

Summary
Human endothelial nitric oxide synthase (eNOS) is a complex enzyme, requiring binding of calmodulin (CaM) for electron transfer. The prevailing view is that calcium-activated CaM binds eNOS at the canonical binding site located at residues 493-510, which induces a conformational change to facilitate electron transfer. Here we demonstrated that the CaM enhances the rate of electron transfer from NADPH to FAD on a truncated eNOS FAD subdomain (residues 682-1204) purified from baculovirus-infected Sf9 cells, suggesting more complicated regulatory mechanism of CaM on eNOS. Metabolically 35S-labeled CaM overlay on fusion proteins spanning the entire linear sequence of eNOS revealed three positive 35S-CaM binding fragments: sequence 66-205, sequence 460-592, and sequence 505-759. Synthetic peptides derived from these fragments are tested for their effects on CaM binding and eNOS catalytic activities. Peptides corresponding to the proximal heme-binding site (E1, residues 174-193) and the CD1 linker connecting FAD/FMN subdomains (E4, residues 729-757) bind CaM at both high Ca2+ (Ca2+CaM) and low Ca2+ (apoCaM) concentrations, whereas peptide of the canonical CaM-binding helix (E2, residues 493- 510) binds only Ca2+CaM. All three peptides E1, E2 and E4 significantly inhibit oxygenase activity in a concentration-dependent manner, but only E2 effectively inhibits reductase activity. Concurrent experiments with human iNOS showed major differences in the CaM binding properties between eNOS and iNOS. The results suggest that multiple regions of eNOS might interact with CaM with differential Ca2+ sensitivity in vivo. A possible mechanism in regulating eNOS activation and deactivation is proposed.
Keywords: CaM overlay, eNOS, iNOS, synthetic peptide inhibition, oxygenase activity, apoCaM
Introduction
Nitric oxide synthase (NOS)1 catalyzes the synthesis of nitric oxide (•NO) from L-arginine in a variety of cells (1-4). Mammalian tissues express three structurally related NOS isoforms referred to as the neuronal NOS (nNOS) (5, 6), macrophage inducible NOS (iNOS) (7, 8), and endothelial NOS (eNOS) (9). All NOSs are homodimers, with each monomer containing a C-terminal reductase domain, and an N-terminal oxygenase domain (10-12). The reductase domain harbors binding sites for NADPH, FAD and FMN cofactors, while the oxygenase domain binds protoporphrin IX heme, tetrahydropterin (H4B) and L-arginine (13). Binding of Ca2+/CaM to a canonical CaM-binding site situated near the junction of oxygenase/reductase domains triggers intradomain electron transfer between flavins as well as interdomain electron transfer from the flavin of one subunit to the heme of adjacent subunit (14-18).
CaM is a ubiquitous Ca2+-binding protein structurally resembling a dumbbell. It contains N-terminal and C-terminal lobes connected by a flexible helical linker (19). Each lobe comprises two E-F hands with two Ca2+ binding sites. The Ca2+ binding sites in the C-terminal lobe (sites 3 and 4) have a higher affinity for Ca2+ than those in the N-terminal lobe (sites 1 and 2). Ca2+ binding triggers conformational changes of CaM, bringing the two lobes together which form a hydrophobic interface to interact with target peptides (20-21). CaM regulates all three NOS isoforms. nNOS and eNOS bind CaM in a Ca2+-dependent manner (9, 22), whereas iNOS binds CaM tightly even at basal cellular Ca2+ concentrations (23). It is generally believed that the CaM-regulated electron flow is mediated through NOS conformational change rather than redox potential change between redox partners (24-25).
Despite extensive studies to elucidate the interaction between CaM and its canonical binding site (26-28), it remains uncertain whether this interaction alone is adequate for the complex CaM-mediated electron transfer among NOS isoforms. Additional contacts are believed to exist. Gachhui et al (29) suggested that CaM's actions on nNOS be completely within the reductase domain, but others indicated that CaM influence the heme domain as well (30-31). Stevens-Truss and coworkers further showed that activations of nNOS and iNOS by CaM require Ca2+, but do not need Ca2+ bound to all four Ca2+-binding sites of CaM. These two isoforms interact with CaM differently and exhibit specific and distinct requirements for Ca2+-bound CaM (32-33). However, study of eNOS activation by CaM is complicated by its attachment to the plasma membrane that affects the Ca2+-dependent eNOS catalysis by multiple cell signaling pathways (34). The exact mechanism of eNOS activation by CaM remains elusive.
Since eNOS and nNOS can reversibly bind CaM in a Ca2+-dependent manner, it has been suggested that CaM maybe associated with nNOS as an inactive complex in cells at resting condition (35). This association would keep CaM available to sense changes in local concentrations of Ca2+ for a rapid and complete response. At an elevated cellular Ca2+ concentration, Ca2+ binds to N- and C-terminal lobes of CaM, thereby inducing conformational changes to facilitate electron transfer and oxygenase activation, while at the basal Ca2+ level, calcium binds only to the high affinity sites at the C-terminal lobe, which allows for CaM binding to eNOS or nNOS without altering its conformation or triggering electron transfer. Thus, a single CaM binding site at the junction between the oxygenase and reductase domains of NOS could not account for the complex binding behavior and function of CaM. We therefore postulated that additional CaM recognition sequences are present on the eNOS molecule that tether CaM to eNOS even at low Ca2+ concentrations. To test this hypothesis, we performed 35S-labeled CaM binding to various eNOS truncated domains and identified two peptide sequences that bound CaM with or without Ca2+ and inhibited eNOS catalytic activity.
Experimental Procedures
Materials
L-[2,3,4,5-3H]arginine (58 Ci/mmol) and Tran 35S-label NO-thaw metabolic labeling reagent were obtained from MP Biochemicals. (6R)-5,6,7,8,-Tetrahydro-L-biopterin was obtained from Research Biochemical International. AG 50W-X8, cation-exchange resin, Bradford protein dye reagent, and electrophoretic chemicals were products of Bio-Rad. Amylose resin and restriction enzymes were from New Englands Biolabs. Peptides were synthesized at the Peptron, Inc. NADPH and other reagents were obtained from Sigma.
