

Abstract
The Salmonella and related bacteria modify the structure of the lipid A portion of their lipopolysaccharide in response to environmental stimuli. Some lipid A modifications are required for virulence and resistance to cationic antimicrobial peptides. We now demonstrate that membranes of Salmonella typhimurium contain a novel hydrolase that removes the 3′-acyloxyacyl residue of lipid A in the presence of 5 mm Ca2+. We have identified the gene encoding the S. typhimurium lipid A 3′-O-deacylase, designated lpxR, by screening an ordered S. typhimurium genomic DNA library, harbored in Escherichia coli K-12, for expression of Ca2+-dependent 3′-O-deacylase activity in membranes. LpxR is synthesized with an N-terminal type I signal peptide and is localized to the outer membrane. Mass spectrometry was used to confirm the position of lipid A deacylation in vitro and the release of the intact 3′-acyloxyacyl group. Heterologous expression of lpxR in the E. coli K-12 W3110, which lacks lpxR, resulted in production of significant amounts of 3′-O-deacylated lipid A in growing cultures. Orthologues of LpxR are present in the genomes of E. coli 0157:H7, Yersinia enterocolitica, Helicobacter pylori, and Vibrio cholerae. The function of LpxR is unknown, but it could play a role in pathogenesis because it might modulate the cytokine response of an infected animal.
Salmonella typhimurium and related organisms are enteric Gram-negative bacteria. S. typhimurium cause gastroenteritis in human hosts but in mice can produce a fatal, typhoid-like sepsis (1, 2). These bacteria invade the epithelial cells and M cells of Peyer patches and then pass into the lymphatic system by colonizing phagocytic cells. Subsequently, the bacteria survive and multiply within modified vacuoles of macrophages that can ultimately produce macrophage apoptosis (3–6).
Lipopolysaccharide (LPS)3 is the principal component of the outer leaflet of the outer membrane of Gram-negative bacteria. Recognition of LPS by the mammalian innate immune system results in the production of cell adhesion proteins in endothelial cells and of pro-inflammatory molecules such as tumor necrosis factor-α and interleukin-1β in monocytes (7, 8). Lipid A, the hydrophobic anchor of LPS, produces most of these responses (9, 10) after its detection by Toll-like receptor 4 (TLR-4) (11–13). Lipid A of S. typhimurium and Escherichia coli is a β1′-6-linked disaccharide of glucosamine, phosphorylated at the 1 and 4′ positions and acylated at the 2, 3, 2′, and 3′ positions with R-3-hydroxymyristate (Fig. 1A) (9, 14, 15). The hydroxyl groups of the R-3-hydroxymyristate residues, attached at positions 2′ and 3′, are further acylated with laurate and myristate, respectively. Key features of the molecule shown to be important for TLR-4 activation include the phosphate groups and the secondary acyl chains (15–17). The biosynthetic enzymes that generate lipid A have been identified and their corresponding structural genes cloned, primarily from E. coli (15).
FIGURE 1. Comparison of S. typhimurium Kdo2-lipid A and its modified forms.

A, nine enzymes are required to produce the constitutive Kdo2-lipid A molecule (15). The numbering scheme is used for the NMR analysis. B, the phosphate groups and the acyl chains of S. typhimurium Kdo2-lipid A can be derivatized in a regulated fashion. The phosphate moieties of lipid A can be substituted with 4-amino-4-deoxy-L-arabinose by L-4-aminoarabinose transferase (ArnT) (18) and/or phosphoethanolamine by phosphoethanolamine transferase (EptA) (PmrC) (19, 20). Formation of the 2-hydroxymyristate group requires an oxygen-dependent hydroxylase, designated LpxO (25). The incorporation of the acyloxyacyl-linked palmitate chain at the 2-position is catalyzed by PagP (26); removal of the ester-linked R-3-hydroxymyristoyl chain at the 3-position is catalyzed by PagL (27).
In response to certain environmental conditions, S. typhimurium synthesizes additional enzymes that covalently alter lipid A (Fig. 1B). The inner membrane enzymes l-4-aminoarabinose transferase (ArnT) (18) and phosphoethanolamine transferase (EptA) (PmrC) (19, 20) can modify the phosphate moieties of lipid A with 4-amino-4-deoxy-l-arabinose or phosphoethanolamine groups, respectively. The addition of 4-amino-4-deoxy-l-arabinose and phosphoethanolamine groups to lipid A has been correlated with resistance to cationic antimicrobial peptides such as polymyxin (21–24). In addition, the inner membrane of S. typhimurium contains an Fe2+/α-ketoglutarate/O2-dependent enzyme, designated LpxO, that hydroxylates position 2 of the 3′ secondary myristate chain (25). Two outer membrane enzymes, PagP (26) and PagL (27), remodel lipid A. PagP catalyzes the addition of a phospholipid-derived palmitate chain to the hydroxyl of the R-3-hydroxymyristate chain at the 2 position of lipid A. PagL catalyzes the removal of the R-3-hydroxymyristate chain at position 3 of lipid A.
We now report a novel, Ca2+-dependent 3′-O-deacylase present in the membranes of S. typhimurium (Fig. 2). The structural gene (lpxR) encoding the 3′-O-deacylase was identified and expressed in E. coli K-12. LpxR, like PagP and PagL, is localized to the outer membrane. Expression of LpxR in the E. coli K-12 strain W3110 results in a significant production of 3′-O-deacylated lipid A species but does not affect growth. Orthologues of S. typhimurium LpxR are found in the genomes of several other Salmonella species, including both typhi and paratyphi, as well as several other Gram-negative pathogens. The function of LpxR is not known; however, partial 3′-O-deacylation of lipid A might be advantageous during infection, allowing S. typhimurium to evade the innate immune response by remodeling its lipid A.
FIGURE 2. LpxR catalyzed modification of Kdo2-lipid A.

LpxR, a Ca2+-requiring outer membrane enzyme, hydrolyzes the ester linkage at the 3′ position of Kdo2-lipid A and liberates the intact 3′-acyloxyacyl group.
Experimental Procedures
Materials
32Pi and [γ-32P]ATP were obtained from PerkinElmer Life Sciences. Silica gel 60 (0.25 mm) thin layer chromatography (TLC) plates were from Merck. Chloroform, ammonium acetate, and sodium acetate were obtained from EM Science. Tryptone and yeast extract were from Difco. The bicinchoninic acid protein determination kit and Triton X-100 were from Pierce. All other chemicals were reagent grade and were purchased from either Sigma or Mallinckrodt.
Bacterial Strains
The bacterial strains used in this study are described in Table 1. Typically, bacteria were grown at 37 °C in LB medium (28). When required for selection of plasmids, cells were grown in the presence of 100 μg/ml ampicillin, 12 μg/ml tetracycline, 25 μg/ml chloramphenicol, or 30 μg/ml kanamycin.
TABLE 1. Relevant bacterial strains and plasmids.
| Strain/Plasmid | Description | Source or reference |
|---|---|---|
| E. coli strains | ||
| XL1 Blue-MR | mcrABC recA1 endA1 gyrA96 relA1 supE44 thi-1 lac | Stratagene |
| W3110 | Wild type, F−, λ− | E. coli Genetic Stock Center (Yale) |
| NovaBlue(DE3) | K-12 strain, DE3 lysogen | Novagen |
| WBB06 | W3110 mtl, Δ(waaC-waaF)::tet6, heptose-deficient | 41 |
| MKV15 | W3110 lpxL::kan, lpxM::Ωcam, lpxP::kan | 37 |
| S. typhimurium strain | ||
| LT2 | Virulent wild type | Salmonella Genetic Stock Center, University of Calgary, Canada |
| Plasmids | ||
| PET23a | Expression vector, T7 promoter, ampr | Novagen |
| pWSK29 | Low copy expression vector, lac promoter, ampr | 33 |
| pLpxR1 | pET23a expressing lpxR (STM1328) | This work |
| pLpxR2 | pWSK29 expressing lpxR (STM1328) | This work |
Molecular Biology Applications
Protocols for handling of DNA samples were those of Sambrook and Russell (29). Transformation-competent cells of E. coli were prepared by the method of Inoue et al. (30). When required, E. coli cells were prepared for electroporation by the method of Sambrook and Russell (29). Plasmids were isolated using the Qiagen Spin Prep kit. DNA fragments were isolated from agarose gels using the QIAquick gel extraction kit. Genomic DNA was isolated using the protocol for bacterial cultures in the Easy-DNA™ kit (Invitrogen). T4 DNA ligase (Invitrogen), restriction endonucleases (New England Biolabs), and shrimp alkaline phosphatase (U. S. Biochemical Corp.) were used according to the manufacturers’ instructions. Double-stranded DNA sequencing was performed with an ABI Prism 377 instrument at the Duke University DNA Analysis Facility. Primers were purchased from MWG-Biotech.
Screening of a S. typhimurium Genomic DNA Library for the 3′-O-Deacylase
A bacterial artificial chromosome (BAC) library generated from genomic DNA of the S. typhimurium strain LT2 and harbored in an E. coli K-12 background was obtained from the Salmonella Genetic Stock Center (University of Calgary). Each of the 71 BAC clones harbors the pBeloBAC11 vector (31) with inserts of S. typhimurium genomic DNA. The BAC clones were grown in 5 ml of LB medium with 25 μg/ml chloramphenicol to an A600 ∼1.0. The membrane fraction of each BAC clone was prepared as previously described (32) and assayed for 3′-O-deacylase activity. Protein concentrations were determined by the bicinchoninic acid method using bovine serum albumin as the standard.
