

Summary
The cancer drug, Imatinib, is a selective Abl kinase inhibitor which does not inhibit the closely related kinase c-Src. This one drug and its ability to selectively inhibit Abl over c-Src has been a guiding principle in virtually all kinase drug discovery efforts in the last fifteen years. A prominent hypothesis explaining the selectivity of Imatinib is that Abl has an intrinsic ability to adopt an inactive conformation (termed DFG-out), whereas c-Src appears to pay a high intrinsic energetic penalty for adopting this conformation effectively excluding Imatinib from its ATP pocket. This explanation of the difference in binding affinity of Imatinib for Abl versus c-Src makes the striking prediction that it would not be possible to design an inhibitor that binds to the inactive conformation of c-Src with high affinity. We now report the discovery of a series of such inhibitors. We use structure-activity relationships and X-ray crystallography to confirm our findings. These studies suggest that small molecules are capable of inducing the generally unfavourable DFG-out conformation in c-Src.
Introduction
Protein kinases represent one of the largest super-families of drug targets across all therapeutic areas. The central challenge in the development of kinase inhibitor drug candidates is in targeting the disregulated kinase while avoiding inhibition of non-disease related kinases containing closely related ATP binding pockets. Imatinib, the first clinically approved kinase inhibitor provided a remarkable example of a highly selective inhibitor of the translocation product Bcr-Abl (Capdeville et al., 2002; Sawyers, 2002). Imatinib potently inhibits Bcr-Abl, the oncogene which drives chronic myelogenous leukaemia, but does not inhibit the cytoplasmic tyrosine kinase, c-Src, despite the fact that the two kinases share almost completely identical amino acids lining the ATP binding pocket which Imatinib contacts (Figure 1A; Schindler et al., 2000; Seeliger et al., 2007). Significant medicinal chemistry, structural biology, and computational modelling efforts have focussed on understanding the differential selectivity of Imatinib for Bcr-Abl and c-Src.
Figure 1.

Figure 1A. A schematic representation of Imatinib contacts identified in its complexes with c-Src (PDB ID 2OIQ) and Abl (PDB ID 1IEP). The upper and lower sequences are aligned based on structural superposition and the numbering scheme in based on c-Src. Residues are grouped based on their location within the N-lobe, hinge region and c-lobe of the kinase domain. The interaction between Imatinib and Tyr253 in Abl (Phe278 in c-Src) was not observed in the c-Src complex and is depicted by a yellow star.
1B. Type I inhibitors, such as PP1, occupy the adenosine pocket forming multiple hydrogen bonds with the hinge region of the kinase and threonine gatekeeper. The molecular envelop of PP1 (depicted by the blue shadow) is not thought to influence the conformation of the DFG motif (shown here in the ‘IN’ conformation) or helix αC (shown in the ‘OUT’ conformation).
1C. Type II inhibitors, such as Imatinib, engage both the hinge binding region and extend into the pocket created by the DFG flip. The extended portion of Imatinib (depicted by the pink shadow) directly senses the ‘OUT’ conformation of the DFG motif. Common features of type II binding are the interaction with the conserved glutamate within helix αC and the backbone amide of the DFG triad (Liu and Gray, 2006).
The first insight into the basis for selectivity of Imatinib was revealed when Kuriyan and co-workers solved the Imatinib-Abl co-crystal structure (Nagar et al., 2002; Schindler et al., 2000). This structure revealed a not-previously observed kinase conformation indicating that Imatinib binds Abl in a catalytically inactive conformation defined by a crank shaft-like displacement of the N-terminal region of the activation loop of the kinase effecting a dramatic change in the conformation of the Asp-Phe-Gly (DFG) triad. This conformational change has been subsequently observed in other protein kinase-drug co-crystal structures (Irk, Kit, Flt3, p38 Mapk and B-Raf; Griffith et al., 2004; Hubbard et al., 1994; Mol et al., 2004; Pargellis et al., 2002; Wan et al., 2004) and has been termed the type-II or DFG-out conformation (ATP competitive inhibitors which bind to kinases in the active conformation are termed type-I or DFG-in binders; Figure 1B and C; Liu and Gray, 2006). The identification of an inactive conformation of Abl bound by the highly selective inhibitor Imatinib has guided many successful medicinal chemistry campaigns in search of selective kinase inhibitors (Angell et al., 2008; Cumming et al., 2004; Gill et al., 2005; Heron et al., 2006; Okram et al., 2006).