FAD subdomain of eNOS
The cDNA fragment of FAD subdomain was generated by PCR method using a pair of primer (5′-2078 CGGCTTCTAGAGATGGGCCAGGGCGACGAG and 5′-3757 GCACCACCTCTAGAGGGGAGG) with human eNOS cDNA as template, and subcloned into the XbaI site of pVL1392 vector. The protein was expressed in a Sf9/baculoviral system and purified by 2′, 5′-ADP Sepharose and AcA34 gel-filtration columns as described previously (36). The purified FAD subdomain (2 μM) was treated with NADPH (20 μM) in a buffer containing 25 mM Tris, pH 7.5, 100 mM NaCl, 10% glycerol with or without CaM (0.5 μM), and then allowed to autoxidize at room temperature. The semiquinone decay and flavin reoxidation were monitored in a Shimadzu-2501 PC for the changes in absorbance at 600 and 485 nm, respectively over time. Concentration-dependent effect of CaM on ferricyanide reduction was evaluated in a reaction mixture containing 25 mM Tris, pH 7.5, 100 mM NaCl, 1 mM ferricyanide, 100-μM β-NADPH, 1 μg of FAD subdomain and varied concentrations of CaM.
Generation and Expression of Fusion Proteins
The specific primers used to amplify the various eNOS fragments by PCR are listed in Table I. Fragments 1, 4, 5, 6, 7 and 8 were cloned into the SalI site; fragments 2 and 3 were cloned into the EcoRI site at a maltose-binding protein vector, pMalc-2× (New England Biolab). The in-frame ligation was confirmed by sequencing at the Lone-Star Labs (Houston, Texas). BL21 (DE3) transformed with these constructs were cultured in 50-ml LB/ampicilin media plus 0.2% glucose and induced with 0.3 mM IPTG at mid-log phase. Bacterial were sonicated 20 s for three times in a buffer containing 20 mM Tris, pH 7.5, 200 mM NaCl, 1 mM EDTA and 1 mM PMSF, then centrifuged at 25,000 × g for 30 min at 4°C. The supernatant was loaded onto a column with amylose resin (1 × 1 cm), washed with 20 volumes of buffer containing 20 mM Tris, pH 7.5 and 200 mM NaCl, and eluted with buffer containing 20 mM Tris, pH 7.5 and 10 mM maltose.
Table I. Fusion proteins of Maltose-Binding Proteins with varied eNOS fragments.
| Fragment | Primers | Amino Acid |
|---|---|---|
| Fr-1 | CAGCCCGTCGACGGGCCCAAGTTCCCTCGT | 66-205 |
| AGATGTAGGTCGACATTTACTGTGCAG | ||
| Fr-2 | CGATGCCCGGGAATTCAGGTCTGCACAG | 203-341 |
| CGGGGAATTCCAGGCCCCCAATTTACAG | ||
| Fr-3 | CCTGGAATTCCCCGCAGCCC | 349-460 |
| ACAGGAAATAGTTGACGAATTCCTAATG | ||
| Fr-4 | CAGGAGATGGTCGACTATTTCCT | 464-592 |
| GTTGTAGGGGGTCGACATCTACATCAG | ||
| Fr-5 | CAACGCCGTCGACATCTCCGCCTCGCT | 505-756 |
| CCTGGTCGACCTACCGCCTGTGCACGTGGATCAG | ||
| Fr-6 | CTGATCCACGTCGACAGGCGGAAG | 755-856 |
| CAGGAAGAAGTCGACAGCCTAGCGCAG | ||
| Fr-7 | CTGCGCCAGGCTGTCGACTTCTTCCTG | 861-1006 |
| CACCAGGATGTCGACCTAGCTGGGATC | ||
| Fr-8 | CTGCCAGTCGACCCCAGCTTGCCCTGC | 1005-1203 |
| AGGGAAAGCCAGTCGACTCTCAGGGGC |
Generation, Expression and Purification of Wild type and Mutant CaMs
Human CaM cDNA was generously provided by Dr. E.E. Strehler. CaM mutant, B1234Q with Glu to Gln substitutions at all four Ca2+ coordinating sites (E32Q, E64Q, E105Q and E141Q) was generated. Wild type and mutant CaM were subcloned into the NdeI and HindIII sites of pT7-7 vector. BL21(DE3) transformed with these constructs were grown and induced with 0.3 mM IPTG. Purification of wild type CaM was carried out essentially as described previously (37). The B1234Q protein was purified with modifications. Bacteria expressing B1234Q were sonicated 30 s for four times in buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, and 1 mM phenylmethylsulfonyl fluoride. The lysate was heated to 70°C, immediately cooled on ice, and centrifuged at 25,000× g for 30 min at 4°C. After gradually bringing the supernatant to a 25% saturation of ammonium sulfate, the precipitate was removed by centrifugation, and the supernatant was then brought to a nearly saturated ammonium sulfate and precipitated. The resuspended solution was applied onto a gel-filtration column (1×120 cm, Ultrogel AcA34) and was eluted with a buffer containing 50 mM MOPS, pH 7.0. The CaM concentration was either determined by Bradford method (38) or estimated spectroscopically using value of Є277 nm =2560 M-1 cm-1.
Expression and Purification of 35S-Labeled CaM
35S-labeled CaM was prepared as described previously (39) with modifications. Briefly, two ml of an overnight culture from a single colony of BL21 (DE3) transformed with CaM/pT7-7 construct was transferred to a 200-ml LB/ampicilin medium, grown at 37°C to mid-log phase, centrifuged, and resuspended in a 200-ml M9/ampicilin medium with 0.3 mM IPTG and 2 mCi of Tran 35S- metabolic labeling reagent, which continued culture overnight at room temperature. The 35S-labeled CaM was purified as described above.
[35S]CaM Overlay assay
Equivalent amount of each fusion proteins was separated on a 10% SDS-PAGE gel and transferred to nitrocellulose membrane. The membranes was first blocked with blocking buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 1 mg/ml ovalbumin, 0.01% Tween-20 and 100 μM CaCl2 for 1 h at room temperature, then incubated with 35S-CaM (106 cpm/ml) in the same buffer for another hour. The membrane was washed with the cold blocking buffer five times (at least 5 min each) at 4°C, air dried and exposed to Biomax MR X-ray film for 7 days.
Gel mobility-shift assay
CaM (200 pmol) was incubated with each peptide at several molar ratios for 1 h in 10 μl of buffer containing 20 mM Tris, pH 7.5 with either 100 μM CaCl2 or1 mM EGTA. The sample was subjected to nondenaturing, non-reducing polyacrylamide gel (18%) electrophoresis (40) at 30 mA under high Ca2+ conditions (100 μM free Ca2+ in all gel buffers) or low Ca2+ conditions (1mM EGTA in all gel buffers). Binding of peptides to CaM was visualized by Coomassie blue R-250 staining.