Construction of lpxR Expression Vectors
S. typhimurium lpxR, formerly STM1328, was cloned into pET23a (Novagen) behind the T7 promoter. The predicted coding region for lpxR was amplified by PCR from S. typhimurium strain LT2 genomic DNA using Pfu DNA polymerase (Stratagene), according to the manufacturer's instructions. The forward primer contained a clamp region, an NdeI site (underlined), and the lpxR-coding region with its start codon (bold). The reverse primer contained a clamp region, a BamHI site (underlined) and the coding region with its stop codon (bold). Sequences of the forward and reverse primers were 5′-GCGCGCCATATGAACAAATACAGCTATTGCGCAACG-3′ and 5′-GCGCGCGGATCCTCAGAAGAAGAAGGTGATGTCTCC-3′, respectively. The PCR product and the vector were both digested with NdeI and BamHI, ligated together, and transformed into XL-1 Blue cells (Stratagene) for propagation of the plasmid, designated pLpxR1. For some experiments, pLpxR1 was used directly for LpxR expression in E. coli NovaBlue(DE3) (Table 1). Alternatively, the lpxR gene was transferred from pET23a to pWSK29 (33), a lac-inducible, low-copy expression vector. The XbaI/BamHI-digested fragment, consisting of the lpxR gene as well as the pET23a-derived ribosome binding site, was ligated to the corresponding restriction sites of pWSK29. This plasmid, designated pLpxR2, was then transformed into competent E. coli W3110 or S. typhimurium LT2.
Heterologous Expression of Salmonella LpxR
E. coli NovaBlue(DE3) harboring either the pET23a vector or pLpxR1 were grown at 37 °C until the A600 reached 0.4. Isopropyl-1-thio-β-d-galactopyranoside at 1 mm was then added, and the cultures were shifted to 25 °C for 4 h more. Washed membranes were prepared as previously described (32) and stored in 50 mm MES, pH 6.5, at −80 °C. Cultures of W3110 harboring either pWSK29 or pLpxR2 were grown at 37 °C with 1 mm isopropyl-1-thio-β-d-galactopyranoside present throughout the growth. Cells were harvested when the A600 of the cultures reached 1.0, and washed membranes were prepared as previously described (32). Membranes were also prepared from LB-grown cultures of S. typhimurium LT2 grown without CaCl2 or in the presence of 5 mm CaCl2 using the methods described above.
Preparation of Radiolabeled and Carrier Substrates
The 4′-32P-labeled lipid IVA was generated from 100 μCi of [γ-32P]ATP and the disaccharide 1-phosphate lipid acceptor using the overexpressed 4′-kinase present in membranes of E. coli BLR(DE3)/pLysS/pJK2 (18, 34, 35). Kdo2-4′-32P-labeled lipid IVA was prepared by adding purified E. coli Kdo transferase (WaaA) immediately after the 4′-kinase reaction (35, 36). Kdo2-[4′-32P-labeled lipid A was synthesized enzymatically from Kdo2-[4′-32P-labeled lipid IVA (32). Carrier lipid IVA (5–10 mg) was isolated from E. coli strain MKV15 (37–39). Carrier Kdo2-lipid IVA was synthesized in vitro by WaaA-catalyzed Kdo additions to lipid IVA (40). Carrier Kdo2-lipid A was isolated from the heptose-deficient E. coli strain WBB06 (41), described previously (42).
Assay of the 3′-O-Deacylase
The 3′-O-deacylase activity of LpxR was assayed under optimized conditions in a 15-μl reaction mixture containing 50 mm MES, pH 6.5, 1% Triton X-100, 5 mm CaCl2, and either 10 μm Kdo2-4′-32P-labeled lipid A, Kdo2-4′-32P-labeled lipid IVA, or 4′-32P-labeled lipid IVA (each at 50,000 cpm/nmol) as the substrate. Washed membranes were employed as the enzyme source. Reactions were incubated at 30 °C for the indicated times. Reactions were stopped by spotting 4-μl portions of the mixtures onto a Silica Gel 60 TLC plate, and the plate was dried under a cool air stream for 20 min. Reaction products were separated using the solvent chloroform, pyridine, 88% formic acid, water (50:50:16:5, v/v), whereas reactions containing Kdo2-4′-32P-labeled lipid IVA were analyzed in the solvent chloroform, pyridine, 88% formic acid, water (30:70:16:10, v/v). The reactions that were carried out with the hexa-acylated substrate, Kdo2-4′-32P-labeled lipid A, were analyzed in the solvent chloroform, methanol, water, acetic acid (25:15:4:4, v/v). After chromatography, the plates were dried, and radioactive substrates were detected using a GE Healthcare PhosphorImager (STORM 840) equipped with ImageQuant software. The enzyme units (nmol/min) were calculated by determining the percentage of the substrate converted to product.
Purification of the Salmonella 3′-O-Deacylase in Vitro Reaction Product
A large scale 3′-O-deacylase reaction mixture was prepared by assembling the following components: 50 mm MES, pH 6.5, 1% Triton X-100, 5 mm CaCl2, 100 μm Kdo2-lipid A, and either 1 mg/ml S. typhimurium strain LT2 membranes or 0.1 mg/ml NovaBlue(DE3)/pLpxR1 membranes, as described below. The reaction was initiated by the addition of the membranes and incubated at 30 °C for 12 h. The reaction mixture was then converted into a two-phase Bligh/Dyer (43) system consisting of chloroform, methanol, and 0.1 m HCl (2:2: 1.8, v/v). The phases were separated by centrifugation at 4000 × g for 8 min. The organic phase was removed, and the resulting aqueous phase was extracted a second time by the addition of pre-equilibrated acidic organic phase. The organic phases were pooled and neutralized by the addition of a few drops of pyridine. The sample was dried by rotary evaporation, and lipids were fractionated using a DEAE cellulose anion-exchange column as previously described (44). Fractions containing the desired lipid species were pooled and converted to an acidic two-phase Bligh/Dyer mixture by the addition of appropriate amounts of chloroform and HCl. The organic phase was dried under a stream of N2 and stored at −80 °C.
Separation of Inner and Outer Membranes
Membranes isolated from either W3110/pWSK29 or W3110/pLpxR2 were separated by isopycnic sucrose gradient centrifugation using previously described methods (45, 46). Each gradient fraction (0.5 ml) was assayed for NADH oxidase (inner membrane marker) and phospholipase A (outer membrane marker) (47). The amount of protein in each fraction was determined using the bicinchoninic acid assay (48). Each fraction was also assayed for 3′-O-deacylase activity using the standard conditions described above.
Protein Microsequencing of LpxR
Inner and outer membranes of W3110/pWSK29 and W3110/pLpxR2 were analyzed by SDS-PAGE on a Bio-Rad Protean II XI apparatus with a 12% polyacrylamide, 1.5 mm thick gel (49). A piece of the gel containing LpxR was excised. The protein was blotted onto an Immobilon-P polyvinylidene fluoride membrane (Millipore) equilibrated in 10 mm CAPS, pH 11, containing 10% methanol at 15 V for 30 min using a Bio-Rad Semi-Dry apparatus. The blot was stained with 0.1% Ponceau S in 1% acetic acid for 1 min followed by destaining with 1% acetic acid for 10 min. The band corresponding to LpxR was excised, rinsed three times with distilled water, and analyzed by high sensitivity protein micro-sequencing at the Harvard Microchemistry and Proteomics Facility (Cambridge, MA).
Large Scale Isolation of Lipid A from LpxR-expressing Cultures
Cultures of E. coli strain W3110 harboring either pWSK29 or pLpxR2 were grown in 1 liter of LB medium as described above. The cultures were harvested by centrifugation at 5000 × g for 20 min when the A600 reached 1.0. The cells were washed once with 40 ml of phosphate-buffered saline, pH 7.4. The final cell pellet was resuspended in 40 ml of phosphate-buffered saline, pH 7.4. Lipid A was released from the LPS, purified as described previously (38, 39), and stored frozen at −80 °C. Lipid A was also prepared from CaCl2 (10 mm)-treated cultures of S. typhimurium strain LT2 harboring either pWSK29 or pLpxR2, as just described.
Mass Spectrometry of Kdo2-Lipid A or Lipid A Samples
Spectra were acquired in the negative-ion linear mode using an AXIMA-CFR (Kratos Analytical, Manchester, UK) matrix-assisted laser desorption ionization/time of flight (MALDI-TOF) mass spectrometer with a 337-nm nitrogen laser. The instrument was operated with a 20-kV extraction voltage and time-delayed extraction, providing a mass resolution of about ±1 atomic mass unit for compounds with a Mr of ∼2, 000. Each spectrum was the average of 100 laser shots. Samples were prepared for MALDI-TOF analysis by depositing 0.3 μl of the lipid dissolved in chloroform/methanol (4:1, v/v) followed by 0.3 μl of a saturated solution of 6-aza-2-thiothymine in 50% acetonitrile and 10% tribasic ammonium citrate (9:1, v/v) as the matrix. The samples were dried at room temperature before the spectra were acquired.
Mass Spectrometry of the Released 3′-Acyloxyacyl Moiety
Spectra were acquired on a QSTAR XL quadrupole TOF tandem mass spectrometer (ABI/MDS-Sciex, Toronto, Canada) equipped with an electrospray ionization (ESI) source. Spectra were acquired in the negative-ion mode and typically were the accumulation of 60 scans collected from 200–2000 atomic mass units. For mass spectrometry (MS) analysis, the extracted lipids were dissolved in 200 μl of chloroform/methanol (2:1, v/v) and infused into the ion source at 5–10 μl/min. The negative-ion ESI was carried out at −4200 V. Data acquisition and analysis was performed using the Analyst QS software.