A wealth of data currently supports the view that the Imatinib bound conformation (DFG-out) of Abl is thermodynamically stable in complex with Imatinib, but that such conformations require energetically unfavourable interactions in c-Src complexes (Levinson et al., 2006; Nagar et al., 2002; Seeliger et al., 2007; Vajpai et al., 2008). Imatinib has been crystallized in both its potent target-Abl (Nagar et al., 2002; Schindler et al., 2000), as well as the poorly inhibited target, c-Src (Seeliger et al., 2007). Surprisingly, the Imatinib/co-crystal structures are virtually identical despite the significantly different affinities of Imatinib for the two protein kinases. Efforts to construct mutant forms of c-Src with the ability to be potently inhibited by Imatinib were only partially successful, which led Kuriyan and co-workers to suggest a distributed thermodynamic penalty for c-Src to adopt the DFG-out conformation (Seeliger et al., 2007). The importance of kinase conformational preference over precise amino acid identity is highlighted by studies with the Imatinib target receptor kinase, c-Kit. Although c-Kit is more closely related to c-Src than Abl in the amino acids lining the ATP binding pocket, c-Kit is more potently inhibited by Imatinib (Deininger et al., 2005). Structural studies of c-Kit in the absence of ligand (ATP or Imatinib) show the kinase adopts the DFG-out conformation, suggesting the Imatinib bound conformation is stable and pre-formed in the absence of Imatinib, thereby explaining its Imatinib sensitivity (Mol et al., 2004).
The resulting widely held explanation of the discrepancy in affinity of Imatinib despite the close similarity in structure of the two drug-protein complexes is based on the relative propensity of the two kinases to adopt the relevant drug-bound (DFG-out/type II) conformation: Abl is predicted to prefer the DFG out conformation relative to c-Src, and since Imatinib binds to the type-II conformation of the kinase, its affinity is higher to Abl than to c-Src. This explanation of the difference in binding affinity of Imatinib for Abl vs. c-Src makes the striking prediction that it would not be possible to design an inhibitor, which binds potently to the type-II conformation of c-Src.
Results
We asked if we could develop a DFG-out binder for c-Src as a test of this prediction. We applied an approach pioneered by Liu, Gray, and co-workers whereby type II (DFG-out) kinase inhibitors can be created by fusing a so-called hinge binding element of a type-I kinase inhibitor to an element capable of binding in the pocket created by the characteristic DFG movement in type II inhibitor bound structures (Liu and Gray, 2006; Okram et al., 2006). We chose the hinge-binding element from the well-characterized pyrazolopyrimidine PP1. We chose PP1 because it has been examined at both the structural and functional level and was first identified as a selective c-Src family tyrosine kinase inhibitor (Hanke et al., 1996; Liu et al., 1999; Schindler et al., 1999).
In order to select the DFG-out binding element for our design, we examined the co-crystal structures of Abl, Raf and p38 in complex with Imatinib, BAY43-9006, and BIRB796, respectively; three chemically distinct type II inhibitors with three different kinase targets (Pargellis et al., 2002; Schindler et al., 2000; Wan et al., 2004). Each inhibitor follows nearly the identical path within the active site pocket, despite their chemical uniqueness (Supplementary Figure 1). A key feature of the observed binding modes is the interaction with a portion of the activation segment termed the DFG motif and a highly conserved glutamic acid residue within helix αC, which are mediated through the amide/urea linker and hydrophobic portions of the inhibitors. Movement of the Asp residue out and the Phe residue in (hence ‘DFG-out’) by a flip of approximately 180 degrees relative to their position in the active state creates the cavity that is filled by these inhibitors. The extended portions of each inhibitor are remarkably similar, and their interactions with the kinase are mediated through highly conserved residues within the ATP pocket, suggesting that the general inhibitor features could be applied to other kinases.