Fluorescence measurements
To assess whether tryptophan-containing peptides interact with CaM, we monitored the spectrum of intrinsic tryptophan fluorescence of candidate peptides (5 μM each) in the absence or presence of a single equivalent of CaM at buffer containing 20 mM Tris, pH 7.5 with either 100 μM Ca2+ or 1 mM EGTA. Fluorescence data were collected on a Shimadzu RF-5301PC fluorophotometer with 2-nm slit width. Excitation wavelength was set at 290 nm and emission spectrum was recorded from 300 to 450 nm.
Analysis of NOS activity
The steady state of eNOS catalysis was carried out as described previously (36). Concentration-dependent inhibition of eNOS oxygenase by peptides was tested at 37 °C for 5 min by measuring L-[3H]citrulline formation in a mixture containing 25 mM Tris, pH 7.5, 100 mM NaCl, 1 μM calmodulin, 0.1 mM EGTA, 0.1 mM EDTA, 0.5 mM Ca2+, 100 μM β-NADPH, 100 μM H4B, 10 μM L-arginine, 1 μCi of L-[3H]arginine, 50 nM eNOS and varied concentrations of each peptide. Cytochrome c reductase activity was determined at 37 °C in a reaction mixture (500 μl) in the presence or absence of 10 μm of each peptide containing 25 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10% glycerol, 100 μm cytochrome c, 100 μm NADPH, and 30 nm enzyme with or without 0.5 μm calmodulin and 100 μM CaCl2. Ferricyanide reduction was carried out in a reaction mixture similar to that for cytochrome c assay except that 1 mm ferricyanide was used.
Results
Effect of CaM on FAD subdomain
We have purified eNOS FAD subdomain (residues 682-1204) from baculovirus-infected Sf9 cells. Absorbance spectra of the purified FAD subdomain displayed a typical oxidized flavin band at 456-485 nm (Fig. 1A). To evaluate the effect of CaM, FAD subdomain was treated with NADPH in the presence or absence of CaM. Semiquinone formation and flavin reoxidation were monitored over time at 600 nm and 485 nm, respectively. The decay of semiquinone after NADPH addition was much faster for FAD subdomain with CaM than that without CaM (Fig. 1B). The absorbance increase at 485 nm once NADPH had become depleted revealed that flavin reoxidation of FAD subdomain was much faster with CaM than without CaM (Fig. 1C). Concentration-dependent enhancement by CaM on the ferricyanide reduction was also observed (Fig. 1D). Although numerous reductase and oxygenase constructs which lack the canonical CaM-binding site lack CaM sensitivity (10-12, 36), this FAD subdomain lacking the canonical CaM-binding site and FMN subdomain displays CaM-dependent stimulation on the rate of electron transfer from NADPH to FAD, indicating that there are additional CaM recognition sequences situated at the FAD subdomain.
Fig.1. Effects of CaM on the rate of electron transfer of eNOS FAD subdomain.
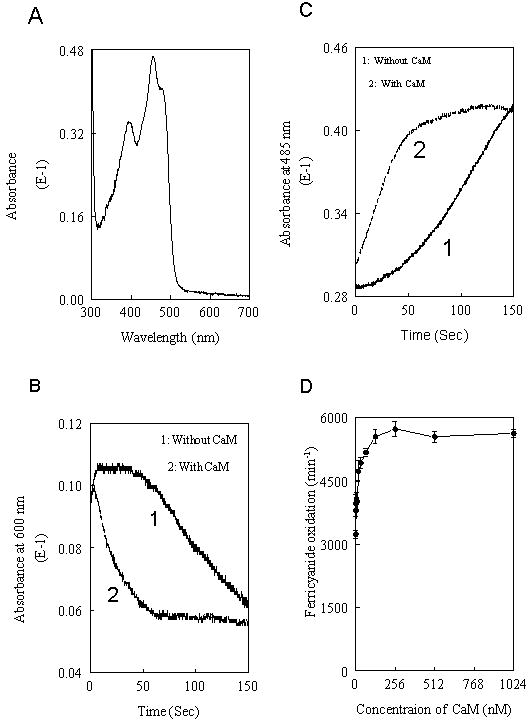
The resting spectrum of 2 μM purified FAD subdomain (Panel A). The formation and decay of semiquinone were monitored at 600 nm after addition of 20 μM NADPH to a solution containing 25 mM Tris, pH 7.5, 100 mM NaCl, 10% glycerol and 2 μM of FAD subdomain with or without 0.5 μM CaM (Panel B). The flavin reoxidation was monitored over time with the absorbance increase at 485 nm once the NADPH had depleted in the mixture of Panel B (Panel C). The concentration-dependent stimulation by CaM on ferricyanide reduction of FAD subdomain was shown in Panel D. Each bar represents mean ± standard deviation of triplicate experiments.
Search for CaM binding sequences by [35S]CaM Overlays
We used 8 fusion proteins (fused to maltose binding protein vector pMALc-2×) spanning the entire eNOS coding sequence except the first 65 amino acids at the N-terminus (Table I) to identify potential CaM binding domains. The fusion proteins partially purified by amylose affinity chromatography were tested for CaM binding by [35S]CaM overlay. To ensure that the protein was loaded at equivalent concentrations, each fusion protein was separated on a SDS-PAGE gel and stained by Coomassie blue R-250. There was no significant difference in the protein level (Fig. 2A). The [35S]CaM overlay analysis showed an expected binding of CaM to fragment 4 (lane 5), which harbors the canonical CaM binding site. Binding was also detected with fragment 1 (sequence 66-205) and fragment 5 (sequence 505-759), which was weaker than that of fragment 4 (Fig. 2B). To confirm this, we searched for CaM binding motifs in eNOS sequence using the proposed 1–8-14 and 1-5-10 rules where numbers refer to the position of hydrophobic residues (41). Five sequences conform to the CaM binding motif rule (Table 2). As expected, E2 (491-510), which harbors the canonical CaM binding site conforms to the 1-8-14 rule. Since fragments 1 and 5 bound 35S-CaM, E1, E3 and E4 could potentially bind CaM. When displayed in a helical wheel plot (42), E1, E2 and E4 have tendency to form basic, amphipathic helices (data not shown) that is typical for CaM recognition sequence.
Fig.2. Identification of putative CaM-binding domains in human eNOS by 35S-CaM overlay.