NMR Spectroscopy of 3′-O-Deacylated Lipid A
The lipid A of E. coli W3110 harboring pLpxR2 was isolated as described above and further purified by DEAE cellulose anion exchange chromatography (44). Fractions containing 3′-O-deacylated lipid A were pooled, and the lipid was extracted by conversion into a two-phase Bligh/Dyer system (43). The lower phase was dried under a stream of N2, and 0.5 mg of the dried lipid was dissolved in 0.35 ml of CDCl3/CD3OD/D2O (2:3:1, v/v) in a 3-mm NMR tube. NMR spectroscopy was carried out at the Duke University NMR Spectroscopy Center. Proton chemical shifts are reported relative to tetramethylsilane at 0.00 ppm. NMR spectra were obtained on Varian Inova 800 or 500 MHz NMR spectrometers, each equipped with a Sun Ultra 10 computer and 5-mm Varian probe. 1H NMR spectra at 800 MHz were obtained with a 7.2-kHz spectral window, a 67° pulse field angle (4.5 μsec), a 4.5-s acquisition time, and a 1-s relaxation delay. The spectra were digitized using 64,000 points to obtain a digital resolution of 0.225 Hz/point. Two-dimensional NMR experiments with solvent suppression (correlation spectroscopy, two-dimensional nuclear Overhauser effect spectroscopy, total correlation spectroscopy, multiple quantum coherence) were performed as previously described (50, 51). Directly detected, 1H-decoupled 31P NMR spectra were recorded at 202.37 MHz with a spectral window of 12143.3 Hz digitized into 25,280 data points (digital resolution of 1 Hz/point or ∼0.005 ppm/point), a 60° pulse flip angle (8 μs), and a 1.6-s repeat time. 31P chemical shifts were referenced to 85% H3PO4 at 0.000 ppm. Inverse decoupled difference spectra were recorded as 1H-detected, 31P-decoupled heteronuclear NMR experiments as previously described (50, 51).
Results
Ca2+-dependent Conversion of Kdo2-lipid A to a 3′-O-Deacylated Product
As shown in Figs. 3, A and B, membranes of S. typhimurium LT2 efficiently convert Kdo2-4′-32P-labeled lipid A to an unknown product, designated A, when 5 mm Ca2+ is included in the assay (lanes 4 and 5 versus lanes 2 and 3). Formation of A is not strictly dependent upon the addition of Ca2+ to the growth medium (lane 4 versus lane 5). The activity is localized primarily to S. typhimurium outer membranes, as discussed in detail below (data not shown).
FIGURE 3. Detection of a novel 3′-O-deacylase in membranes of wild type S. typhimurium strain LT2.

A, membranes (0.02 mg/ml) from wild type S. typhimurium LT2 grown with or without 5 mm CaCl2 were assayed with 10 μm Kdo2-4′-32P-labeled lipid A in the absence or presence of 5 mm CaCl2. The reactions were incubated at 30 °C for 60 min. Products were separated by TLC in the solvent chloroform/methanol/water/acetic acid (25:15:4:4, v/v) and analyzed with a PhosphorImager. Lane 1, no enzyme; lane 2, S. typhimurium grown without added Ca2+; lane 3, S. typhimurium grown with added Ca2+ (5 mm); lane 4, S. typhimurium grown without added Ca2+; lane 5, S. typhimurium grown with added Ca2+ (5 mm). B, S. typhimurium LT2 membranes (0.1 mg/ml) from the “no Ca2+” culture were assayed with 10 μm Kdo2-4′-32P-labeled lipid A for the indicated times. C, Kdo2-lipid A and its deacylated product were partially purified from a large-scale reaction mixture, prepared with 1 mg/ml S. typhimurium LT2 membranes. The lipids were analyzed by negative-ion mode MALDI-TOF mass spectrometry. amu, atomic mass units.
Product A (Fig. 3A), generated in vitro with S. typhimurium membranes and Kdo2-lipid A, was partially purified by chromatography on DEAE cellulose, as described under “Experimental Procedures” and was analyzed by low resolution MALDI-TOF mass spectrometry in the negative ion mode. As shown in Fig. 3C, the predominant peak at m/z 1801.7 atomic mass units can be interpreted as [M – H]− of a 3′-O-deacylated Kdo2-lipid A derivative, which has a molecular weight of 1802.01. The smaller peak at m/z 1361.5 atomic mass units is due to loss of both Kdo residues from 3′-O-deacylated Kdo2-lipid A during MALDI-TOF mass spectrometry (44). However, the minor peak at m/z 2040.3 atomic mass units is attributed to [M – H]− of a 3′-O-deacylated Kdo2-lipid A derivative to which an additional palmitate residue was attached in vitro by the outer membrane enzyme PagP (26).
Positive-ion MALDI-TOF mass spectrometry of product A (not shown) confirmed the loss of the 3′-acyloxyacyl group. The spectrum revealed peaks at m/z 1825.1 atomic mass units, interpreted as [M + Na]+ of 3′-O-deacylated Kdo2-lipid A, m/z 1704.8 atomic mass units, interpreted as the B2+ ion of 3′-O-deacylated Kdo2-lipid A, and m/z 1091.1 atomic mass units, interpreted as the B1+ ion of 3′-O-deacylated Kdo2-lipid A. The mass of the B1+ oxonium ion, derived from the distal lipid A unit by cleavage of the β1′-6 glycosidic linkage (52), provides additional direct support for the proposed 3′-O-deacylation.
Identification of the S. typhimurium 3′-O-Deacylase Structural Gene
To find the gene encoding the S. typhimurium 3′-O-deacylase, an expression-cloning strategy was employed. Extracts of E. coli K-12 do not exhibit detectable 3′-O-deacylase activity, allowing us to utilize E. coli K-12 as the host. A BAC library of S. typhimurium LT2 genomic DNA harbored in E. coli K-12 was acquired from the Salmonella Genetic Stock Center (Calgary, Canada). Assays of the membranes from each of the 71 BAC clones revealed only one (BAC 23) that exhibited 3′-O-deacylase activity (data not shown). BAC 23 contained a DNA fragment encompassing bases 1379597–1496842 of the S. typhimurium chromosome (53).
To identify the gene encoding the enzyme, we assumed that an orthologue of the S. typhimurium 3′-O-deacylase was not present in E. coli K-12 and that it was annotated as an outer membrane protein in the S. typhimurium data base. Only one open reading frame, STM1328, within the relevant segment of the S. typhimurium genome met both criteria. The product of STM1328 has a predicted N-terminal signal peptide, consistent with outer membrane targeting (54). A tBLASTn (55) search with the predicted protein product of STM1328 also did not reveal any significant orthologues in E. coli K-12.
STM1328 (renamed lpxR) was subcloned into pET23a to create pLpxR1 and expressed in E. coli NovaBlue(DE3). As shown in Fig. 4, membranes derived from LpxR-expressing E. coli NovaBlue(DE3) exhibited robust in vitro 3′-O-deacylase activity (lane 4) qualitatively similar to that seen with membranes of S. typhimurium LT2 (lane 2). Membranes of E. coli NovaBlue(DE3) harboring the vector control showed no activity (lane 3). The specific activity of S. typhimurium LT2 membranes (0.48 nmol/min/ mg) and of the LpxR-expressing E. coli NovaBlue(DE3) membranes (434 nmol/min/mg) were determined using 10 μm Kdo2-4′-32P-labeled lipid A as the substrate.
FIGURE 4. Presence of 3′-O-deacylase in membranes of E. coli NovaBlue(DE3)-expressing LpxR.

Membranes (0.1 mg/ml) of S. typhimurium LT2, E. coli NovaBlue(DE3) harboring pET23a, and E. coli NovaBlue(DE3) harboring pLpxR1 (pET23a into which STM1328 was subcloned) were assayed for 3′-O-deacylase activity with 10 μm Kdo2-4′-32P-labeled lipid A at 30 °C for 30 min. Products were separated by TLC in chloroform/methanol/water/acetic acid (25:15:4:4, v/v) and analyzed with a PhosphorImager. Lane 1, no enzyme; lane 2, S. typhimurium LT2; lane 3, E. coli NovaBlue (N.B.)(DE3)/pET23a; lane 4, E. coli NovaBlue (N.B.)(DE3)/pLpxR1.
Table 2 lists some of the closest orthologues of LpxR present in the non-redundant data base. Helicobacter pylori and Yersinia enterocolitica, which have orthologues of LpxR, have been previously reported to contain lipid A species that lack the 3′-acyloxyacyl moiety (56, 57). Ca2+-dependent 3′-O-deacylase activity is present in the membranes of several Gram-negative pathogens that contain LpxR orthologues, including E. coli 0157:H7, Y. enterocolitica, and Vibrio cholerae.4 The complete nucleotide sequence of lpxR has been submitted to GenBank™ under accession number DQ272513.
TABLE 2. Selected proteins related to S. typhimurium LpxR.
Possible orthologues in the NCBI database as of March 24, 2006 were identified with the PSI-Blast algorithm using the predicted S. typhimurium LpxR protein sequence of 319 residues as the probe.
| Organism | Homology (gaps)a | Approximate E values |
|---|---|---|
| S. typhi | 318/318/319 | 8 × 10−178 |
| S. paratyphi | 308/308/310 | 2 × 10−171 |
| E. coli O157:H7 | 228/272/318 (1) | 1 × 10−133 |
| Y. enterocolitica | 230/269/320 (1) | 2 × 10−124 |
| Rhodospirillum rubrum | 76/117/254 (20) | 8 × 10−15 |
| Gluconobacter oxydans | 60/102/236 (9) | 2 × 10−11 |
| Xanthomonas campestris | 59/94/254 (35) | 3 × 10−7 |
| V. cholerae V51 | 74/130/338 (61) | 5 × 10−4 |
| H. pylori J99 | 46/103/250 (15) | 7 × 10−3 |
Homology is given as the number of identities/number of positives/number of residues (including gaps) in the related segment when compare with S. typhimurium LpxR, a hypothetical protein of 319 amino acid residues.
