

Abstract
Von Willebrand factor (VWF) is a large multimeric glycoprotein produced in endothelial cells and megakaryocytes and present in subendothelial matrix, blood plasma and platelets. VWF mediates adhesion and aggregation of platelets at sites of vascular injury, processes that are critical for both haemostasis and thrombosis. Thrombus formation involves complex events that are influenced by different environmental conditions. Progress in understanding the structure and function of VWF and the mechanisms that underlie its interactions with platelets has led to important insight into the differentiation between normal haemostasis and pathological arterial thrombosis. The conventional view of signalling-induced platelet aggregation has recently been extended to include activation-independent aggregation. A novel mechanism has been demonstrated for initiating thrombus formation under high haemodynamic forces that involves aIIbβ3-independent platelet aggregation at the interface between immobilised and soluble VWF. This VWF-mediated process may be a key determinant of platelet accumulation in stenotic arteries leading to acute thrombotic occlusion.
Keywords: Shear stress, Platelet adhesion, Platelet activation, Platelet aggregation, Thrombus formation, von Willebrand factor, VWF
Introduction
Mature von Willebrand factor (VWF) is a multimeric protein composed of a variable number of identical subunits, each consisting of 2050 amino acid residues and up to 22 carbohydrate side chains [1]. Subunits are disulphide-bonded into dimers of approximately 500 kilodaltons, which in turn are disulphide-linked into multimers of increasing size that can reach a molecular mass as high as 20 million Daltons. Our understanding of the structure and function of VWF and the mechanisms that underlie its involvement in haemostasis are developing constantly. The essential physiological functions of VWF have been elucidated, including binding and transportation of the pro-coagulant factor VIII (FVIII), mediating platelet adhesion to reactive surfaces, and mediating platelet aggregation and thrombus growth. Discrete domains within the VWF subunit exhibit specific functions that have been defined [2]. The major functional domains of VWF are A1, which contains the only binding site for the platelet receptor glycoprotein (GP) Iba, A3, through which VWF binds to collagen, C1, which contains the RGD sequence recognized by the β3 integrins (aIIbβ3 and avβ3) and D′–D3, which form the site that binds FVIII [2].
Many details regarding the biosynthesis and secretion of VWF are well established [3]. VWF is synthesised exclusively in endothelial cells and megakaryocytes. Following synthesis, the VWF produced in endothelial cells is secreted via one of two distinct pathways: a constitutive pathway directly linked to synthesis (i.e. molecules are released as soon as their synthesis is completed), and a regulated pathway involving storage of mature molecules for release after stimulation by secretagogues [3]. Because released VWF undergoes a process of regulated reduction of multimer size, the availability of a source of uncleaved larger multimers from cellular storage sites permits maximal function in areas where rapid platelet adhesion and aggregation is required. The storage organelles for VWF that are found in endothelial cells and megakaryocytes are the rod-shaped Weibel-Palade bodies and the a-granules, respectively [3, 4]. In megakaryocyte-derived circulating platelets, only the regulated pathway of VWF secretion is effectively operative in vivo.
Therefore, the VWF circulating in plasma is essentially all of endothelial cell origin, as platelets release their a-granule content only when activated [6]. The VWF secreted from endothelial cells, through either the constitutive or regulated pathway, is directed towards both the lumen and the subendothelial matrix.
Von Willebrand factor is initially uncleaved, and the physiological reduction in the size of the multimers occurs through a controlled proteolytic cleavage event. The metalloproteinase, ADAMTS13 [7], cleaves specifically the Tyr842–Met843 peptide bond [8]. This cleavage has effects on the size of circulating multimers and consequently modulates proadhesive functions [9]. The largest VWF multimers display enhanced thrombogenic functions, possibly because multiple interactive sites for vessel wall components and platelets support more efficient adhesion. Thus, the controlled release of the largest multimers at the time of injury allows their presence at sites of tissue damage, and physiological regulatory mechanisms cause their disappearance from the circulation, possibly to prevent excessive thrombus formation [2].
In the present review article, recent advances in the role of VWF in the complex events of platelet thrombus formation are reviewed, with an emphasis on the processes influenced by shear rates occurring in pathological conditions.