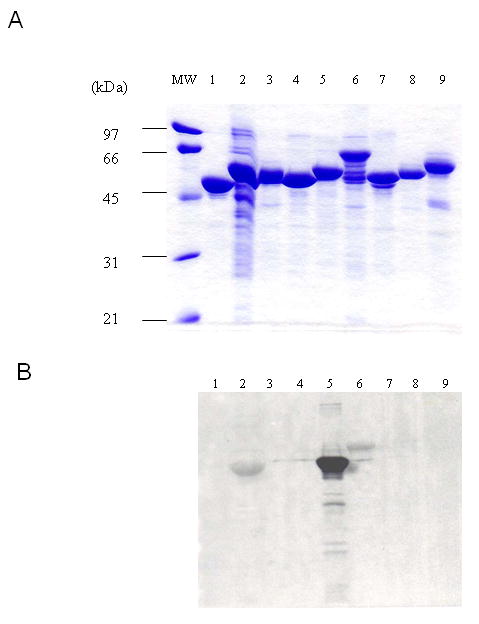
The pMALc-2× fused with varied eNOS fragments as well as pMALc-2× without an insert was expressed in BL21 and the expressed proteins were partially purified by amylose resin. Equivalent amount of each fusion protein was separated by SDS-PAGE and transferred to nitrocellulose membrane. The gel was stained by Coomassie blue R-250 (Panel A). The membrane was overlaid by 35S-CaM and autoradiographed (Panel B). Lane 1, pMALc-2× without an insert; lane 2, pMALc-2× with fragment 1; lane 3, with fragment 2; lane 4, with fragment 3; lane 5, with fragment 4; lane 6, with fragment 5; lane 7, with fragment 6; lane 8, with fragment 7; lane 9, with fragment 8. The amino acid sequences of each fragment are shown in Table I. The size of molecular mass standard is shown at the left in kDa.
Table II. Sequence of synthetic peptides of putative CaM-recognition sites.
| Human eNOS | Residues | Region | |
|---|---|---|---|
| E0 | PKFPRVKNWEVGSITYDTLS | 67-86 | |
| E1 | AKQA1WRNA5PRC8VG10RIQW14GKL | 174-193 | E1 spans the cystein that is the heme ligand |
| E2 | TRKKTF1KEVA5NAV8KISASL14M | 491-510 | E2 is the canonical CaM-binding site |
| E3 | YNSSPRPEQHKSY1KIRF5NSI8SCSDPL14 | 597-622 | E3 is located in the autoregulatory region |
| E4 | KRSWKRQRYRL1SAQA5EGL8QLLPGL14IHVHR | 729-757 | E4 is in the hinge region connecting FMN and FAD domains |
| E5 | VTSRIRTQSF1SLQERQL8RGAVPW14 | 1170-1192 | E5 is in the C-terminal regulatory element |
| Human iNOS | |||
| M1 | TKQA1WRNA5PRC8IG10RIQW14SNL | 190-209 | M1 spans the cystein that is the heme ligand |
| M2 | RRPKRREIPL1KVLV5KAV8LFACML14M | 506-529 | M2 is the canonical CaM-binding site |
| M4 | NVTWDPHHYRL1VQDSQPL8DLSKAL14SSMHA | 701-729 | M4 is in the hinge region connecting FMN and FAD domains |
The sequences of synthetic peptides derived from human eNOS and iNOS are shown, and the hydrophobic residues based on the 1-8-14 rules are indicated
Interaction of E1 and E4 with CaM evaluated by Gel mobility-shift assay
Peptides E1-E5 were synthesized based on the eNOS sequences (Table 2). A peptide sequence corresponding to residues 67-86 (in fragment 1, E0) was also included for comparison. Each peptide at increasing concentrations (0-2000 pmol) was incubated with purified CaM at a fixed concentration (200 pmol) in the presence of 100 μM Ca2+ or 1 mM EGTA, and the mixtures were electrophoresed on nonreducing, nondenaturing gels. Peptide-CaM complexes and free CaM were evaluated. Free peptides are not detected because all six peptides are positively charged at neutral pH that will have an upward mobility toward cathode and do not enter the gel. Most peptide-CaM complexes migrate as higher molecular weight bands than free CaM at high Ca2+, while at low Ca2+, CaM-peptide complex does not move as a discrete band. It has been suggested that the extent of CaM-peptide interaction is more reliably assessed by measuring attenuation of free CaM bands at increasing peptide to CaM molar ratios (43-44). As shown in a representative set of gels, in the presence of 100 μM Ca2+, the canonical E2 peptide exhibited increases in CaM-peptide complex bands and reciprocal attenuation of free CaM bands as the peptide/CaM molar ratios increased (Fig. 3A). E1 and E4 also exhibited CaM binding but the affinity was lower than that of E2. Neither E3 nor E5 had detectable binding activity, despite the 1-8-14 motif in these two peptides. The control peptide (E0) did not have detectable binding activity. In the presence of EGTA, the canonical E2 had no detectable binding to CaM while E1 and E4 retained the binding activity at high peptide/CaM molar ratios (Fig. 3B). The results suggest that contrast to requirement of a high Ca2+ concentration for binding of CaM to canonical site; two non-canonical peptides can bind CaM at the low Ca2+ concentration.
Fig.3. The interaction of peptides based on eNOS sequence with CaM determined by gel mobility shift.
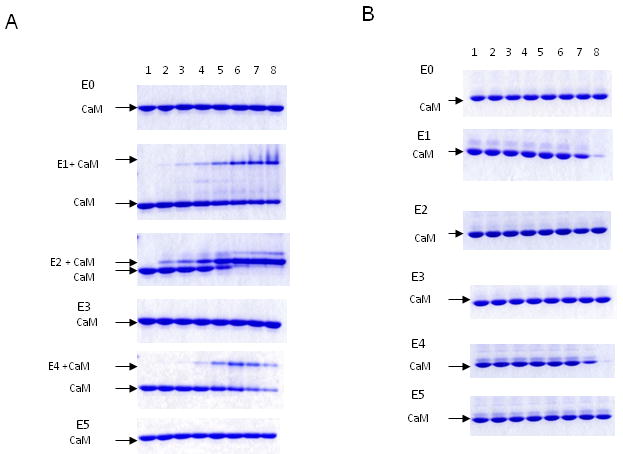
CaM (200 pmole) was incubated with each peptide (E0, E1, E2, E3, E4, E5) by increasing CaM:peptide molar ratio for 1 h. The samples were electrophoresed on 18% nondenaturing gels in the presence of 100 μM Ca2+ (Panel A), or 1 mM EGTA (Panel B). The free CaM and CaM/peptide complex were visualized by Coomassie-blue R250 and indicated. The lane 1 in each gel contains CaM alone. CaM:peptide ratios used were: lane 2 (1:0.15), lane 3 (1:0.3), lane 4 (1:0.5), lane 5 (1:1), lane 6 (1:2), lane 7 (1: 5) and lane 8 (1:10).
