIP on up-regulating Bcl-2 expression in CE cells (Fig. 7) and in CE cells that have sustained the oxidative stress injury (Fig. 8), indicating Bcl-2 played a role in mediating the VIP effect. Furthermore, a recent report (Koh et al., 2008) has demonstrated the effect of VIP on up-regulating N-cadherin expression in CE cells and the present study demonstrated the this function of VIP remained in CE cells injured by oxidative stress (Fig. 8). As an integral component of the cell-cell junctional complex, N-cadherin not only mediates intercellular adhesion strengthening but also functions as an adhesion-activated receptor capable of initiating distinctive signaling pathways leading to cell differentiation and survival (reviewed by Derycke and Bracke, 2004).
Trophic factors have been shown to rescue cells that are dying of apoptosis but not, necrosis (Fujita and Ueda, 2003a; Kim et al., 2004; Ueda et al., 2007; Ueda,2008). At the present time whether VIP pretreatment, resulting in up-regulation of Bcl-2 and N-cadherin in severe oxidative stress-injured CE cells, rescued those apoptotic cells by interrupting the progression of the death program is not known. The results of VIP-promoted long-term (64 h) survival (Fig. 6) suggested such a possibility.
In the CE cells, only the VPAC1 but not VPAC2 receptor was expressed (Figure 10). A 64 kD VPAC1-immunoreactive molecule was detected in CE cell extract in the present study, whereas a 63-kD VIP receptor, which after N-glycanase digestion generated a 50 kD core protein, was identified in the ciliary process of the eye (Horio et al., 1995). Like the corneal endothelium, the ciliary process borders the anterior chamber, an immune-privileged site. Both VPAC1 and VPAC2 mediate the VIP effect of neuroprotection against the excito-toxicity (Favrais, 2007; Brenneman, 2007).
Although we have hypothesized that VIP- promoted necrosis to apoptosis switch is a prerequisite for the anti-apoptotic effects of VIP (in up-regulation of Bcl-2 and N-cadherin) to have beneficial effects on CE cell survival and presented sequence of events in the present study in support of this hypothesis, the anti-apoptotic effect of VIP has been reported previously in lung injury (Said and Dickman, 2000) and in hippocampal stem cells (Antonawich and Said,, 2002). In particular, a dual function of VIP as anti-apoptotic and anti-inflammatory in the lung injury model has been hypothesized (Said and Dickman, 2000).
In conclusion, the present study demonstrated the role of VIP in promoting survival of CE cells ex vivo against the killing effect of H2O2 and in the meantime down-regulating the inflammatory potential of the dying CE cells by switching the inflammatory-causing necrotic death mode to an apoptotic one. VIP-promoted necrosis to apoptosis switch played a key role in promoting the long-term survival of CE cells, since the pathway leading to VIP-up-regulated Bcl-2 and N-cadherin expression survived the severe oxidative stress-induced injury. Thus, it is the first time that VIP was demonstrated to be both protective and anti-inflammatory.
Acknowledgments
The present study was supported in part by National Institutes of Health Grant RO1EY-11607, Pangborn Award from the University of Maryland, Baltimore, and Research to Prevent Blindness, Inc., New York, NY.