The role of VWF in platelet adhesion and aggregation
VWF works to support thrombus formation not only with respect to maintaining platelet adhesion to sites of injury but also as platelet-platelet cohesion or aggregation. Platelets respond rapidly to alterations of endothelial cells by attaching firmly to the site of lesion where exposure of subendothelial components may have occurred. The first layer of platelets is in contact with the thrombogenic surface (adhesion), whereas subsequent growth of the haemostatic plug depends on platelet-to-platelet interactions (aggregation). Both aspects of platelet function are influenced by VWF interactions with specific platelet membrane receptors. Multiple domains of VWF are involved in securing initiation and growth of platelet plugs.
Mechanisms of platelet adhesion
In a flow field with shear rate above a threshold value (in human circulation around 1000 s−1), only GP Iba interaction with immobilised VWF multimers (for example bound to collagen) can initiate the tethering of circulating platelets to the vessel wall (Fig. 1) [10]. The binding of GP Iba to the A1 domain of VWF occurs rapidly and is the essential adhesive interaction that can tether platelets to a surface when the shear rate is elevated. This interaction supports initially transient bonds, and translocation of the tethered platelets may occur. Activation of the integrin aIIbβ3 occurs during the transient tethering, mainly through signalling initiated by membrane receptors that bind collagen or other components of exposed thrombogenic surfaces or respond to stimulation by agonists released (e.g. ADP) or generated (e.g. a-thrombin) locally. The final result is the stable adhesion of recruited platelets to the surface. At lower shear rates, the adhesive functions of VWF are no longer indispensable for the initial attachment to a thrombogenic surface, and collagen receptors (among others) can permit stable adhesive interactions to form rapidly [2, 10].
Figure 1.
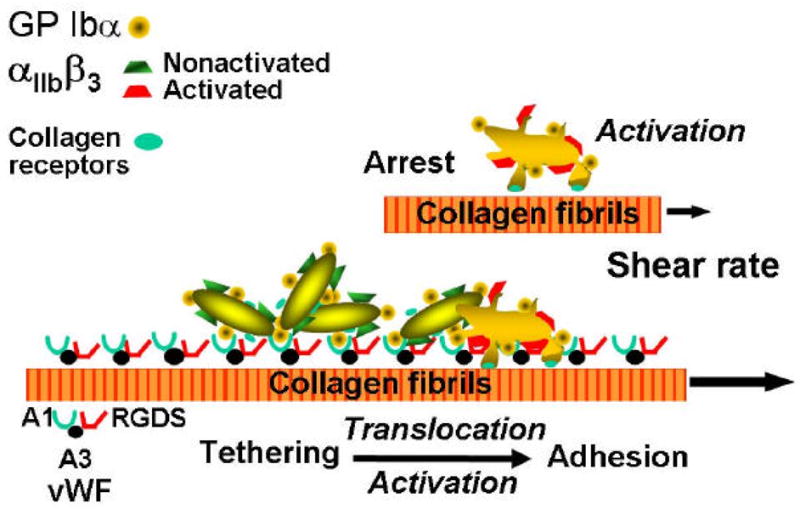
Schematic representation of the mechanisms of platelet adhesion in flowing blood. At the time of vascular injury, particularly in vessels of high shear stress, the platelets initially adhere transiently to subendothelial von Willebrand factor (VWF) through the glycoprotein (GP) Ib receptor. This contact significantly slows the movement of the platelets and leads to transient arrests, which then allow the engagement of other adhesive receptors, such as the collagen receptors shown here, resulting in a stable attachment. Modified from [4].
Mechanisms of platelet aggregation
The first layer of activated platelets that are firmly attached to a reactive surface becomes the substrate for accumulation of more platelets and thrombus growth (Fig. 2) [10]. Adhesive ligands, mainly fibrinogen and VWF, bind via activated aIIbβ3 on the membrane of the adherent platelets and become the substrate for the additional recruitment and attachment of incoming platelets. In this phase of thrombus growth (or platelet aggregation), the shear rate at the surface of the growing thrombus imposes the same constraint on the function of adhesive bonds as during the initial platelet adhesion to the thrombogenic surface. Therefore, when the shear rate is elevated, the bridging effect of fibrinogen, which is required to stabilise the platelet plug, occurs only after the initial tethering of platelets to one another through the interaction of VWF and GP Iba [2, 9]
Figure 2.