We hypothesized that derivatization on the phenyl ring in PP1 with a m-trifluoromethyl phenylurea group would create an inhibitor that could engage the DFG-out pocket. The pyrazolopyrimidine core of PP1 occupies the portion of the active site within which the adenosine ring of ATP normally sits, forming key hydrogen bonds with the backbone of the kinase hinge region (Figure 1B). Since this portion of the active site is not subject to allosteric control, PP1 and type I inhibitors should bind to their kinase targets irrespective of their activation state. We synthesized a panel of molecules with this design searching for an inhibitor with tight (nM) binding affinity for c-Src. Because of the known sensitivity of PP1 derivatives to sterically bulky phenyl replacements, we reasoned that if we identified a potent binder it would be likely to adopt a type-II orientation in order to accommodate the bulky phenyl substituent.
Our modelling suggested that addition of a methylene group between the pyrazolopyrimidine core and the phenyl ring would provide flexibility in guiding the m-trifluromethyl phenyl urea substitution into the DFG pocket. We therefore synthesized compounds 1–4, in which the phenyl group of PP1 has been replaced with a benzyl functionality and the N1 position of the pyrazole ring has been varied with different alkyl groups (Figure 2 middle). In our design strategy, we also anticipated that a direct link between the pyrazolopyrimidine core and the derivatized phenyl could avoid a potential negative interaction with the threonine gatekeeper, and thus we created compound 5 (Figure 2 bottom). Each molecule was prepared based on previously established routes for generating pyrazolopyrimidines (Bishop et al., 1999; Bishop et al., 1998; Blethrow et al., 2004) with the exception that the urea linker was appended through inclusion of a nitro group in the starting material, which in the final synthetic steps was reduced and coupled to m-trifluoromethyl phenyl isocyanate to generate the type II analogues.
Figure 2.

IC50 values of Imatinib, and compounds 1–5 for both c-Src and Abl. The values adjacent to the bar graph represent the mean calculation and uncertainty in μM units.
To ascertain the potency of our designed compounds, we examined their ability to inhibit kinase domain fragments of c-Src and Abl that were expressed and purified identically from bacteria in their unphosphorylated forms. We measured half maximal inhibitory concentrations (IC50) utilizing an in vitro assay in which the kinase catalyses phosphorylation of a synthetic peptide substrate in the presence of 100 μM ATP and varying amounts of inhibitor (Figure 2). From this analysis, we determined IC50 values for Imatinib of 24,370 and 11 nM for c-Src and Abl, respectively. These values are in close agreement to published values and highlight the inherent selectivity of Imatinib for Abl with respect to c-Src (Seeliger et al., 2007).
Compound 1 was found to inhibit c-Src with an IC50 of approximately 6.2 μM, whereas a control compound in which the urea linker was placed at the para position of the benzyl ring lacked any detectable inhibitory activity (data not shown). In measuring the IC50 values for 1–4, we observed an interesting correlation between the size of the alkyl group substitutions and selectivity for c-Src and Abl (Figure 2). The methyl derivative 1 was the weakest inhibitor against both c-Src and Abl, followed by the isopropyl 2 and t-butyl 3 compounds which gained moderate potency, with the optimal derivative appearing to be the cyclopentyl substitution 4, with an IC50 of 480 nM for c-Src (Figure 2). Curiously, while most compounds in this set equally inhibited both c-Src and Abl, the cyclopentyl derivative showed a reproducible selectivity towards c-Src over Abl of approximately 5 fold. Although small, this modest degree of selectivity appeared significant in comparison to the yet smaller IC50 value differences between c-Src and Abl for compounds 1, 2, and 4. Compound 5 was the most potent inhibitor that we identified, with IC50 values of 25 and 41 nM for c-Src and Abl, respectively (Figure2). Interestingly, the potency of 5 approaches that of Imatinib for Abl, but without any significant discrimination against c-Src. In our small test of compounds we identified two interesting features: compound 3 with unexpected selectivity for c-Src, and compound 5 with extremely high potency for both c-Src and Abl. These intriguing features made us wonder if we had achieved our designed mode of binding, and to resolve this issue we determined co-crystal structures of c-Src bound to inhibitors 3 and 5.