LpxR-catalyzed Production of 3′-O-deacylated Kdo2-lipid A and Release of the Intact 3′-Acyloxyacyl Moiety
To confirm that the recombinant LpxR catalyzes the same 3′-O-deacylation of Kdo2-lipid A seen with Salmonella membranes (Fig. 3), a large-scale in vitro 3′-O-deacylase reaction mixture was prepared using E. coli NovaBlue(DE3)/pLpxR1 membranes as the enzyme source. After purification over DEAE-cellulose, negative-ion MALDI-TOF mass spectrometry was used to analyze the products eluting with 240 to 480 mm ammonium acetate. The major peak, interpreted as [M – H]− of 3′-O-deacylated Kdo2-lipid A, at m/z 1801.9 atomic mass units (data not shown) was the same within experimental error as that seen with wild-type Salmonella membranes (Fig. 3C).
High-resolution electrospray ionization (ESI) mass spectrometry was used to determine whether or not the 3′-acyloxyacyl moiety was released intact from Kdo2-lipid A by recombinant LpxR. An analysis of the LpxR-derived fatty acids, which elute with 30 mm ammonium acetate from the DEAE cellulose column, revealed a major species at m/z 453.387 atomic mass units, consistent with the predicted [M – H] − of the cleaved 3′-acyloxyacyl moiety of E. coli lipid A (exact mass of 453.394 atomic mass units). The peak at m/z 453.387 atomic mass units was then subjected to tandem ESI-MS/MS analysis to confirm the structural assignment. Collisional activation of the parent ion at m/z 453.387 generated a product ion at m/z 227.193 atomic mass units (Fig. 5B), as expected for the [M – H] − ion of myristic acid (predicted exact mass of 227.201).
FIGURE 5. LpxR releases the intact 3′-acyloxyacyl moiety from Kdo2-lipid A.
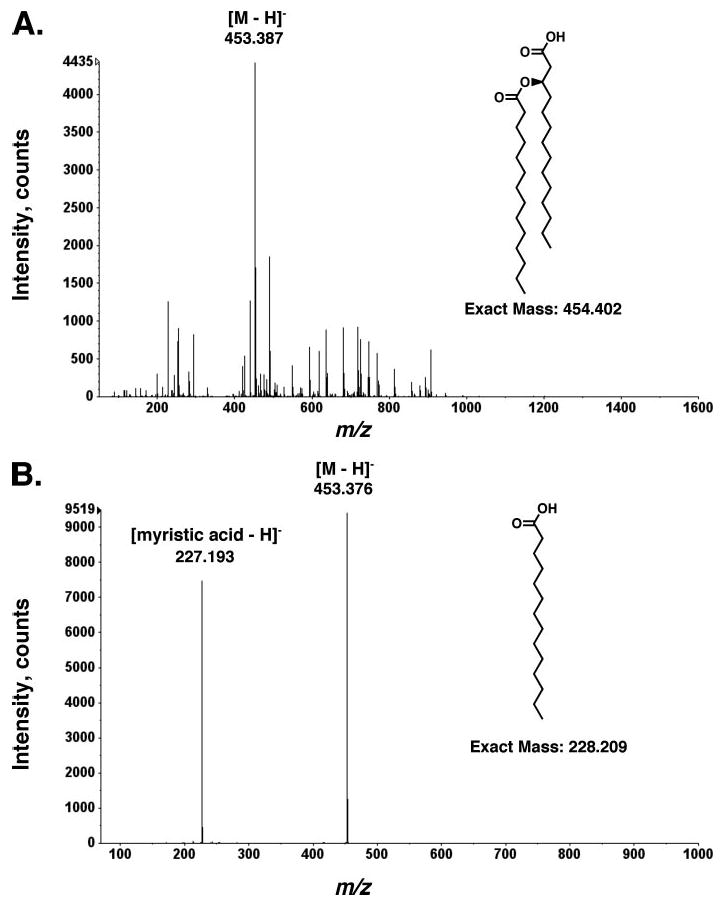
A large scale 3′-O-deacylase reaction using E. coli NovaBlue(DE3)/pLpxR1 membranes (0.1 mg/ml) as enzyme was carried out as described under “Experimental Procedures.” Lipids were fractionated by DEAE cellulose chromatography (44). A, negative-mode ESI-MS analysis of the 30 mm ammonium acetate fraction revealed a major species (453.387 atomic mass units) that could be interpreted as [M – H]− of the intact 3′-acyloxyacyl moiety of lipid A (exact molecular mass = 454.402 atomic mass units). Most of the minor species in this fraction are contaminating phospholipids from the membranes of the LpxR over-producing strain. B, the peak at m/z 453.387 atomic mass units was subjected to tandem MS/MS in the negative mode to confirm the structural assignment. The product ion (227.193 atomic mass units) is interpreted as [myristic acid – H]− (the exact mass of myristic acid = 228.209 atomic mass units). Mass spectrometry of the Kdo2-lipid A present in the 240 mm ammonium acetate step confirmed the production of 3′-O-deacylated Kdo2-lipid A by LpxR-expressing E. coli NovaBlue (DE3) membranes (data not shown).
Substrate Specificity of S. typhimurium LpxR
In vitro 3′-O-deacylation of Kdo2-4′-32P-labeled lipid A by LpxR required the presence of the nonionic detergent Triton X-100 with optimal activity at 1%. LpxR also required Ca2+ for activity, with an optimum at 5 mm (data not shown). Product formation by E. coli NovaBlue(DE3) membranes over-expressing LpxR, assayed with 10 μm Kdo2-4′-32P-labeled lipid A under optimized conditions, was linearly dependent upon both protein concentration and time (data not shown). The ability of LpxR to hydrolyze (Kdo2-4′-32P-labeled lipid A, Kdo2-4′-32P-labeled lipid IVA and 4′-32P-labeled lipid IVA) in vitro was compared. Assays with each of these substrates at 10 μm revealed that 4′-32P-labeled lipid IVA was a relatively poor substrate (1 nmol/min/mg of membrane protein) when compared with Kdo2-4′-32P-labeled lipid IVA (132 nmol/min/mg) or Kdo2-4′-32P-labeled lipid A (434 nmol/min/mg). The data show that the 3′-O-deacylase activity is strongly Kdo-dependent.
Divalent Cation Dependence and Thermal Stability of the 3′-O-Deacylase
Membranes of E. coli strain NovaBlue(DE3)/ pLpxR1 were assayed for LpxR activity in the presence of various divalent cations, each held at 5 mm (Fig. 6). Ca2+ (lane 5) and Sr2+ (lane 9) produced the greatest stimulation. Cd2+ (lane 15) showed partial stimulation, whereas Mg2+, Zn2+ and Ba2+ were inactive.
FIGURE 6. Comparison of divalent cations to stimulate 3′-O-deacylase activity.

Various metals were tested for their ability to support the 3′-O-deacylase activity of LpxR in vitro. Membranes (0.02 mg/ml) from NovaBlue(DE3) harboring either pET23a or pLpxR1 were assayed in the absence of added metal (lanes 2 and 3) or in the presence of 5 mm Ca2+ (lanes 4 and 5), Mg2+ (lanes 6 and 7), Sr2+ (lanes 8 and 9), Ba2+ (lanes 10 and 11), Zn2+ (lanes 12 and 13), or Cd2+ (lanes 14 and 15). Lane 1, no-enzyme control; even lanes, membranes of NovaBlue (DE3)/pET23a; odd lanes after lane 1, membranes of NovaBlue (DE3)/pLpxR1.
The outer membrane enzymes PagP and PagL have been previously shown to resist heat denaturation (26, 27). Similarly, E. coli membranes over-expressing LpxR retained significant activity (∼67%) after a 10-min pre-incubation at 100 °C (data not shown).
Outer Membrane Localization of Recombinant LpxR
Inner and outer membranes of E. coli W3110/pLpxR2 were separated by isopycnic sucrose gradient centrifugation (Fig. 7). The heavier outer membranes were detected by their phospholipase A activity, whereas the lighter inner membranes were detected by assaying for NADH oxidase (Fig. 7B). LpxR activity was predominantly localized to the outer membrane (Fig. 7A), although a portion was also recovered in the intermediate region (fractions 9 to 13), which was depleted of both marker enzymes (Fig. 7B).
FIGURE 7. Isopycnic sucrose density gradient centrifugation of LpxR-containing membranes.

Inner and outer membranes, isolated from W3110/ pLpxR2, were separated as described previously (45, 46), and ∼0.5-ml fractions were collected. A, protein concentration (open circles) and the 3′-O-deacylase activity of LpxR (closed circles) were determined. B, outer membrane phospholipase A activity (open squares) and inner membrane NADH oxidase activity (closed squares) were measured.
Total membranes, outer membranes, and inner membranes of W3110, harboring either the vector pWSK29 or pLpxR2, were analyzed by 12% SDS-PAGE. As shown in Fig. 8, a band corresponding to processed LpxR (32.4 kDa) was present in the un-fractionated membranes of W3110/pLpxR2 (lane 3) and absent in the vector control (lane 2). The LpxR band was greatly enriched in the outer membranes (lane 5) versus the inner membranes (lane 7).
FIGURE 8. Outer membrane localization of LpxR protein in membranes of E. coli W3110/pLpxR2.