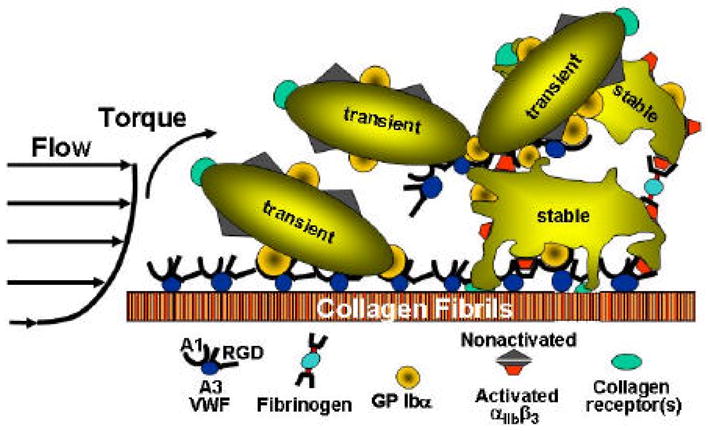
Schematic representation of the mechanisms of platelet aggregation in flowing blood. After stable adhesion, platelets are activated, secrete the contents of their granules and bind adhesive proteins from plasma, such as fibrinogen and von Willebrand factor (VWF), which form the substrate onto which additional platelets are recruited to form a thrombus. Modified from [10].
Influence of shear rate on thrombus formation
The concept that VWF is required for thrombus formation on collagen when the shear rate is high, but not necessarily when it is low, can be demonstrated in real time perfusion studies [10]. In one such experiment, thrombi formed when control blood was perfused over collagen type 1 fibrils at shear rates of 500 s−1 and 1500 s−1 (Fig. 3) [10]. However, when the same blood was treated before perfusion with an anti-VWF A3 antibody that blocks binding to collagen, thrombus formation was abolished when the shear rate was 1500 s−1 but minimally affected when the shear rate was 500 s−1 [10].
Figure 3.

Single frames from a real-time recording of perfusion studies demonstrating how inhibiting von Willebrand factor (VWF) binding to collagen using a monoclonal antibody against the VWF A3 domain obliterates thrombus formation when the wall shear rate is 1500 s−1 but not 500 s−1. Adapted from [10].
As previously mentioned, the complex interactions during a normal haemostatic response are influenced by the flow of blood. The velocity of blood near the vessel wall is lower than at the centre of the vessel, and this difference creates a shearing effect between adjacent layers of fluid that is greatest at the luminal surface. Shear rate – a difference in flow velocity as a function of distance from the wall – is expressed in cm/second per cm (cm·s−1·cm−1) or the equivalent inverse seconds (s−1). The highest wall shear rate in the normal circulation occurs in small arterioles of 10–50 μm diameter where levels have been estimated to vary between 500 and 5000 s−1 [2]. However, shear rates are considerably higher in stenosed arteries. For example, a 90% lumen reduction in a coronary artery may cause shear rates of 20,000–40,000 s−1 at, or just upstream of the stenosis [11].
Platelet adhesion and aggregation under elevated shear stress
Haemostasis and pathological arterial occlusion occur in distinct hydrodynamic environments. While platelet aggregation is conventionally thought to initiate after signalling-induced activation, it now appears that additional mechanisms can operate under blood flow conditions comparable to those existing in stenotic coronary arteries.
Distinct platelet adhesion and aggregation mechanisms as a function of shear rate
A key function of VWF is to initiate platelet aggregation under elevated shear stress conditions, independent of activation [11]. This form of aggregation may precede, and is necessary for, stable adhesion to thrombogenic surfaces. We have recently demonstrated platelet aggregation independent of activation [11]. Thrombus formation was examined using model reactive surfaces composed of collagen type I fibrils or immobilised VWF in a flow chamber perfused with blood containing an a-thrombin inhibitor as the anticoagulant. Real-time visualisation was used to examine platelet interactions with the substrates and with one another. After 7 seconds of flow, single platelets adhered and aggregated one by one when the wall shear rate was 3000 s−1. However, when the shear rate was 24,000 s−1, groups of platelets had already aggregated even before stable adhesion had taken place. Thus, under high shear-stress conditions, a process of platelet cohesion (aggregation) appears to link platelets even before stable adhesion is established. This process is actually what makes adhesion possible under these conditions, otherwise the flow rates are such that the resulting hydrodynamic forces would be sweeping the surface and not allowing the platelets to attach there.