References
- Antonawich FJ, Said SI. Vasoactive intestinal peptide attenuates cytochrome c translocation, and apoptosis, in rat hippocampal stem cells. Neurosci Lett. 2002;325:151–4. doi: 10.1016/s0304-3940(02)00254-9. [DOI] [PubMed] [Google Scholar]
- Atlante A, Giannattasio S, Bobba A, Gagliardi S, Petragallo V, Calissano P, Marra E, Passarella S. An increase in the ATP levels occurs in cerebellar granule cells en route to apoptosis in which ATP derives from both oxidative phosphorylation and anaerobic glycolysis. Biochim Biophys Acta. 2005;1708:50–62. doi: 10.1016/j.bbabio.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Brenneman DE, Gozes I. A femtomolar-acting neuroprotective peptide. J Clin Invest. 1996;97:2299–307. doi: 10.1172/JCI118672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE. Neuroprotection: A comparative view of vasoactive intestinal peptide and ituitary adenylate cyclase-activating poypeptide. Peptides. 2007:281720–1726. doi: 10.1016/j.peptides.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Brown AM, Tekkok BS, Ransom BR. Energy transfer from astrocytes to axons: the role of CNS glycogen. Neurochem Int. 2004;45:529–536. doi: 10.1016/j.neuint.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. Am J Physiol Cell Physiol. 2008;294:C1509–20. doi: 10.1152/ajpcell.00456.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- Cocco RE, Ucker DS. Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol Biol Cell. 2001;12:919–930. doi: 10.1091/mbc.12.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conconi MT, Spinazzi R, Nussdorfer GG. Endogenous ligands of PACAP/VIP receptors in the autocrine-paracrine regulation of the adrenal gland. Int Rev Cytol. 2006;249:1–51. doi: 10.1016/S0074-7696(06)49001-X. [DOI] [PubMed] [Google Scholar]
- Cursiefen C. Immune privilege and angiogenic privilege of the cornea. Chem Immunol Allergy. 2007;92:50–7. doi: 10.1159/000099253. [DOI] [PubMed] [Google Scholar]
- Cvelanovic M, Ucker DS. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J Immunol. 2004;172:880–889. doi: 10.4049/jimmunol.172.2.880. [DOI] [PubMed] [Google Scholar]
- Delgado M, Pozo D, Ganea D. The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev. 2004;56:249–290. doi: 10.1124/pr.56.2.7. [DOI] [PubMed] [Google Scholar]
- Delgado M, Varela N, Gonzalez-Rey E. Vasoactive intestinal peptide protects against beta-amyloid-induced neurodegeneration by inhibiting microglia activation at multiple levels. Glia. 2008;56:1091–103. doi: 10.1002/glia.20681. [DOI] [PubMed] [Google Scholar]
- Derycke LDM, Bracke ME. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signaling. Int J Dev Biol. 2004;48:463–476. doi: 10.1387/ijdb.041793ld. [DOI] [PubMed] [Google Scholar]
- Erwig LPand Henson PM. Immunological consequences of apoptotic cell phagocytosis. Am J Pathol. 2007;17:2–8. doi: 10.2353/ajpath.2007.070135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2008;15:243–50. doi: 10.1038/sj.cdd.4402184. [DOI] [PubMed] [Google Scholar]
- Favrais G, Couvineau A, Laburthe M, Gressens P, Lelievre V. Involvement of VIP and PACAP in neonatal brain lesions generated by a combined excitotoxic/inflammatory challenge. Peptides. 2007;8:1727–1737. doi: 10.1016/j.peptides.2007.06.022. [DOI] [PubMed] [Google Scholar]
- Fernández M, Sanchez-Franco F, Palacios N, Sánchez I, Cacicedo L. IGF-I and vasoactive intestinal peptide (VIP) regulate cAMP-response element-binding protein (CREB)-dependent transcription via the mitogen-activated protein kinase (MAPK) pathway in pituitary cells: requirement of Rap1. J Mol Endocrinol. 2005;34:699–712. doi: 10.1677/jme.1.01703. [DOI] [PubMed] [Google Scholar]