Binding Mode Revealed by Co-crystallography
Purified c-Src kinase domain in complex with 3 and 5 yielded crystals that diffracted to 2.8 and 2.3 Angstrom, respectively. Both structures were determined by molecular replacement, finding a single copy of c-Src within the asymmetric unit of the P21 crystal form for the c-Src-3 complex and two copies of c-Src in the P1 crystal form of the c-Src-5 complex. Interestingly, only one kinase molecule within the c-Src-5 complex appeared to contain inhibitor. This feature was observed previously in the co-crystal structure of c-Src with Imatinib, where only one kinase within the asymmetric unit was found to be in a drug complex despite molar equivalents of the protein and inhibitor at a concentration well above their binding constant (Seeliger et al., 2007). The structures of c-Src in complex with 3 and 5 are shown in Figure 3, with corresponding magnification of the active site.
Figure 3.
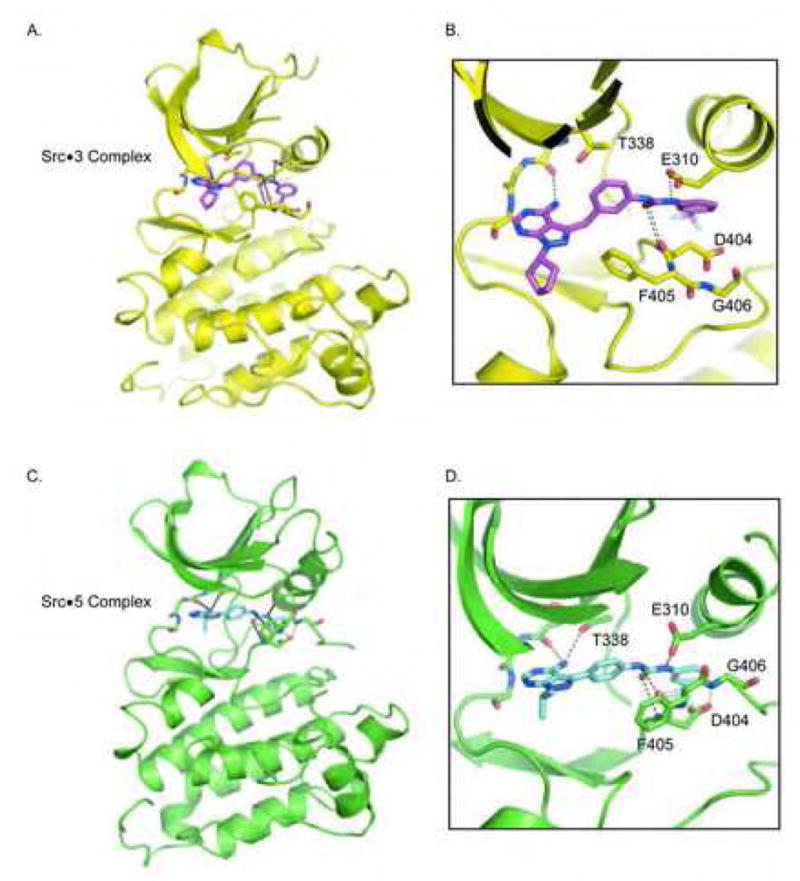
Crystal structures of compounds 3 and 5 bounds to c-Src.
A. Cartoon representation of c-Src in complex with 3. The side chains of Thr338, Glu310, main chain atoms within the hinge region, and both main chain and side chain atoms of the DFG triad are shown as sticks. Hydrogen bonding interactions are shown as dashed lines. Panels B to D are similarly labelled.