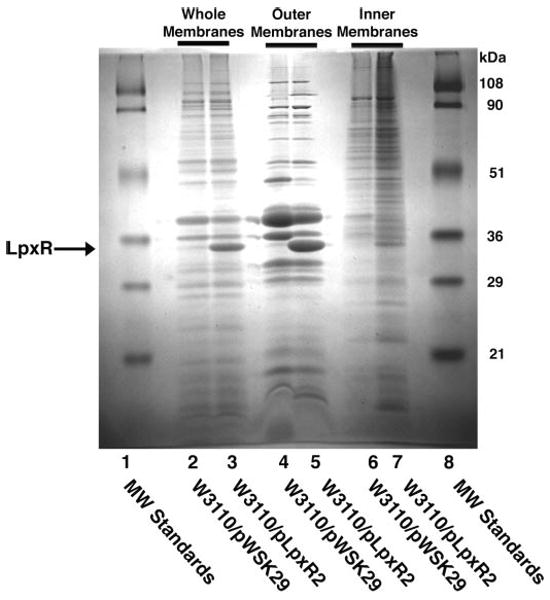
The inner and outer membranes from W3110 harboring either the empty vector pWSK29 or pLpxR2 were separated by isopycnic sucrose density ultracentrifugation. Next, 30-μg samples of protein from the unfractionated membranes, the outer membranes, and the inner membranes were analyzed by 12% SDS-PAGE and Coomassie Blue staining. The molecular mass standards are indicated to the right of the gel.
To verify the presence of the signal peptide predicted by Signal-P (54) (http://www.cbs.dtu.dk/services/SignalP/) and to establish the exact cleavage site, LpxR from the outer membranes of W3110/ pLpxR2 was blotted to a polyvinylidene difluoride membrane and subjected to N-terminal micro-sequencing. The first ten amino acid residues were SSLAISVAND, indicating that the cleavage of the signal peptide occurred between residues 22 and 23 of the full-length protein, consistent with the prediction of the Signal-P program. Thus, mature LpxR is comprised of 296 amino acids with a molecular weight of 32,400.
Deacylation of Lipid A by LpxR in Living Cells
Although wild-type S. typhimurium membranes contain measurable LpxR activity (Fig. 3A), the lipid A of Salmonella is not appreciably 3′-O-deacylated under the commonly used growth conditions (58–61). In contrast, heterologous over-expression of Salmonella LpxR in E. coli K-12 resulted in extensive 3′-O-deacylation of endogenous lipid A (Fig. 9). The lipid A species, released from cells by hydrolysis at pH 4.5, were purified and analyzed by low-resolution MALDI-TOF mass spectrometry. The endogenous lipid A of W3110/pWSK29 consisted primarily of the hexa-acylated bis-phosphate form, typically seen with wild-type E. coli K-12 strains. The peak at m/z 1798.4 atomic mass units in the negative-ion mode was interpreted as [M – H] − of lipid A (Fig. 9A). Upon over-expression of LpxR, an additional peak was seen at m/z 1361.6 atomic mass units in the negative-ion mode (Fig. 9B), interpreted as [M – H]− of 3′-O-deacylated lipid A. Heterologous expression of LpxR in E. coli did not slow the growth rate (data not shown).
FIGURE 9. Presence of 3′-O-deacylated lipid A in living cells of E. coli W3110/pLpxR2.

A, lipid A isolated from E. coli strain W3110 harboring the empty vector pWSK29 consists mainly of hexa-acylated lipid A. B, the lipid A of W3110/pLpxR2 contains a significant portion of an additional component with the mass expected for a 3′-O-deacylated lipid A species, as indicated. amu, atomic mass units.
A similar result was seen upon overexpression of the 3′-O-deacylase in S. typhimurium strain LT2. As shown in Fig. 10A, analysis of the lipid A species from S. typhimurium/pWSK29 grown in the presence of 10 mm CaCl2 yielded a major peak at m/z 1798.3 atomic mass units, interpreted as [M – H] − of lipid A. Calcium was included in the growth media to suppress the PhoP/PhoQ-and/or PmrA/PmrB-regulated lipid A modifications (62). Over-expression of LpxR resulted in an additional peak at m/z 1361.5 atomic mass units, interpreted as [M – H] − of the 3′-O-deacylated variant (Fig. 10B).
FIGURE 10. Presence of 3′-O-deacylated lipid A in living cells of S. typhimurium LT2/pLpxR2.
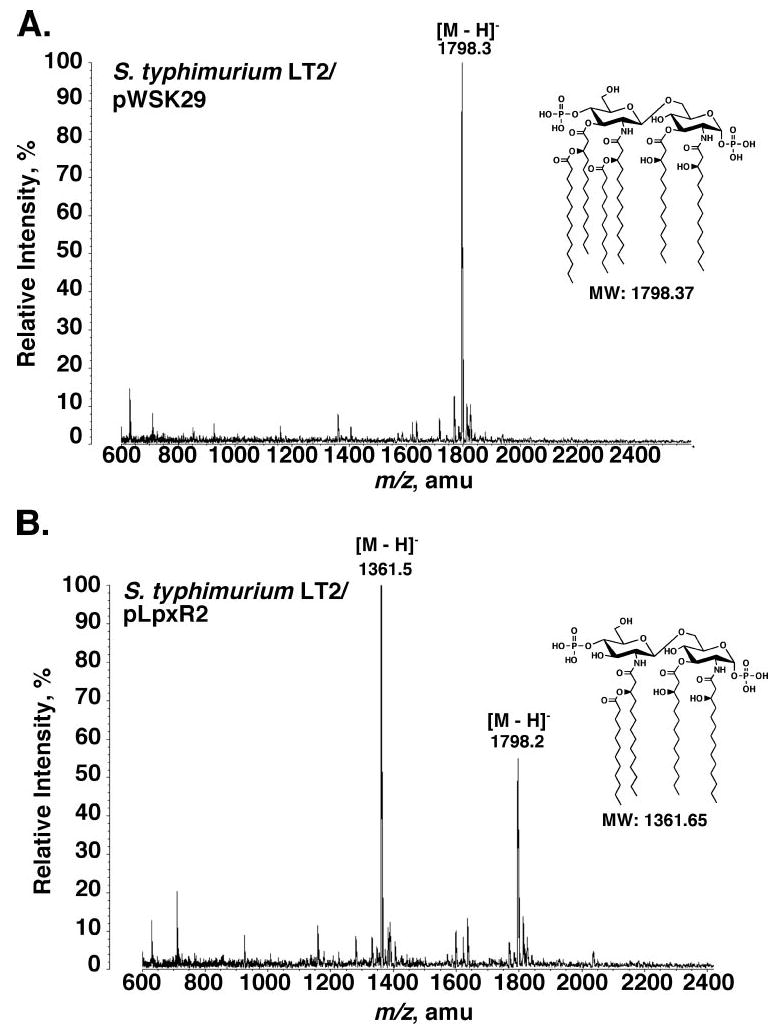
A, lipid A isolated from CaCl2 (10 mm)-treated S. typhimurium strain LT2 harboring the empty vector pWSK29 consists mainly of hexa-acylated lipid A. B, lipid A of CaCl2-treated S. typhimurium LT2/pLpxR2 contains a significant portion of an additional component with the mass expected for a 3′-O-deacylated lipid A species, as indicated. amu, atomic mass units.
NMR Spectroscopy of 3′-O-deacylated Lipid A Isolated from W3110/pLpxR2
The partial 800 MHz 1H NMR spectrum of 3′-O-deacylated lipid A (Fig. 11), purified from W3110/pLpxR2, revealed well resolved resonances in the sugar and acyl chain regions (Table 3), similar to those previously reported for wild-type E. coli K-12 lipid A dissolved in CDCl3/CD3OD/D2O (2:3:1 v/v) (50, 51). The sugar region was comprised of eight resolved single-proton resonances, three 2-proton envelopes (5.2, 3.97 and 3.65 ppm) and one 3-proton envelope (3.82 ppm), consistent with fourteen sugar and three β-hydroxymethine protons (Fig. 11A). The α CH2 resonances integrated to eight protons (Fig. 11B), indicating the existence of four acyl chains. Comparison with the α CH2 resonances of wild-type E. coli lipid A (50) showed the lack of the 2.7 ppm multiplet, previously assigned to the 3′ acyl chain, and the loss of one α CH2 resonance near 2.3 ppm. The NMR data are therefore consistent with the removal of the 3′ -acyloxyacyl moiety by LpxR.
FIGURE 11. 1H NMR spectroscopy of 3′-O-deacylated lipid A.
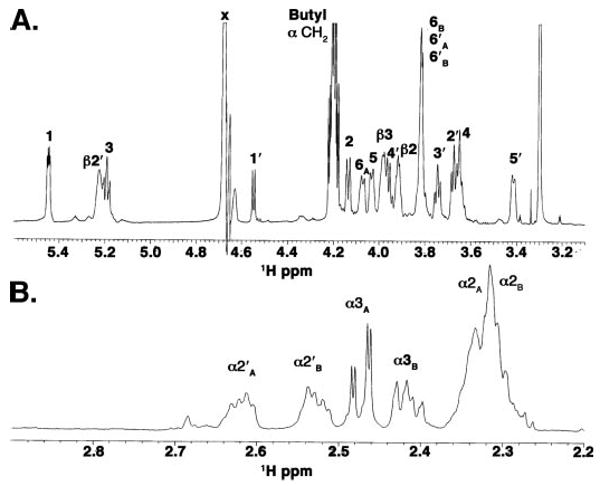
A, partial 800 MHz 1H NMR spectrum of 3′-O-deacylated lipid A in CDCl3:CD3OD:D2O (2:3:1, v/v) showing the sugar proton region. The sugar proton resonances integrate to 14 sugar and three β-hydroxymethines, consistent with the presence of two hexoses and three β-hydroxyacyl chains. The intensity of the H-1′ doublet is reduced due to a presaturation pulse used to eliminate the water solvent signal. The sharp signal at 4.2 ppm (∼1.2 Hz line width) arises from the butyl group of a solvent impurity in the sample preparation. The broadened lines of the lipid A resonances (3–8 Hz) are consistent with lipid self-aggregation. B, partial 800 MHz 1H NMR spectrum of 3′-O-deacylated lipid A showing the acyl α-methylene proton resonances, with resolution of the α2′ and α3 methylene signals as eight-line multiplets. The α-methylene region integrates to eight protons, indicating the existence of four acyl chains.