- Fujita R, Ueda H. Protein kinase C-mediated cell death mode switch induced by high glucose. Cell Death Differ. 2003a;10:1336–1347. doi: 10.1038/sj.cdd.4401300. [DOI] [PubMed] [Google Scholar]
- Fujita R, Ueda H. Protein kinase C-mediated necrosis-apoptosis switch of cortical neurons by conditioned medium factors secreted under the serum-free stress. Cell Death Differ. 2003b;10:782–790. doi: 10.1038/sj.cdd.4401239. [DOI] [PubMed] [Google Scholar]
- Furman S, Steingart RA, Mandel S, Hauser JM, Brenneman DE, Gozes I. Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron Glia Biol. 2004;1:193–199. doi: 10.1017/S1740925X05000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazi H, Osborne NN. Agonist-induced glycogenolysis in rabbit retinal slices and cultures. Br J Pharmacol. 1989;96:895–905. doi: 10.1111/j.1476-5381.1989.tb11900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rey E, Chorny A, Delgado M. Regulation of immune tolerance by anti-inflammatory neuropeptides. Nat Rev Immunol. 2007;7:52–63. doi: 10.1038/nri1984. [DOI] [PubMed] [Google Scholar]
- Gomes P, Srinivas SP, Van Driessche W, Vereecke J, Himpens B. ATP release through connexin hemichannels in corneal endothelial cells. Invest ophthalmol Vis Sci. 2005;46:1208–18. doi: 10.1167/iovs.04-1181. [DOI] [PubMed] [Google Scholar]
- Grunenfelder J, Miniati DN, Murata S, Falk V, Hoyt EG, Kown M, Koransky ML, Robbins RC. Upregulation of Bcl-2 through caspase-3 inhibition ameliorates ischemia/reperfusion injury in rat cardiac allografts. Circulation. 2001;104:I202–I206. doi: 10.1161/hc37t1.094833. [DOI] [PubMed] [Google Scholar]
- Gutiérrez-Cañas I, Rodríguez-Henche N, Bolaños O, Carmena MJ, Prieto JC, Juarranz MG. VIP and PACAP are autocrine factors that protect the androgen-independent prostate cancer cell line PC-3 from apoptosis induced by serum withdrawal. Br J Pharmacol. 2003;139:1050–1058. doi: 10.1038/sj.bjp.0705317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Sueishi K, Tanaka K, et al. Immunohistochemical evidence of the origin of human corneal endothelial cells and keratocytes. Graefes Arch Clin Exp Ophthalmo. 1986;224:452–456. doi: 10.1007/BF02173362. [DOI] [PubMed] [Google Scholar]
- Hokari R, Lee H, Crawley SC, Yang SC, Gum JR, Jr, Miura S, Kim YS. Vasoactive intestinal peptide upregulates MUC2 intestinal mucin via CREB/ATF1. Am J Physiol Gastrointest Liver Physiol. 2005;289:G949–959. doi: 10.1152/ajpgi.00142.2005. [DOI] [PubMed] [Google Scholar]
- Hori J, Joyce NC, Streilein JW. Immune privilege and immunogenicity reside among different layers of the mouse cornea. Ocul Immunol Inflamm. 2007;15:225–39. doi: 10.1080/09273940701382374. [DOI] [PubMed] [Google Scholar]
- Horio B, Law NM, Rosenzweig SA. Characterization of vasoactive intestinal peptide receptors in rabbit ciliary processes. Invest Ophthalmol Vis Sci. 1995;36:192–199. [PubMed] [Google Scholar]
- Johnston MC, Noden DM, Hazelton RD, Coulombre JL, Coulombre AJ. Origins of avian ocular and periocular tissues. Exp Eye Res. 1979;29:27–43. doi: 10.1016/0014-4835(79)90164-7. [DOI] [PubMed] [Google Scholar]
- Joselin AP, Schulze-Osthoff K, Schwerk C. Loss of Acinus inhibits oligonucleosomal DNA fragmentation but not chromatin condensation during apoptosis. J Biol Chem. 2006;281:12475–12484. doi: 10.1074/jbc.M509859200. [DOI] [PubMed] [Google Scholar]
- Joyce NC. Cell cycle status in human corneal endothelium. Exp Eye Res. 2005;81:629– 638. doi: 10.1016/j.exer.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology. 2003;124:494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- Kim DH, Zhao X, Tu CH, Casaccia-Bonnefil P, Chao MV. Prevention of apoptotic but not necrotic cell death following neuronal injury by neurotrophins signaling through the tyrosine kinase receptor. J Neurosurg. 2004;100:79–87. doi: 10.3171/jns.2004.100.1.0079. [DOI] [PubMed] [Google Scholar]