B. Magnification of the active site of c-Src in complex with 3.
C. Cartoon representation of c-Src in complex with 5.
D. Magnification of the active site of c-Src in complex with 5.
As shown, the pyrazolopyrimidine core for both inhibitors lie deep within the adenosine pocket that is lined by the hinge region of the kinase. In comparison to PP1, the plane of the pyrazolopyrimidine rings of both 3 and 5 deviate slightly with respect to each other. As a result of the altered geometry, both 3 and 5 only form a single hydrogen bond to the main chain carbonyl of Glu339. Compound 3 is shifted away from the side chain hydroxyl of the Thr338 gatekeeper, and as a result does not form the hydrogen bond seen in PP1 or in 5 with this residue. As anticipated, both the benzyl group of 3 and the phenyl group in 5 lie juxtaposed to the gatekeeper; both of which are twisted out of plane relative to the pyrazolopyrimidine ring. In both 3 and 5, the urea extension forms the designed hydrogen bond with the side chain of Glu310 within helix αC, while the m-trifluoromethyl phenyl portion of both compounds lie within a pocket lined by residues Leu317, Leu322, Val402, Met314, His384. As a result of occupying this space, Asp404 and Phe405 are flipped near 180 degrees relative to their active state positions. In the c-Src-5 complex, the side chain carbonyl of Glu404 forms a hydrogen bond to the mainchain amide of Gly406 (Figure 3B). To our knowledge, this precise configuration has not been observed in crystal structures of DFG-out kinases, but has been hypothesized to occur during the DFG flip as revealed in molecular dynamic simulations (Levinson et al., 2006). Interestingly, the configuration of the aspartic acid side chain through to the glycine amide is strikingly similar to the structure of a beta bend (Fersht, 1999). In a classic beta bend, a nine atom turn along the mainchain separates a carbonyl acceptor from an amide donor, and often contains a –CH2- glycine between the donor-acceptor pairs. Here the side chain of Asp404 appears to supply both the carbonyl acceptor and intervening –CH2- group.
In both structures of c-Src described here, the configuration of the DFG triad and the position of Glu310 of helix αC adopt conformations that deviate from what was previously observed in either apo c-Src or the PP1-bound form of the closely related enzyme HCK (Schindler et al., 1999; Xu et al., 1997). Rather 3 and 5 recognize the DFG-out configuration of c-Src that is similarly engaged by Imatinib.
Discussion
In their hybrid design approach, the set of type II inhibitors that were successfully developed for Abl by Liu, Gray, and co-workers started from four different type I scaffolds (Okram et al., 2006). It is noteworthy to mention that each of the designed inhibitors was tested against a panel of protein kinases including c-Src. Interestingly, each type II variant exhibited decreased affinity for c-Src relative to the starting scaffolds, whereas they gained potency and selectivity for Abl. While these experiments suggested that a hybrid design approach is feasible, it also hinted at the restricted effectiveness of new type II inhibitors towards certain kinases. The discovery of compounds 3 and 5 open up the very real possibility of further developing potent DFG-out binders for c-Src as an effective inhibitor design strategy. In many cases inhibitors that share the general features of DFG-out binders may already exist (for eg Dimauro et al., 2006), but have been ruled out as type II inhibitors of c-Src because it would have seemed highly unlikely based on the precedence that has been set by Imatinib. Further exploration of c-Src in complex with DFG-out binders will provide a greater understanding of the molecular recognition principles of remarkable drugs such as Imatinib and may provide a basis for predictive modelling of kinase conformational dynamics and the relationship to inhibitor potency.
Assuming that each inhibitor must overcome the same energetic barrier needed to induce the DFG-out conformation, we can begin to speculate on the increased affinity of 3 and 5 versus Imatinib for c-Src keeping in mind that any region alone of the inhibitors or protein likely contribute a fraction of the distributive function that forms the basis for the bimolecular interaction. One of the more significant differences between the c-Src complexes and the Abl-Imatinib structure is in the path of the P-loop (Figure 4A); the region defined by the GXGXXG motif of kinases within the β1-β2 linker and that forms the top shelf of the ATP pocket. Notably, in the Abl-Imatinib complex, the P-loop tightly encloses the drug binding site in large part through residue Tyr253, which folds back onto the lip of the pyrimidine core. In the c-Src-Imatinib complex, the region occupied by Tyr253 of Abl is left unoccupied, whereas in the c-Src-3 complex, the cyclopentyl group of the inhibitor itself fills this space. Experimentally, it would be ideal to test if one could increase the potency of Imatinib for c-Src by derivatizing its pyridine ring with a group similar to the cyclopentyl of 3 to determine if this missing interaction is partially responsible for the weak binding of Imatinib to c-Src. Unfortunately it would be near impossible to maintain the co-planar nature of the phenyl-pyrimidine rings in Imatinib with such an analogue due to intramolecular steric interactions that would twist the rings out of planarity. Since the overall binding conformation of compounds 1–4 are less sensitive to the influence of substitutions at the R-1 position on the pyrazole ring this series of inhibitors could be a reliable measure of engaging the Tyr253 pocket through varying steric bulk of the inhibitor. Indeed, the structure and activity of compounds 1–4 could be explained based on the potential role of the Tyr253 region as an affinity pocket, since there is a distinct structure activity relationship when this substituent is varied.