TABLE 3. 1H NMR chemical shifts of 3′-O-deacylated lipid A versus E. coli lipid A.
1H chemical shifts at 25 °C in CDCl3:CD3OD:D2O (2:3:1, v/v) relative to internal tetramethylsilane (TMS).
| Position | 3′-O-deacylated lipid A | E. coli lipid A |
|---|---|---|
| δH, [(mult), J(Hz)] | δH, [(mult), J(Hz)] | |
| 1 | 5.449 [dd, 3.3, 7.0] | 5.450 [dd, 3.3, 6.8] |
| 2 | 4.137 [ddd, 3.3, 10.9] | 4.147 [ddd, 3.3, 10.6] |
| 3 | 5.192 [dd, 10.9,9.3] | 5.213 [dd, 10.8,9.3] |
| 4 | 3.650 [dd, 9.3] | 3.679 [dd, 9.3] |
| 5 | 4.031 [m] | ∼4.03 [m] |
| 6a | 4.071 [m,12.0] | 4.103 [dd, 2.0, 12.3] |
| 6b | ∼3.82 [m] | 3.847 [dd, 5.1, 12.3] |
| 1′ | 4.549 [d, 8.4] | 4.613 [d, 8.8] |
| 2′ | 3.674 [m] | ∼3.83 [dd, 8.8, 10.8] |
| 3′ | 3.745 [m] | 5.18 [dd, 10.8,9.3] |
| 4′ | 3.958 [m] | 4.166 [dd, 9.3] |
| 5′ | 3.415 [m] | 3.460 [m] |
| 6′a | ∼3.82 [m] | ∼3.94 [dd, 2.0, 12.3] |
| 6′b | ∼3.82 [m] | 3.774 [dd, 5.1, 12.3] |
| α2a | 2.33 [m] | ∼2.33 |
| α2b | 2.29 [m] | |
| β2 | ∼3.92 [m] | ∼3.95 [m] |
| γ2 | ∼1.42 [m] | ∼1.45 [m] |
| α3a | 2.472 [dd, 2.3, 15.3] | ∼2.45 |
| α3b | 2.423 [dd, 9.6, 15.3] | |
| β3 | ∼3.98 [m] | ∼4.03 [m] |
| γ3 | ∼1.45 [m] | ∼1.50 [m] |
| α2′a | 2.618 [dd, 7.4, 14.7] | ∼2.561 |
| α2′b | 2.526 [dd, 6.4, 14.7] | |
| β2′ | ∼5.23 [m] | ∼5.18 [m] |
| γ2′ | ∼1.62 [m] | ∼1.61 [m] |
| α3′a | 2.727 [dd, 7.2, 16.3] | |
| α3′b | 2.665 [dd, 5.5, 16.3] | |
| β3′ | ∼5.25 [m] | |
| γ3′ | ∼1.61 [m] | |
| α CH2 | ∼2.32 [m] | ∼2.34 [m] |
| β CH2 | ∼1.62 [m] | ∼1.61 [m] |
| (CH2)n | ∼1.254 [m] | ∼1.24 [m] |
| ω – 2 CH2 | ∼1.87 [m] | ∼1.87 [m] |
| ω –1 CH2 | 1.279 [m] | 1.279 [m] |
| ω CH3 | 0.886 [t, ∼7] | 0.891 [t, ∼7] |
The anomeric H-1 of the proximal sugar (5.45 ppm) and the H-1′ of the distal sugar (4.54 ppm) (Fig. 11A) served as convenient entry points for full 2D NMR assignments (Table 3), which were based on correlation spectroscopy and total correlation spectroscopy analysis (not shown). The proximal sugar protons (H-1 to H-6) of the 3′-O-deacylated lipid A showed similar chemical shifts to those of wild-type E. coli lipid A (50, 51) (Table 3). Significantly, H-3′ of the 3′-O-deacylated lipid A at 3.75 ppm (Table 3) was 1.44 ppm upfield compared with H-3′ of wild-type E. coli lipid A (5.18 ppm). Other protons of the distal unit were not shifted by more than several tenth of a ppm (Table 3), thus confirming the selective deacylation of the 3′-position by LpxR.
Discussion
Lipid A is present in virtually all Gram-negative bacteria and is detected by the innate immune system receptor TLR-4 (9, 10). Nine constitutive enzymes of lipid A biosynthesis have been identified in E. coli (15), and with few exceptions, single copies of the corresponding structural genes are present in all Gram-negative organisms. Despite the conservation of the biosynthetic pathway, which generates the lipid A 1, 4′-bis phosphate shown in Fig. 1A, the lipid A moiety of LPS displays significant structural diversity in many important pathogens. For instance, in Salmonella lipid A is covalently modified in response to environmental stimuli that activates the two-component regulatory systems, PhoP/PhoQ (63, 64) and PmrA/PmrB (59, 65, 66) (Fig. 1B). Modification of lipid A is correlated with bacterial virulence, antimicrobial peptide resistance, and attenuation of TLR-4 activation (21–24, 66–70). The enzymes catalyzing the covalent modifications of Salmonella lipid A (Fig. 1B) have recently been identified and characterized (18–20, 25–27).
Here we report the discovery of LpxR, an outer membrane enzyme catalyzing a novel deacylation of lipid A at the 3′-position. Utilizing an in vitro assay system, we determined that membranes of wild-type S. typhimurium convert Kdo2-lipid A to 3′-O-deacylated Kdo2-lipid A (Fig. 3) in the presence of Ca2+. That membranes of S. typhimurium could catalyze 3′-O-deacylation of Kdo2-lipid A was surprising given that 3′-O-deacylated lipid A species have not been reported for this organism. However, 3′-O-deacylated lipid A species have been isolated from H. pylori (56), Y. enterocolitica (57), Francisella tularensis (71), and Porphyromonas gingivalis (72). Our data demonstrate that a lipid A-3′-O-deacylase is present in the outer membrane of S. typhimurium but that it is latent in vivo under all growth conditions examined to date. The previously reported 3-O-deacylase PagL, which is also found in the outer membrane, displays a similar latency (73). Endogenous inhibitors present in stoichiometric quantities relative to LpxR and PagL might account for these findings. We found that it is possible to generate significant amounts of 3′-O-deacylated lipid A in S. typhimurium LT2 through pWSK29-mediated overexpression of LpxR (Fig. 10B). We can, thus, hypothesize that an up-regulation of lpxR expression in wild type Salmonella would lead to detectable 3′-O-deacylated lipid A species.
LpxR-catalyzed 3′-O-deacylase activity is absolutely dependent upon the presence of Ca2+ in our in vitro system (Fig. 6). Sr2+ and to a lesser extent Cd2+ could also support LpxR activity. LpxR is the second example of an LPS-modifying enzyme that exhibits a requirement for Ca2+. Recently, our laboratory reported that EptB, a phosphoethanolamine transferase specific for the outer Kdo moiety of LPS, is a Ca2+-dependent enzyme (32). LpxR also shares some characteristics with the outer membrane phospholipase A. Outer membrane phospholipase A is an integral Ca2+-dependent membrane enzyme that participates in secretion of colicins and has been implicated in bacterial virulence (74–76). The activity of outer membrane phospholipase A is regulated by a Ca2+-induced, reversible dimerization (74). It is possible that Ca2+ may likewise be required to facilitate LpxR dimerization. Interestingly, PagL, which catalyzes a related hydrolysis reaction (3-O-deacylation), does not require Ca2+ or other divalent cations in vitro (27). PagL and LpxR may utilize different mechanisms to deacylate the lipid A 3 or 3′ positions, respectively. Studies of purified LpxR should provide insights into how Ca2+ modulates the activity of this enzyme.
Mass spectrometry of the reaction products generated during the in vitro assay revealed that LpxR selectively cleaves the ester linkage at the 3′ position of lipid A, as no 3-O-deacylated species was detected. NMR spectroscopy of the LpxR-modified lipid A confirmed the selective loss of the 3′-acyloxyacyl group (Fig. 11 and Table 3). Our data further demonstrate that LpxR releases the intact 3′-acyloxyacyl moiety (Fig. 5) and does not further cleave the acyloxyacyl-linkage. Accordingly, it is reasonable that LpxR does not share any sequence similarity with the acyloxyacyl hydrolase (77, 78) of mammalian cells, which removes only the secondary acyl chains of lipid A.