Parallel experiments with iNOS
We synthesized M1, M2 and M4 peptides based on iNOS sequences corresponding to those of E1, E2 and E4 in eNOS respectively (Table 2) and compared their CaM binding property. As expected, the canonical CaM binding peptide M2 bound CaM with high affinity at high or low Ca2+ concentration (27). It is interesting to note that multiple bands of M2-CaM complex were detected in the presence of 100 μM Ca2+, suggesting that each Ca2+/CaM binds more than one M2 peptide. M1 was also able to bind CaM in the presence or absence of Ca2+. M4 had no detectable binding activity in the presence of Ca2+ or EGTA (Fig. 4A & 4B).
Fig.4. The interaction of peptides derived from human iNOS with CaM evaluated by gel mobility shift.
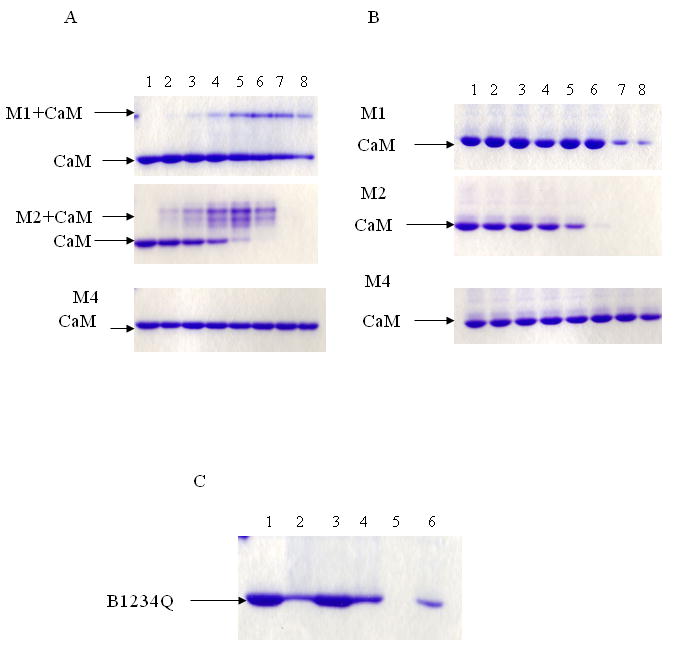
CaM (200 pmole) was incubated with peptide in increasing CaM:peptide molar ratio for 1 h. The sample was electrophoresed on 18% nondenaturing gels with either 100 μM Ca2+ (Panel A), or 1 mM EGTA (Panel B). The Coomassie-blue stained gels containing CaM and increasing molar ratios of M1, M2, and M4 are shown. The lane 1 in each gel contains CaM alone. CaM:peptide ratios used were: lane 2 (1:0.15), lane 3 (1:0.3), lane 4 (1:0.5), lane 5 (1:1), lane 6 (1:2), lane 7 (1: 5) and lane 8 (1:10). Panel C denotes the interaction of B1234Q with E1, E2, E4, M1 and M2 at ratio 1:10 in 100 μM Ca2+. The Lane 1 contains B1234Q alone. The lane 2 is B1234Q with E1, lane 3 is B1234Q with E2, lane 4 is B1234Q with E4, lane 5 is B1234Q with M2 and lane 6 is B1234Q with M1.
The interaction of peptides with B1234Q
CaM mutant B1234Q with mutations at all four Ca2+ binding sites is unable to bind Ca2+ even at high Ca2+ concentrations and thus can be used as a model of apoCaM. To confirm the above data, the peptides were incubated with B1234Q at 1:10 of CaM:peptide ratio under 100 μM Ca2+. Disappearance of free B1234Q band was analyzed. The results showed that canonical E2 was unable to bind B1234Q while all other peptides E1, E4, M1 as well as canonical M2 in iNOS were able to bind B1234Q. It is notable that M2 (lane 5) binds B1234Q with the highest affinity (Fig. 4C).
Assessment of interaction between peptides and CaM by Intrinsic tryptophan fluorescence
As gel mobility-shift assay might be influenced by solubility of peptides at high concentrations; we confirmed the interaction of the non-canonical peptides E1, E4 and M1 with CaM by measuring changes in intrinsic tryptophan fluorescence of peptides. In the presence of Ca2+, CaM induced a 2-fold increase in the fluorescence intensity with emission peak shift from 354 nm to 335 nm (Fig. 5A). In the presence of EGTA, CaM caused a slight shift to 348 nm (Fig. 5B). Canonical E2 and M2 do not comprise tryptophan residues and therefore their binding was not measured by this fluorescence shift. The results provide strong evidence to support binding of E1, E4 and M1 to CaM.
Fig.5. Effect of CaM on the intrinsic tryptophan fluorescenc of the synthetic peptides containing tryptophan residues (E1, E4 and M1).
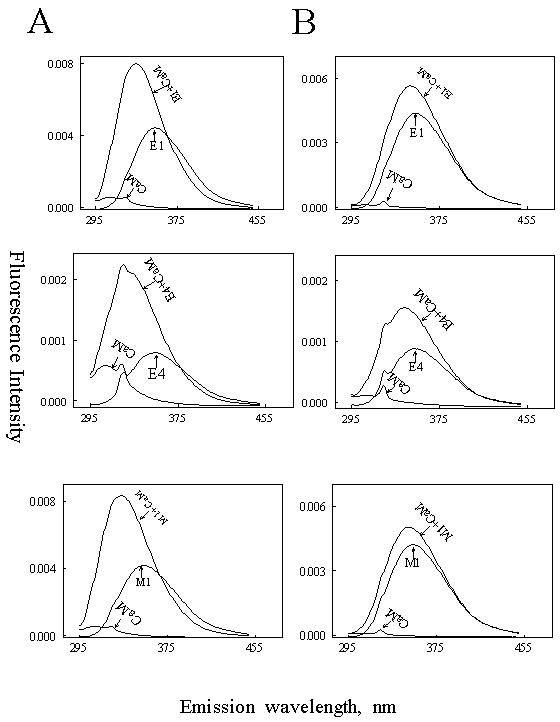
The fluorescence emission spectra of E1, E4 and M1 were obtained in a solution containing 20 mM Tris, pH 7.5 and 5 μM of each peptide with or without 5 μM CaM in the presence of 100 μM Ca2+ (Panel A) or 1 mM EGTA (Panel B). The excitation wavelength was set at 290 nm. Peptide alone, CaM alone and CaM-peptide complex are indicated in each plot. The fluorescence data were obtained by subtracting CaM and buffer effects.