- Koh SW. VIP stimulation of polarized macromolecule secretion in cultured chick embryonic retinal pigment epithelium. Exp Cell Res. 1991;197:1–7. doi: 10.1016/0014-4827(91)90472-7. [DOI] [PubMed] [Google Scholar]
- Koh SW, Waschek JA. Corneal endothelial cell survival in organ cultures under acute oxidative stress: effect of VIP. Invest Opthamol Vis Sci. 2000;41:4085–4092. [PubMed] [Google Scholar]
- Koh SW. Ciliary neurotrophic factor released by corneal endothelium surviving oxidative stress ex vivo. Invest Ophthalmol Vis Sci. 2002;43:2887–2896. [PubMed] [Google Scholar]
- Koh SW, Yue BY. VIP stimulation of cAMP production in corneal endothelial cells in tissue and organ cultures. Cornea. 2002;21(3):270–274. doi: 10.1097/00003226-200204000-00007. [DOI] [PubMed] [Google Scholar]
- Koh SW, Rutzen A, Coll T, Hemady R, Higginbotham E. VIP immunoreactivity in human aqueous humor. Curr Eye Res. 2005;30:189–194. doi: 10.1080/02713680490908715. [DOI] [PubMed] [Google Scholar]
- Koh SW, Guo Y, Bernstein SL, Waschek JA, Liu X, Symes AJ. Vasoactive intestinal peptide induction by ciliary neurotrophic factor in donor human corneal endothelium in situ. Neurosci Lett. 2007;423:89–94. doi: 10.1016/j.neulet.2007.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SW, Chandrasekara K, Abbondandolo CJ, Coll TJ, Rutzen AR. VIP and VIP gene silencing modulation of differentiation marker N-cadherin and cell shape of corneal endothelium in human corneas ex vivo. Invest Ophthalmol Vis Sci. 2008;49:3491–3498. doi: 10.1167/iovs.07-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer I, Robberecht P. Molecular mechanisms involved in vasoactive intestinal peptide receptor activation and regulation:current knowledge, similarities to and differences from the A family of G-protein-coupled receptors. Biochem Soc Trans. 2007;35:724–728. doi: 10.1042/BST0350724. [DOI] [PubMed] [Google Scholar]
- Lee HS, Tomarev SISI. Optimedin induces expression of N-cadherin and stimulates aggregation of NGF-stimulated PC12 cells. Exp Cell Res. 2007;313:98–108. doi: 10.1016/j.yexcr.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeLievre CS, LeDouarin NM. Mesenchymal derivatives of the neural crest: Analysis of chimaeric quail and chick embryos. J Embryo Exp Morphol. 1975;34:125–154. [PubMed] [Google Scholar]
- Li M, Carpio DF, Zheng Y, Bruzzo P, Singh V, Ouaaz F, Medzhitov RM, Beg AA. An essential role of the NF-kappa B/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J Immunol. 2001;166:7128–7135. doi: 10.4049/jimmunol.166.12.7128. [DOI] [PubMed] [Google Scholar]
- Lorenzo MJ, Sanchez-Franco F, de los Frailes MT, Reichlin S, Fernandez G, Cacicedo L. Synthesis and secretion of vasoactive intestinal peptide by rat fetal cerebral cortical and hypothalamic cells in culture. Endocrinology. 1989;125:1983–1990. doi: 10.1210/endo-125-4-1983. [DOI] [PubMed] [Google Scholar]
- Mabuchi T, Kitagawa K, Kuwabara K, Takasawa K, Ohtsuki T, Xia Zhengui, Storm D, Yanagihara T, Hori Masatsugu H, Matsumoto M. Phosphorylation of cAMP responsive element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vitro. J Neurosci. 2001;21:9204–9213. doi: 10.1523/JNEUROSCI.21-23-09204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Morrison JH, Shoemaker WJ, Sapin V, Blooom FE. Vasoactive intestinal polypeptide induces glycogenolysis in mouse cortical slices: a possible regulatory mechanism for the local control of energy metabolism. Proc Natl Acad Sci U S A. 1981;78:6535–65399. doi: 10.1073/pnas.78.10.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006;209:2304–2311. doi: 10.1242/jeb.02208. [DOI] [PubMed] [Google Scholar]