Like the palmitoyltransferase PagP (26) and the 3-O-deacylase PagL (27), LpxR is associated with the outer membrane. Subcellular fractionation studies of membranes derived from LpxR-expressing E. coli W3110 cultures revealed that 3′-O-deacylase activity was present mostly in the outer membrane fractions (Fig. 7). SDS-polyacrylamide gel electrophoresis provided further evidence of LpxR localization within the outer membrane (Fig. 8). The overexpressed LpxR protein was missing its type I signal peptide, as shown by N-terminal sequencing of the outer membrane-associated band. LpxR (∼32 kDa) is larger than either PagP (∼20 kDa) (26) or PagL (∼20 kDa) (27). The crystal structure of PagP revealed that this protein is an eight-stranded, inside-out β-barrel (79). Topology analysis predicts a 14-stranded β-barrel structure for LpxR.
All S. typhimurium lipid A-modifying enzymes, with the exception of LpxO, are transcriptionally regulated by PhoP/PhoQ and/or PmrA/PmrB. Recently, SlyA (80, 81), a transcription factor that is under the control of PhoP, was shown to regulate STM1328(lpxR) expression. Microarray analysis of a slyA deletion mutant of S. typhimurium revealed that the expression of STM1328 was reduced 3-fold (82). However, it is unclear whether or not SlyA regulation of lpxR via PhoP is functionally significant at the level of protein synthesis, because membranes isolated from a Salmonella phoP null mutant (83) display similar levels of 3′-O-deacylase activity as wild type (data not shown). Other SlyA-regulated genes have functions associated with the bacterial cell envelope, and some genes have been implicated in virulence and resistance to antimicrobial peptides (84–87). SlyA mutants are profoundly attenuated for virulence, unable to replicate in host phagocytes, and sensitive to oxidative stress (80, 87, 88).
Orthologues of LpxR are found in the genomes of E. coli 0157:H7, Y. enterocolitica, H. pylori, and V. cholerae (Table 2). However, only Y. enterocolitica and H. pylori synthesize 3′-O-deacylated lipid A species (56, 57). The lipid A of H. pylori is completely deacylated at the 3′-position, resulting in a tetraacylated lipid A structure (56). In comparison to the lipid A of the Enterobacteriaceae family, H. pylori lipid A shows very little immunological activity, presumably helping the organism to evade the innate immune response and contributing to its persistence (89). Our work suggests that LpxR may be important during H. pylori infection by reducing the number of lipid A acyl chains, thereby resulting in decreased TLR-4 activation. Removal of the R-3-hydroxymyristate chain at the 3-position of S. typhimurium lipid A by PagL likewise attenuates TLR-4 activation (69).
Surprisingly, we were unable to identify orthologues of LpxR in the genomes of F. tularensis and P. gingivalis even though these bacteria synthesize 3′-O-deacylated lipid A species (71, 72). It is, therefore, likely that F. tularensis and P. gingivalis contain additional, structurally distinct 3′-O-deacylases.
LpxR is the sixth enzyme identified to date that is capable of modifying the lipid A component of Salmonella LPS (Figs. 1B and 2). Construction of lpxR null mutants in Salmonella and other pathogens will be necessary to evaluate the functions of LpxR in pathogenesis and/or outer membrane remodeling. The intriguing mechanisms by which LpxR and PagL are rendered latent in wild type Salmonella also need to be investigated.
Acknowledgments
Acknowledgments—We thank Dr. Steven D. Breazeale for assistance with the ESI-MS analysis and Dr. David A. Six, Christopher M. Stead, An X. Tran, and Jessica V. Hankins for helpful discussions. The Duke NMR Center is partially supported by National Institutes of Health NCI P30-CA-14236. NMR instrumentation in the Duke NMR Center was funded by the National Science Foundation, the National Institutes of Health, the North Carolina Biotechnology Center and Duke University.
Footnotes
This research was funded by National Institutes of Health Grants AI-064184 (to M. S. T.), GM-51310 (to C. R. H. R.), and GM-64402 (to R. J. C.).
The nucleotide sequence(s) reported in this paper has been submitted to the GenBank™/EBI Data Bank with accession number(s) DQ272513.
The abbreviations used are: LPS, lipopolysaccharide; BAC, bacterial artificial chromosome; Kdo, 3-deoxy-D-manno-octulosonic acid; MES, 4-morpholineethanesulfonic acid; MALDI-TOF, matrix-assisted laser desorption ionization-time of flight; ESI, electrospray ionization; MS, mass spectrometry; TLR-4, Toll-like rececptor 4; CAPS, 3-(cyclohexylamino)propanesulfonic acid; PagP, PhoP-activated gene P; PagL, PhoP-activated gene L.
C. M. Stead and M. S. Trent, unpublished data.
References
- 1.Jones BD, Falkow S. Annu Rev Immunol. 1996;14:533–561. doi: 10.1146/annurev.immunol.14.1.533. [DOI] [PubMed] [Google Scholar]
- 2.Scherer CA, Miller SI. In: Principles of Bacterial Pathogenesis. Groisman EA, editor. Academic Press, Inc.; San Diego, CA: 2001. pp. 229–243. [Google Scholar]
- 3.Garcia-Del Portillo F. Trends Microbiol. 1999;7:467–469. doi: 10.1016/s0966-842x(99)01618-2. [DOI] [PubMed] [Google Scholar]
- 4.Lindgren SW, Stojiljkovic I, Heffron F. Proc Natl Acad Sci U S A. 1996;93:4197–4201. doi: 10.1073/pnas.93.9.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monack DM, Raupach B, Hromockyj AE, Falkow S. Proc Natl Acad Sci U S A. 1996;93:9833–9838. doi: 10.1073/pnas.93.18.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richter-Dahlfors A, Buchan AMJ, Finlay BB. J Exp Med. 1997;186:569–580. doi: 10.1084/jem.186.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beutler B, Cerami A. Annu Rev Biochem. 1988;57:505–518. doi: 10.1146/annurev.bi.57.070188.002445. [DOI] [PubMed] [Google Scholar]
- 8.Dinarello CA. Blood. 1991;77:1627–1652. [PubMed] [Google Scholar]
- 9.Raetz CRH. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. 2nd. Neidhardt FC, editor. American Society for Microbiology; Washington, D.C.: 1996. pp. 1035–1063. [Google Scholar]
- 10.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Di Padova F, Schreier M, Brade H. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 11.Aderem A, Ulevitch RJ. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 12.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 13.Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 14.Raetz CRH, Dowhan W. J Biol Chem. 1990;265:1235–1238. [PubMed] [Google Scholar]
- 15.Raetz CRH, Whitfield C. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loppnow H, Brade H, Dürrbaum I, Dinarello CA, Kusumoto S, Rietschel ET, Flad HD. J Immunol. 1989;142:3229–3238. [PubMed] [Google Scholar]
- 17.Steeghs L, Berns M, ten Hove J, de Jong A, Roholl P, van Alphen L, Tommassen J, van der Ley P. Cell Microbiol. 2002;4:599–611. doi: 10.1046/j.1462-5822.2002.00214.x. [DOI] [PubMed] [Google Scholar]
- 18.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2001;276:43122–43131. doi: 10.1074/jbc.M106961200. [DOI] [PubMed] [Google Scholar]
- 19.Lee H, Hsu FF, Turk J, Groisman EA. J Bacteriol. 2004;186:4124–4133. doi: 10.1128/JB.186.13.4124-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trent MS, Raetz CRH. J Endotoxin Res. 2002;8:158. [Google Scholar]
- 21.Baker SJ, Gunn JS, Morona R. Microbiology. 1999;145:367–378. doi: 10.1099/13500872-145-2-367. [DOI] [PubMed] [Google Scholar]
- 22.Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI. Infect Immun. 2000;68:6139–6146. doi: 10.1128/iai.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helander IM, Kato Y, Kilpelainen I, Kostiainen R, Lindner B, Nummila K, Sugiyama T, Yokochi T. Eur J Biochem. 1996;237:272–278. doi: 10.1111/j.1432-1033.1996.0272n.x. [DOI] [PubMed] [Google Scholar]
- 24.Vaara M, Vaara T, Jensen M, Helander I, Nurminen M, Rietschel ET, Makela PH. FEBS Lett. 1981;129:145–149. doi: 10.1016/0014-5793(81)80777-6. [DOI] [PubMed] [Google Scholar]
- 25.Gibbons HS, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2000;275:32940–32949. doi: 10.1074/jbc.M005779200. [DOI] [PubMed] [Google Scholar]
- 26.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CRH. EMBO J. 2000;19:5071–5080. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trent MS, Pabich W, Raetz CRH, Miller SI. J Biol Chem. 2001;276:9083–9092. doi: 10.1074/jbc.M010730200. [DOI] [PubMed] [Google Scholar]