Inhibition of eNOS oxygenase activity by CaM-binding peptides
We further tested peptides (E0 to E5) for their effects on eNOS oxygenase activity by measuring the conversion of L-arginine to L-citrulline. The canonical E2 peptide was included as positive control, and E0, E3 and E5, which did not bind CaM, were included as negative controls. As shown in Fig. 6, the canonical CaM binding peptide E2 inhibited L-citrulline formation in a concentration-dependent manner with a half-maximal inhibition at 0.38μM. E1 and E4 also concentration-dependently inhibited citrulline formation with a slightly higher half-maximal inhibition i.e. 0.72 μM and 0.96 μM, respectively. E5 and E0 peptides did not interfere with eNOS catalytic activity, and E3 located at the autoinhibitory element loop showed a weak inhibition on citrulline formation. This inhibition was associated with E3 binding to NOS, rather than to CaM as suggested by Salerno et al (45).
Fig.6. Concentration-dependent inhibition on eNOS activity by synthetic peptides derived from eNOS.
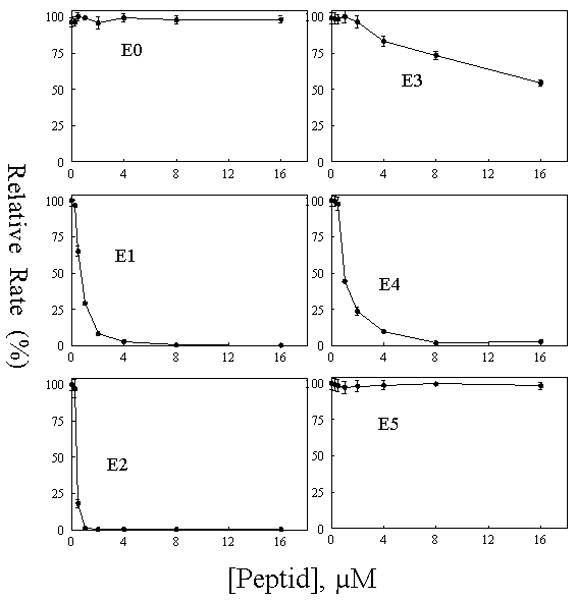
Synthetic peptides, E0, E1, E2, E3, E4 and E5 were tested for their ability to inhibit Ca2+/CaM-dependent eNOS oxygenase activity. Reaction was carried out as described in “EXPERIMENTAL PROCEDURES’. The catalytic activity was normalized to give percentages relative to the reaction rate in the absence of each peptide. Each bar represents mean ± standard deviation of triplicate experiments.
Effects of CaM-binding peptides on intra-reductase domain electron transfer
To determine whether E1 and E4 peptides interfere electron transfer within the reductase domain of eNOS, we performed ferricyanide and cytochrome c reduction assays. Assay in the absence of CaM was performed to serve as baseline reference. Both reductase activities were only slightly inhibited by E1 and E4 (Table 3). At 10 μM, E1 inhibits cytochrome c reduction and ferricyanide reduction by 13% and 28%, respectively and E4, by 20% and 21% respectively, while E2 inhibits the reduction activities completely. Control peptide (E3) has no effect on electron transfer within reductase domain. These results suggest that the binding of E1 and E4 to CaM can effectively block eNOS inter-domain rather than intra-domain electron transfer.
Table III. Effect of peptides on activity of cytochrome c and ferricyanide reduction.
| -CaM (min-1) | +CaM (min-1) | Residues | |
|---|---|---|---|
| Cytochrome c reduction | |||
| None | 133±5.1 | 665±29 | |
| +E1 | 139±8.3 | 586±34 | 174-193 |
| +E2 | 187±12 | 160±10 | 491-510 |
| +E3 | 136±4.8 | 645±32 | 597-622 |
| +E4 | 128±4.6 | 538±19 | 729-757 |
| Ferricyanide reduction | |||
| None | 2044±123 | 6451±245 | |
| +E1 | 2213±95 | 4532±244 | 174-193 |
| +E2 | 2559±99 | 2399±108 | 491-510 |
| +E3 | 1773±106 | 5945±356 | 597-622 |
| +E4 | 2069±112 | 5118±201 | 729-757 |
Each bar represents mean ± standard deviation of triplicate experiments.
Discussion
Although extensive work has attempted to characterize the nature of CaM binding to the NOSs, the regulatory mechanism of CaM on NOS catalysis remains elusive. In this study, 35S-CaM overlay was used to identify possible CaM binding domains in the full eNOS linear coding sequence. We identified three fragments, two of which coincide with the location of the canonical CaM binding site (Fr4, sequence 460-592) and FMN subdomain (Fr5, sequence 505-759) showed positive 35S-CaM binding. Binding was also detected with Fragment 1 (sequence 66-205). We next searched the entire eNOS sequence for CaM binding signature and identified five peptide segments that fulfill the CaM binding motif. Four of the five sequences are confined to the three fragments that exhibit positive 35S-CaM overlay: E1 resides in Fr1, E3 and E4 in FR 5, and the canonical site, E2 in Fr4 (Table I). One sequence (E5) that does not reside in the positive fragments has no CaM binding activity. One of the two Fr 5 sequences with CaM binding motif (E3) also does not have binding activity. Two peptides corresponding to a heme-binding helix in the oxygenase domain (E1, residues 174-193) and a CD1 linker (46) connecting the FMN/FAD subdomains (E4, residues 729-757) possess direct CaM binding property. In comparison to the peptide containing the canonical CaM binding sequence (E2, residue 491-510), these two peptides bind CaM with lower affinity but are capable of CaM binding in the low concentration of Ca2+ (in the presence of EGTA).
Peptides E1 and E4 are active in inhibiting eNOS oxygenase activity with an EC50 close to that of the canonical E2 peptide. By contrast, only E2 peptide effectively blocks intra-reductase domain electron transfer. It is unlikely that E1 and E4 inhibit catalytic activity simply by binding and sequestering CaM as either peptide binds CaM weakly and should not have a striking difference in blocking L-arginine conversion to L-citrulline vs. cytochrome c and ferricyanide reduction. It is possible that E1 and E4 may exert their actions on CaM topographically to influence electron transfer from reductase to oxygenase domain.