Evaluation of activation-independent platelet adhesion and aggregation
To enable a better visualisation of the process of activation-independent adhesion and aggregation, we prevented platelet activation, which would normally be nearly instantaneous following adhesion, by adding prostaglandin (PG) E1 to the blood, and prevented ligand binding to integrins by adding EDTA. Single platelets adhered to immobilised VWF at a shear rate of 3000 s−1, but at 24,000 s−1 rolling platelets aggregated within 100–200 μm from the boundary where immobilised VWF was exposed to blood, which is equivalent to a few seconds from the initial surface contact (Fig. 4A). The formation of these aggregates was strictly dependent on the presence of soluble VWF multimers in addition to surface-immobilized VWF (Fig. 4B, C) [11].
Figure 4.
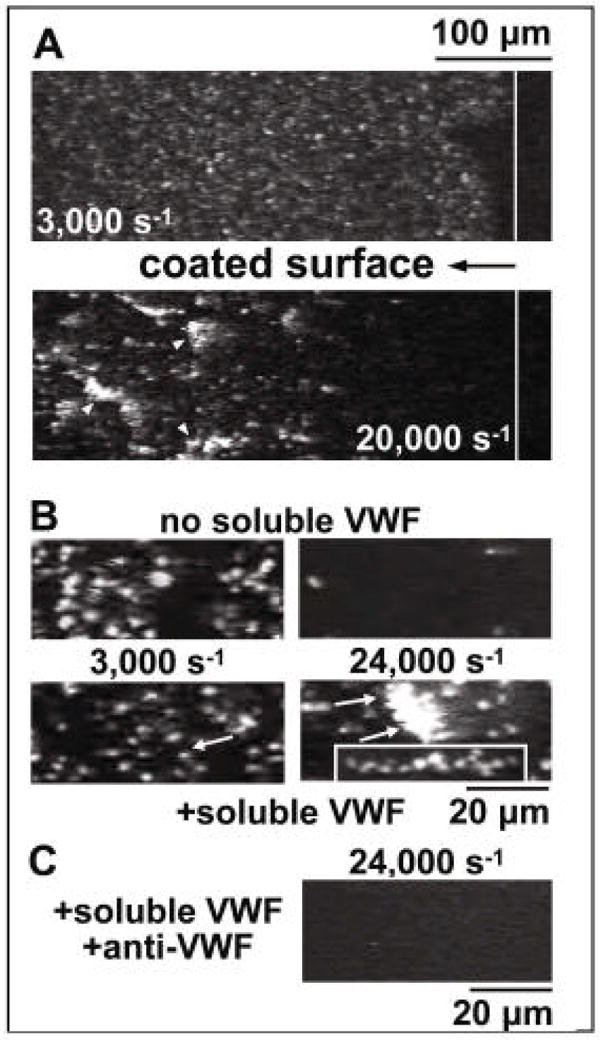
Activation-independent platelet aggregation at the interface of immobilised and soluble VWF. A. Blood containing PPACK (anticoagulant), mepacrine (fluorescent dye), prostaglandin E1 (platelet inactivator) and EDTA (to prevent ligand binding to integrins) was perfused over immobilised von Willebrand factor (VWF). The vertical solid white line distinguishes VWF-coated (left) from uncoated (right) glass. Upper A panel illustrates single platelet adhesion at a shear rate of 3,000 s−1. Lower A panel illustrates rolling aggregates at a shear rate of 20,000 s−1. B. Perfusion over immobilised VWF of washed blood cells. Upper B panels illustrate the results in the absence of added purified VWF, i.e. efficient single platelet adhesion at a shear rate of 3,000 s−1 (left) and adhesion of only a few single platelets at 24,000 s−1 (right). Lower B panels illustrate the results after addition of purified VWF, i.e. single platelet adhesion at a shear rate of 3,000 s−1 (left), but formation of adhering aggregates at 24,000/s (right). The lower right-hand box of panel B illustrates a stretched aggregate during stationary adhesion. C. Perfusion over immobilised VWF of washed blood cells in the presence of added soluble VWF and anti-VWF A1 domain monoclonal antibody [11]. This research was originally published in Blood. Ruggeri ZM, Orje JN, Habermann R, Federici AB, Reininger AJ. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood 2006;108:1903–10. © the American Society of Hematology.