- Malinin GI, Bernstein H. Histochemical demonstration of glycogen in corneal endothelium. Exp Eye Res. 1979;28:381–385. doi: 10.1016/0014-4835(79)90113-1. [DOI] [PubMed] [Google Scholar]
- Martin JL, Magistretti PJ. Release of vasoactive intestinal peptide in mouse cerebral cortex: evidence for a role of arachidonic acid metabolites. J Neurosci. 1989;9:2536–2542. doi: 10.1523/JNEUROSCI.09-07-02536.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrhof FB, Muller FU, Bergmann MW, Li P, Wang Y, Schmitz W, Dietz R, von Harsdorf R. In cardiomyocyte hypoxia, insulin-like growth factor-I-induced antiapoptotic signaling requires phosphatidylinositol-3-OH-kinase-dependent and mitogen-activated protein kinase-dependent activation of the transcription factor cAMP response element-binding protein. Circulation. 2001;23:2088–2094. doi: 10.1161/hc4201.097133. [DOI] [PubMed] [Google Scholar]
- Miyoshi N, Oubrahim H, Chock PB, Stadtman ER. Age-dependent cell death and the role of ATP in hydrogen peroxide-induced apoptosis and necrosis. Proc Natl Acad Sci U S A. 2006;103:1727–1731. doi: 10.1073/pnas.0510346103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody TW, Leyton J, Chan D, Brenneman DC, Fridkin M, Gelber E, Levy A, Gozes I. VIP receptor antagonists and chemotherapeutic drugs inhibit growth of breast cancer cells. Breast Cancer Res Treat. 2001;68:55–64. doi: 10.1023/a:1017994722130. [DOI] [PubMed] [Google Scholar]
- Moody TW, Hill JM, Jensen RT. VIP as a trophic factor in the CNS and cancer cells. Peptides. 2003;24:163–177. doi: 10.1016/s0196-9781(02)00290-5. [DOI] [PubMed] [Google Scholar]
- Niederkorn JY. The immune privilege of corneal grafts. J Leukoc Biol. 2003;74:167–171. doi: 10.1189/jlb.1102543. [DOI] [PubMed] [Google Scholar]
- Niederkorn JY. See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol. 2006;7:354–359. doi: 10.1038/ni1328. [DOI] [PubMed] [Google Scholar]
- Noden DM. The control of avian cephalic neural crest cytodifferentiation: I. skeletal and connective tissues. Dev Biol. 1978;67:296–312. doi: 10.1016/0012-1606(78)90201-4. [DOI] [PubMed] [Google Scholar]
- Onoue S, Ohmori Y, Endo K, Yamada S, Kimura R, Yajima T. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide attenuate the cigarette smoke extract-induced apoptotic death of rat alveolar L2 cells. Eur J Biochem. 2004;271:1757–1767. doi: 10.1111/j.1432-1033.2004.04086.x. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Stolz M, Sorg O, Martin JL, Deschepper CF, Magistretti PJ. Regulation of energy metabolism by neurotransmitters in astrocytes in primary culture and in an immortalized cell line. Glia. 1997;21:74–83. doi: 10.1002/(sici)1098-1136(199709)21:1<74::aid-glia8>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Pocar P, Nestler D, Risch M, Fischer B. Apoptosis in bovine cumulus-oocyte complexes after exposure to polychlorinated biphenyl mixtures during in vitro maturation. Reproduction. 2005;130:857–868. doi: 10.1530/rep.1.00761. [DOI] [PubMed] [Google Scholar]
- Prothmann C, Wellard J, Berger J, Hamprecht B, Verleysdonk S. Primary cultures as a model for studying ependymal functions: glycogen metabolism in ependymal cells. Brain Res. 2001;920:74–83. doi: 10.1016/s0006-8993(01)03021-9. [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Miller E, Sable C, Young P, Heidenreich KA, Boxer LM, Reusch JE. Insulin-like growth factor-I induces Bcl-2 promotor through the transcription factor cAMP-responsive element-binding protein. J Biol Chem. 1999;274:27529–27535. doi: 10.1074/jbc.274.39.27529. [DOI] [PubMed] [Google Scholar]