Examinations of available crystal structures of truncated NOS domains (47-50) show that the E1 and E4 regions unlike E2 are only partially exposed to solvent. A significant interaction between CaM and any target proteins would require substantial unfolding of the protein. As peptide E1 (residues 174-193) carries heme axial ligand and two hydrogen bond partners for the peripheral propionic acid group, any considerable interaction between E1 and CaM would seriously perturb the heme center with possible disruption of heme-binding pocket. Furthermore, many studies conducted over the last decade on calmodulin binding to NOSs fail to demonstrate any effect on heme (24-25). However, recent studies revealed that mutation of a conserved proximal W409 residue in nNOS, which forms hydrogen bond with the heme thiolate cysteine increases the NO synthesis, suggesting that this hydrogen bond is involved in regulating NO turnover. Removing this hydrogen bond affects but not dramatically destroys heme pocket (51).
Peptide E4 forms part of the linker (CD1) connecting the FMN- and FAD-binding subdomains. Analysis of crystal structures showed that this region is exposed to solvent but lies in a crevasse and is not readily approached by CaM. Conformational change which would make this sequence accessible would make it impossible to align the FMN and FAD isoalloxazines for electron transfer. However, an eNOS FAD subdomain which contains E4 sequence but lacks the canonical CaM binding site and FMN subdomain showed the CaM enhancement on the rate of electron transfer from NADPH to FAD of (Fig. 1). The result might imply that multiple regions of eNOS interact with CaM for effective regulation of eNOS catalysis. Certain regions are necessary for CaM contact but this binding is not sufficient for conformational changes and enzyme activation. These regions are different from the regions responsible for activation. Based on the published reports and our result, we proposed that the canonical site, E2 serves as the main CaM binding switch. Full CaM actions on NOSs require CaM binding to the canonical CaM-binding region. Other binding regions would be as a partner with the canonical site in different states of cell. It is possible that at resting Ca2+ levels, a catalytically inactive eNOS forms complex with the C-lobe of CaM. Upon cellular activation with Ca2+ elevation, Ca2+ binding to the tethered CaM promotes movement of Ca2+/CaM to the canonical CaM-binding helix (E2), resulting in the displacement of autoinhibitory element and eNOS activation. Once the intracellular Ca2+ returns to basal level, an interaction might occur between CaM and heme center (E1) with a possible loss of the axial thiolate ligand and NOS deactivation.
Despite a conserved CaM binding site in iNOS, CaM binds tightly to iNOS in the absence of elevated Ca2+. Structural and molecular basis for the different CaM binding behavior between iNOS and eNOS or nNOS is largely unknown. In this study, we synthesized M1, M2 and M4 peptides based on iNOS sequences corresponding to those of E1, E2 and E4 in eNOS respectively. Binding experiments with these peptides reveal significant differences between iNOS peptides and eNOS peptides. Notably, M4 peptide corresponding to sequence 701-729 located within iNOS FAD subdomain did not bind CaM. As iNOS does not contain the autoinhibitory element at the reductase domain, it might be speculated that a CaM recognition sequences in this region is not necessary. It is interesting to note that in the presence of Ca2+, the canonical M2 peptide forms multiple molecular complexes with CaM. This was not observed with E2. Taken together, these findings suggest a fundamental difference in CaM binding topology and dynamics between eNOS and iNOS.
In summary, we have used the [35S]CaM overlays to identify two additional CaM-recognition sequences other than the canonical site in the linear sequences of eNOS. Synthetic peptides derived from these two sites are able to bind CaM with or without Ca2+ and potently inhibit eNOS oxygenase activity. The available crystal structures of NOS domains show that the two identified sequences (E1 and E4) appear unlikely to bind CaM in these truncated eNOS domains. However, a possible physiological role of these two sites in regulating eNOS activation and deactivation at different states of cell can not be ruled out. Solving the three-dimensional structures of full-length eNOS and iNOS will be indispensable for understanding the topology and dynamics of CaM binding and its function.
Acknowledgments
The work was supported in part by National Institutes of Health Grants NS-23327 (K.K.W.) and HL-50675 (K.K.W.).
Footnotes
The abbreviations used are: H4B, (6R)-5,6,7,8- tetrahydro-L-biopterin; CaM, calmodulin; DTT, dithiothreitol; NO, nitric oxide; NOS, nitric oxide synthase; eNOS, endothelial NOS; iNOS, inducible NOS; nNOS, neuronal NOS; IPTG, isopropy-β-D-thiogalactopyranoside.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Förstermann U, Schmidt HHW, Pollock JS, Sheng H, Mitchell JA, Warner TD, Nakane M, Murad F. Biochem Pharmacol. 1991;42:1849–1857. doi: 10.1016/0006-2952(91)90581-o. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 3.Marletta MA. J Biol Chem. 1993;268:12231–12234. [PubMed] [Google Scholar]
- 4.Nathan C, Xie QW. Cell. 1994;8:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt HHW, Wilke P, Evers B, Bohme E. Biochem Biophys Res Commun. 1989;165:813–819. doi: 10.1016/0006-291x(89)91067-x. [DOI] [PubMed] [Google Scholar]
- 6.Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, Snyder SH. Nature. 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 7.Stuehr DJ, Cho HJ, Kwon NS, Weise M, Nathan C. Proc Natl Acad Sci U S A. 1991;88:7773–7777. doi: 10.1073/pnas.88.17.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Ding A, Troso T, Nathan C. Science. 1992;256:225–228. doi: 10.1126/science.1373522. [DOI] [PubMed] [Google Scholar]