Activation-independent platelet aggregation in vivo
To evaluate whether VWF-mediated platelet aggregation occurs within the vasculature in vivo, we performed experiments in the mesenteric circulation of anaesthetised mice [11]. Platelets that had been treated with PGE1 were labelled with a red fluorochrome and non-treated platelets were labelled with a green fluorochrome. Platelets were then injected back concurrently into the mouse and their accumulation monitored at a site of injury. The green-labelled platelets formed thrombi and attached in a stable manner. In contrast, however, the red-labelled platelets, which could not be activated, linked to one another (aggregated) and also attached to the platelets that were already activated, but not in a stable manner. Therefore, this is an aggregation process that precedes activation, but develops only under very high flow conditions such as may occur in areas where the vascular lumen is restricted by a growing thrombus.
Conclusions
Pathological arterial blood flow generates fluid shear stresses that directly cause platelets to aggregate. At shear rates up to 10,000 s−1, initial adhesion to a reactive substance and subsequent aggregation follow the generally accepted pattern of progressive accrual of single platelets. However, the studies described here have shown that at shear rates greater than 10,000 s−1, activation-independent platelet aggregation mediated by soluble VWF facilitates adhesion and precedes stable aggregation. We have to think that VWF, in addition to being essential for the initial adhesion, is also crucial for linking platelets to one another. In other words, this molecule acts to hold platelets together transiently until activation makes all these events stable. But without it, under high flow conditions, the process could not even begin. And that is why VWF is absolutely essential in thrombus formation, haemostasis and pathological thrombosis.
Acknowledgments
The author wishes to acknowledge the contribution of Dr Sandra Cox who provided writing assistance in the preparation of this manuscript, which is part of a proceedings supplement from a CSL-Behring-sponsored symposium in which the author received an honorarium.
Role of the funding source
The original work referred to in this article was supported by grants from the National Institutes of Health of the United States (National Heart, Lung and Blood Institute grants HL-31950, HL-42846, HL-75736, HL-78784 and HL-80718).
Footnotes
Conflict of interest
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Titani K, Kumar S, Takio K, Ericsson LH, Wade RD, Ashida K, et al. Amino acid sequence of human von Willebrand factor. Biochemistry. 1986;25:3171–84. doi: 10.1021/bi00359a015. [DOI] [PubMed] [Google Scholar]
- 2.Mendolicchio GL, Ruggeri ZM. New perspectives on von Willebrand factor functions in hemostasis and thrombosis. Semin Hematol. 2005;42:5–14. doi: 10.1053/j.seminhematol.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Ruggeri ZM. Structure and function of von Willebrand factor. Thromb Haemost. 1999;82:576–84. [PubMed] [Google Scholar]
- 4.Wagner DD, Olmsted JB, Marder VJ. Immunolocalization of von Willebrand protein in Weibel-Palade bodies of human endothelial cells. J Cell Biol. 1982;95:355–60. doi: 10.1083/jcb.95.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowie EJ, Solberg LA, Jr, Fass DN, Johnson CM, Knutson GJ, Stewart ML, et al. Transplantation of normal bone marrow into a pig with severe von Willebrand’s disease. J Clin Invest. 1986;78:26–30. doi: 10.1172/JCI112560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dent JA, Berkowitz SD, Ware J, Kasper CK, Ruggeri ZM. Identification of a cleavage site directing the immunochemical detection of molecular abnormalities in type IIA von Willebrand factor. Proc Natl Acad Sci USA. 1990;87:6306–10. doi: 10.1073/pnas.87.16.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–94. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 8.Dent JA, Galbusera M, Ruggeri ZM. Heterogeneity of plasma von Willebrand factor multimers resulting from proteolysis of the constituent subunit. J Clin Invest. 1991;88:774–82. doi: 10.1172/JCI115376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruggeri ZM. Platelet and von Willebrand factor interactions at the vessel wall. Hamostasiologie. 2004;24:1–11. doi: 10.1267/hamo04010001. [DOI] [PubMed] [Google Scholar]
- 10.Ruggeri ZM. Old concepts and new developments in the study of platelet aggregation. J Clin Invest. 2000;105:699–701. doi: 10.1172/JCI9604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruggeri ZM, Orje JN, Habermann R, Federici AB, Reininger AJ. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood. 2006;108:1903–10. doi: 10.1182/blood-2006-04-011551. [DOI] [PMC free article] [PubMed] [Google Scholar]