- Rangon CM, Goursaud S, Medja F, Lelievre V, Mounien L, Husson I, Brabet P, Jegou S, Janet T, Gressens P. VPAC2 receptors mediate vasoactive intestinal peptide-induced neuroprotection against neonatal excitotoxic brain lesions in mice. J Pharmacol Exp Ther. 2005;314:745–752. doi: 10.1124/jpet.105.086405. [DOI] [PubMed] [Google Scholar]
- Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008;3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin M, Saxena A, Joost P, Lewerenz J, Methner A. Induction of Bcl-2 by functional regulation of G-protein coupled receptors protects from oxidative glutamate toxicity by increasing glutathione. Free Radical Research. 2006;40:1113–1123. doi: 10.1080/10715760600838191. [DOI] [PubMed] [Google Scholar]
- Said SI, Dickman KG. Pathways of inflammation and cell death in the lung: modulation by vasoactive intestinal peptide. Regul pept. 2000;93(1–3):21–9. doi: 10.1016/s0167-0115(00)00174-9. [DOI] [PubMed] [Google Scholar]
- Sastry KSR, Smith AJ, Karpova Y, Datta SR, Kulik G. Diverse antiapoptotic signaling pathways activated by vasoactive intestinal polypeptide, epidermal growth factor, and phosphatidylinositol 3-kinase in prostate cancer cells converge on BAD. J Biol Chem. 2006;281:20891–20901. doi: 10.1074/jbc.M602928200. [DOI] [PubMed] [Google Scholar]
- Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prevost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkok SB, Brown AM, Westenbroek R, Pellerin L, Ransom BR. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J Neurosci Res. 2005;81:644–652. doi: 10.1002/jnr.20573. [DOI] [PubMed] [Google Scholar]
- Ueda H, Fujita R. Cell death mode switch from necrosis to apoptosis in brain. Biol Pharm Bull. 2004;27:950–955. doi: 10.1248/bpb.27.950. [DOI] [PubMed] [Google Scholar]
- Ueda H, Fujita R, Yoshida A, Matsunaga H, Ueda M. Identification of prothymosin-α1, the necrosis-apoptosis switch molecule in cortical neuronal cultures. J Cell Biol. 2007;176:853–862. doi: 10.1083/jcb.200608022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H. Prothymosin alpha plays a key role in cell death mode-switch, a new concept for neuroprotective mechanisms in stroke. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:315–323. doi: 10.1007/s00210-007-0254-7. [DOI] [PubMed] [Google Scholar]
- Van Valen F, Jorgen’s H, Winkelmann W, Keck E. Vasoactive intestinal peptide receptor regulation of cAMP accumulation and glycogen hydrolysis in the human Ewing’s sarcoma cell line WE-68. Glia. 1989;1:435–446. doi: 10.1016/0898-6568(89)90029-6. [DOI] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Virag L, Szabo C. Purines inhibit poly(ADP-ribose) polymerase activation and modulate oxidant-induced cell death. FASEB J. 2001;15:99–107. doi: 10.1096/fj.00-0299com. [DOI] [PubMed] [Google Scholar]
- Wilson BE, Mochon E, Boxer LM. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol. 1996;16:5546–5556. doi: 10.1128/mcb.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, Wang J, Boxer LM. Role of the cyclic AMP response element in the bcl-2 promoter in the regulation of endogenous Bcl-2 expression and apoptosis in murine B cells. Mol Cell Biol. 2006;26:8599–8606. doi: 10.1128/MCB.01062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang NC, Ho WM, Chen YH, Hum ML. A convenient one-step extraction of cellular ATP using boiling water for the luciferin-luciferase assay of ATP. Anal Biochem. 2002;306:323–327. doi: 10.1006/abio.2002.5698. [DOI] [PubMed] [Google Scholar]