- 28.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 29.Sambrook JG, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd. Cold Spring Harbor; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 30.Inoue H, Nojima H, Okayama H. Gene (Amst) 1990;96:23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- 31.Shizuya H, Birren B, Kim UJ, Mancino V, Slepak T, Tachiiri Y, Simon M. Proc Natl Acad Sci U S A. 1992;89:8794–8797. doi: 10.1073/pnas.89.18.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reynolds CM, Kalb SR, Cotter RJ, Raetz CRH. J Biol Chem. 2005;280:21202–21211. doi: 10.1074/jbc.M500964200. [DOI] [PubMed] [Google Scholar]
- 33.Wang RF, Kushner SR. Gene (Amst) 1991;100:195–199. [PubMed] [Google Scholar]
- 34.Garrett TA, Kadrmas JL, Raetz CRH. J Biol Chem. 1997;272:21855–21864. doi: 10.1074/jbc.272.35.21855. [DOI] [PubMed] [Google Scholar]
- 35.Basu SS, York JD, Raetz CRH. J Biol Chem. 1999;274:11139–11149. doi: 10.1074/jbc.274.16.11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belunis CJ, Raetz CRH. J Biol Chem. 1992;267:9988–9997. [PubMed] [Google Scholar]
- 37.Vorachek-Warren MK, Ramirez S, Cotter RJ, Raetz CRH. J Biol Chem. 2002;277:14194–14205. doi: 10.1074/jbc.M200409200. [DOI] [PubMed] [Google Scholar]
- 38.Odegaard TJ, Kaltashov IA, Cotter RJ, Steeghs L, van der Ley P, Khan S, Maskell DJ, Raetz CRH. J Biol Chem. 1997;272:19688–19696. doi: 10.1074/jbc.272.32.19688. [DOI] [PubMed] [Google Scholar]
- 39.Zhou Z, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 1999;274:18503–18514. doi: 10.1074/jbc.274.26.18503. [DOI] [PubMed] [Google Scholar]
- 40.Brozek KA, Hosaka K, Robertson AD, Raetz CRH. J Biol Chem. 1989;264:6956–6966. [PubMed] [Google Scholar]
- 41.Brabetz W, Muller-Loennies S, Holst O, Brade H. Eur J Biochem. 1997;247:716–724. doi: 10.1111/j.1432-1033.1997.00716.x. [DOI] [PubMed] [Google Scholar]
- 42.Doerrler WT, Raetz CRH. J Biol Chem. 2002;277:36697–36705. doi: 10.1074/jbc.M205857200. [DOI] [PubMed] [Google Scholar]
- 43.Bligh EG, Dyer JJ. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 44.Kanipes MI, Lin S, Cotter RJ, Raetz CRH. J Biol Chem. 2001;276:1156–1163. doi: 10.1074/jbc.M009019200. [DOI] [PubMed] [Google Scholar]
- 45.Guy-Caffey JK, Rapoza MP, Jolley KA, Webster RE. J Bacteriol. 1992;174:2460–2465. doi: 10.1128/jb.174.8.2460-2465.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Osborn MJ, Munson R. Methods Enzymol. 1974;31:642–653. doi: 10.1016/0076-6879(74)31070-1. [DOI] [PubMed] [Google Scholar]
- 47.Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CRH. J Biol Chem. 1998;273:12466–12475. doi: 10.1074/jbc.273.20.12466. [DOI] [PubMed] [Google Scholar]
- 48.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 49.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 50.Ribeiro AA, Zhou Z, Raetz CRH. Magn Res Chem. 1999;37:620–630. [Google Scholar]
- 51.Zhou Z, Ribeiro AA, Raetz CRH. J Biol Chem. 2000;275:13542–13551. doi: 10.1074/jbc.275.18.13542. [DOI] [PubMed] [Google Scholar]
- 52.Costello CE, Vath JE. Methods in Enzymology. 1990;193:738–768. doi: 10.1016/0076-6879(90)93448-t. [DOI] [PubMed] [Google Scholar]
- 53.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 54.Nielsen H, Brunak S, von Heijne G. Protein Eng. 1999;12:3–9. doi: 10.1093/protein/12.1.3. [DOI] [PubMed] [Google Scholar]
- 55.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moran AP, Lindner B, Walsh EJ. J Bacteriol. 1997;179:6453–6463. doi: 10.1128/jb.179.20.6453-6463.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rebeil R, Ernst RK, Gowen BB, Miller SI, Hinnebusch BJ. Mol Microbiol. 2004;52:1363–1373. doi: 10.1111/j.1365-2958.2004.04059.x. [DOI] [PubMed] [Google Scholar]
- 58.Gibbons HS, Kalb SR, Cotter RJ, Raetz CRH. Mol Microbiol. 2005;55:425–440. doi: 10.1111/j.1365-2958.2004.04409.x. [DOI] [PubMed] [Google Scholar]
- 59.Gunn JS, Miller SI. J Bacteriol. 1996;178:6857–6864. doi: 10.1128/jb.178.23.6857-6864.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, Hackett M, Miller SI. Science. 1997;276:250–253. doi: 10.1126/science.276.5310.250. [DOI] [PubMed] [Google Scholar]
- 61.Wosten MM, Kox LF, Chamnongpol S, Soncini FC, Groisman EA. Cell. 2000;103:113–125. doi: 10.1016/s0092-8674(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 62.Garcia Vescovi E, Soncini FC, Groisman EA. Cell. 1996;84:165–174. doi: 10.1016/s0092-8674(00)81003-x. [DOI] [PubMed] [Google Scholar]
- 63.Miller SI, Kukral AM, Mekalanos JJ. Proc Natl Acad Sci U S A. 1989;86:5054–5058. doi: 10.1073/pnas.86.13.5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Groisman EA, Chiao E, Lipps CJ, Heffron F. Proc Natl Acad Sci U S A. 1989;86:7077–7081. doi: 10.1073/pnas.86.18.7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Groisman EA, Kayser J, Soncini FC. J Bacteriol. 1997;179:7040–7045. doi: 10.1128/jb.179.22.7040-7045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI. Mol Microbiol. 1998;27:1171–1182. doi: 10.1046/j.1365-2958.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- 67.Gunn JS. J Endotoxin Res. 2001;7:57–62. [PubMed] [Google Scholar]
- 68.Kawasaki K, Ernst RK, Miller SI. J Endotoxin Res. 2004;10:439–444. doi: 10.1179/096805104225006264. [DOI] [PubMed] [Google Scholar]
- 69.Kawasaki K, Ernst RK, Miller SI. J Biol Chem. 2004;279:20044–20048. doi: 10.1074/jbc.M401275200. [DOI] [PubMed] [Google Scholar]
- 70.Kawasaki K, Ernst RK, Miller SI. J Endotoxin Res. 2005;11:57–61. doi: 10.1179/096805105225006696. [DOI] [PubMed] [Google Scholar]
- 71.Vinogradov E, Perry MB, Conlan JW. Eur J Biochem. 2002;269:6112–6118. doi: 10.1046/j.1432-1033.2002.03321.x. [DOI] [PubMed] [Google Scholar]
- 72.Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, Howald WN, Way SS, Hajjar AM. Infect Immun. 2004;72:5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kawasaki K, Ernst RK, Miller SI. J Bacteriol. 2005;187:2448–2457. doi: 10.1128/JB.187.7.2448-2457.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dekker N, Tommassen J, Lustig A, Rosenbusch JP, Verheij HM. J Biol Chem. 1997;272:3179–3184. doi: 10.1074/jbc.272.6.3179. [DOI] [PubMed] [Google Scholar]
- 75.Dekker N. Mol Microbiol. 2000;35:711–717. doi: 10.1046/j.1365-2958.2000.01775.x. [DOI] [PubMed] [Google Scholar]
- 76.Snijder HJ, Kingma RL, Kalk KH, Dekker N, Egmond MR, Dijkstra BW. J Mol Biol. 2001;309:477–489. doi: 10.1006/jmbi.2001.4675. [DOI] [PubMed] [Google Scholar]
- 77.Hagen FS, Grant FJ, Kuijper JL, Slaughter CA, Moomaw CR, Orth K, O'Hara PJ, Munford RS. Biochemistry. 1991;30:8415–8423. doi: 10.1021/bi00098a020. [DOI] [PubMed] [Google Scholar]
- 78.Erwin AL, Munford RS. J Biol Chem. 1990;265:16444–16449. [PubMed] [Google Scholar]
- 79.Ahn VE, Lo EI, Engel CK, Chen L, Hwang PM, Kay LE, Bishop RE, Prive GG. EMBO J. 2004;23:2931–2941. doi: 10.1038/sj.emboj.7600320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Buchmeier N, Bossie S, Chen CY, Fang FC, Guiney DG, Libby SJ. Infect Immun. 1997;65:3725–3730. doi: 10.1128/iai.65.9.3725-3730.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ludwig A, Tengel C, Bauer S, Bubert A, Benz R, Mollenkopf HJ, Goebel W. Mol Gen Genet. 1995;249:474–486. doi: 10.1007/BF00290573. [DOI] [PubMed] [Google Scholar]
- 82.Navarre WW, Halsey TA, Walthers D, Frye J, McClelland M, Potter JL, Kenney LJ, Gunn JS, Fang FC, Libby SJ. Mol Microbiol. 2005;56:492–508. doi: 10.1111/j.1365-2958.2005.04553.x. [DOI] [PubMed] [Google Scholar]
- 83.Miller SI, Mekalanos JJ. J Bacteriol. 1990;172:2485–2490. doi: 10.1128/jb.172.5.2485-2490.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carlson SA, McCuddin ZP, Wu MT. Microb Pathog. 2005;38:181–187. doi: 10.1016/j.micpath.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 85.Shi Y, Latifi T, Cromie MJ, Groisman EA. J Biol Chem. 2004;279:38618–38625. doi: 10.1074/jbc.M406149200. [DOI] [PubMed] [Google Scholar]
- 86.Stapleton MR, Norte VA, Read RC, Green J. J Biol Chem. 2002;277:17630–17637. doi: 10.1074/jbc.M110178200. [DOI] [PubMed] [Google Scholar]
- 87.Watson PR, Paulin SM, Bland AP, Libby SJ, Jones PW, Wallis TS. Infect Immun. 1999;67:4950–4954. doi: 10.1128/iai.67.9.4950-4954.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kaneko A, Mita M, Sekiya K, Matsui H, Kawahara K, Danbara H. Microbiol Immunol. 2002;46:109–113. doi: 10.1111/j.1348-0421.2002.tb02666.x. [DOI] [PubMed] [Google Scholar]
- 89.Suda Y, Kim YM, Ogawa T, Yasui N, Hasegawa Y, Kashihara W, Shimoyama T, Aoyama K, Nagata K, Tamura T, Kusumoto S. J Endotoxin Res. 2001;7:95–104. [PubMed] [Google Scholar]