- 9.Pollock JS, Förstermann U, Mitchell JA, Warner TD, Schmidt HHW, Nakane M, Murad F. Proc Natl Acad Sci U S A. 1991;88:10480–10484. doi: 10.1073/pnas.88.23.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheta EA, McMillan K, Masters BS. J Biol Chem. 1994;269:15147–15153. [PubMed] [Google Scholar]
- 11.Ghosh DK, Stuehr DJ. Biochemistry. 1995;34:801–807. doi: 10.1021/bi00003a013. [DOI] [PubMed] [Google Scholar]
- 12.Chen PF, Tsai AL, Berka V, Wu KK. J Biol Chem. 1996;271:14631–14635. [PubMed] [Google Scholar]
- 13.McMillan K, Bredt DS, Hirsch DJ, Snyder SH, Clark JE, Masters BS. Proc Natl Acad Sci U S A. 1992;89:11141–11145. doi: 10.1073/pnas.89.23.11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abu-Soud HM, Stuehr DJ. Proc Natl Acad Sci U S A. 1993;90:10769–10772. doi: 10.1073/pnas.90.22.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abu-Soud HM, Yoho LL, Stuehr DJ. J Biol Chem. 1994;269:32047–32050. [PubMed] [Google Scholar]
- 16.Siddhanta U, Presta A, Fan BC, Wolan D, Rousseau DL, Stuehr DJ. J Biol Chem. 1998;273:18950–18958. doi: 10.1074/jbc.273.30.18950. [DOI] [PubMed] [Google Scholar]
- 17.Panda K, Ghosh S, Stuehr DJ. J Biol Chem. 2001;276:233349–23356. doi: 10.1074/jbc.M100687200. [DOI] [PubMed] [Google Scholar]
- 18.Sagami I, Daff S, Shimizu T. J Biol Chem. 2001;276:30036–30042. doi: 10.1074/jbc.M104123200. [DOI] [PubMed] [Google Scholar]
- 19.Babu YS, Bugg CE, Cook WJ. J Mol Biol. 1988;204:191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- 20.Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- 21.Meador WE, Means AR, Quiocho FA. Science. 1992;257:1251–1255. doi: 10.1126/science.1519061. [DOI] [PubMed] [Google Scholar]
- 22.Bredt DS, Snyder SH. Proc Natl Acad Sci U S A. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho HJ, Xie QW, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Nathan C. J Exp Med. 1992;176:599–604. doi: 10.1084/jem.176.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry JM, Moon N, Zhao Y, Dunham WR, Marletta MA. Chem Biol. 1998;5:355–364. doi: 10.1016/s1074-5521(98)90069-2. [DOI] [PubMed] [Google Scholar]
- 25.Noble MA, Munro AW, Rivers SL, Robledo L, Daff SN, Yellowlees LJ, Shimizu T, Sagami I, Guillemette JG, Chapman SK. Biochemistry. 1999;38:16413–16418. doi: 10.1021/bi992150w. [DOI] [PubMed] [Google Scholar]
- 26.Matsubara M, Hayashi N, Titani K, Taniguchi H. J Biol Chem. 1997;272:23050–23056. doi: 10.1074/jbc.272.37.23050. [DOI] [PubMed] [Google Scholar]
- 27.Yuan T, Vogel HJ, Sutherland C, Walsh MP. FEBS Lett. 1998;431:210–214. doi: 10.1016/s0014-5793(98)00750-9. [DOI] [PubMed] [Google Scholar]
- 28.Aoyagi M, Arvai AS, Tainer JA, Getzoff ED. Embo J. 2003;22:766–775. doi: 10.1093/emboj/cdg078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gachhui R, Presta A, Bentley DF, Abu-Soud HM, McArthur R, Brudvig G, Ghosh DK, Stuehr DJ. J Biol Chem. 1996;271:20594–20602. doi: 10.1074/jbc.271.34.20594. [DOI] [PubMed] [Google Scholar]
- 30.Lee SJ, Stull JT. J Biol Chem. 1998;273:27430–27437. doi: 10.1074/jbc.273.42.27430. [DOI] [PubMed] [Google Scholar]
- 31.Rozhkova EA, Fujimoto N, Sagami I, Daff S, Shimizu T. J Biol Chem. 2002;277:16888–16894. doi: 10.1074/jbc.M200642200. [DOI] [PubMed] [Google Scholar]
- 32.Stevens-Truss R, Beckingham K, Marletta MA. Biochemistry. 1997;36:12337–12345. doi: 10.1021/bi970973k. [DOI] [PubMed] [Google Scholar]
- 33.Gribovskaja I, Brownlow KC, Dennis SJ, Rosko AJ, Marletta MA, Stevens-Truss R. Biochemistry. 2005;44:7593–7601. doi: 10.1021/bi0474517. [DOI] [PubMed] [Google Scholar]
- 34.Govers R, Rabelink TJ. Am J Physiol Renal Physiol. 2001;280:F193–F206. doi: 10.1152/ajprenal.2001.280.2.F193. [DOI] [PubMed] [Google Scholar]
- 35.Weissman BA, Jones CL, Liu Q, Gross SS. Eur J Pharmacol. 2002;435:9–18. doi: 10.1016/s0014-2999(01)01560-6. [DOI] [PubMed] [Google Scholar]
- 36.Chen PF, Wu KK. J Biol Chem. 2000;275:13155–13163. doi: 10.1074/jbc.275.17.13155. [DOI] [PubMed] [Google Scholar]
- 37.Pitt GS, Zűhlke RD, Hudmon A, Schlman H, Reuter H, Tsien RW. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- 38.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 39.Lydan MA, O'Day DH. Methods Mol Biol. 1994;31:389–396. doi: 10.1385/0-89603-258-2:389. [DOI] [PubMed] [Google Scholar]
- 40.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 41.Rhoads AR, Friedberg F. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 42.Erickson-Viitanen S, DeGrado W. Methods Enzymol. 1987;139:455–478. doi: 10.1016/0076-6879(87)39106-2. [DOI] [PubMed] [Google Scholar]
- 43.Romanin C, Gamsjaeger R, Kahr H, Schaufler D, Carlson O, Abernethy DR, Soldatov NM. FEBS letters. 2000;487:301–306. doi: 10.1016/s0014-5793(00)02361-9. [DOI] [PubMed] [Google Scholar]
- 44.Tang W, Halling DB, Black DJ, Pate P, Zhang JZ, Pedersen S, Altschuld RA, Hamilton SL. Biophys J. 2003;85:1538–1547. doi: 10.1016/s0006-3495(03)74586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salerno JC, Harris DE, Irizarry K, Patel B, Morales AJ, Smith SM, Martasek P, Roman LJ, Masters BS, Jones CL, Weissman BA, Lane P, Liu Q, Gross SS. J Biol Chem. 1997;272:29769–29777. doi: 10.1074/jbc.272.47.29769. [DOI] [PubMed] [Google Scholar]
- 46.Knudsen GM, Nishida CR, Mooney SD, Ortiz de Montellano PR. J Biol Chem. 2003;278:31814–31824. doi: 10.1074/jbc.M303267200. [DOI] [PubMed] [Google Scholar]
- 47.Raman CS, Li H, Martásek P, Král V, Masters BSS, Poulos TL. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 48.Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Nat Struct Biol. 1999;6:233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- 49.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 50.Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- 51.Adak S, Stuehr DJ. J Inorg Biochem. 2001;83:301–308. doi: 10.1016/s0162-0134(00)00176-8. [DOI] [PubMed] [Google Scholar]