Figure 4.
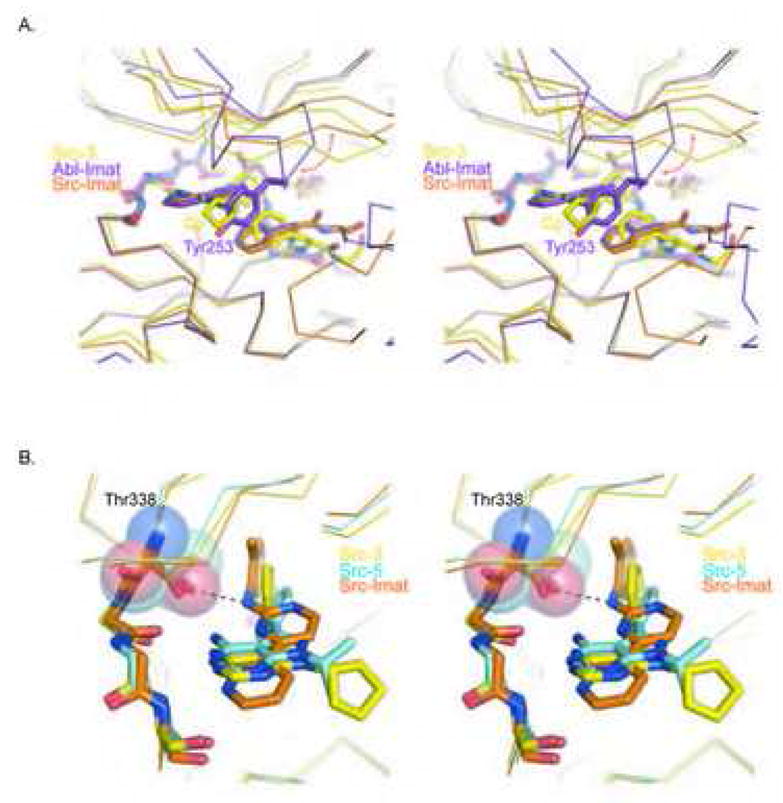
Structural Differences in the binding of 3, 5, and Imatinib to c-Src.
A. Stereo figure of a structural superposition of 3 in complex with c-Src (yellow) and Imatinib in complex with Abl (purple; PDB 1IEP) or c-Src (orange; PDB 2OIQ). The red arrow highlights deviations in the path of the respective P-loops. Tyr253 in Abl and the cyclopentyl (Cp) group of 3 fill a space that is unoccupied within the c-Src-Imatinib complex.
B. Stereo figure of 3, 5, and Imatinib in complex with c-Src. The gatekeeper is highlighted as a semi-transparent surface. The relative position of the inhibitors could make them differentially sensitive to gatekeeper mutations, such as the clinically relevant Thr315Ile mutation found in Abl.
Interestingly, one other distinguishing feature between the 3-, 5-, and Imatinib complexes with c-Src is in the approach of these inhibitors towards the gatekeeper pocket (Figure 4B). Notably, the benzyl group of 3 and the phenyl ring of 5 are rotated away from Thr338 relative to o-methyl-phenylamino portion of Imatinib in a rank order that reflects the relative affinity of the drugs for c-Src. This extra distance from the gatekeeper Thr suggests compounds 1–5 may bind to mutant kinases such as the clinically relevant Imatinib resistant Abl Thr315Ile kinase (Shah et al., 2002).
What have we learned from the discovery of potent DFG-out binders to c-Src? We can conclude that the relative energy differences between favoured and disfavoured conformational states of particular kinases can be overcome by small molecules. This finding implies that DGF-out binders and other such conformation specific binders will not necessarily be kinase specific. Support for this view comes from recent studies on the development of DFG out binders of the p38 Map kinase where it was found that a series of biphenyl amides containing DFG-out kinase inhibitors are in fact less selective than traditional DFG-in binders based on the same scaffold (Angell et al., 2008). Thus, conformation specific kinase inhibitors are not de facto more selective based on intrinsic kinase conformational preferences.
Significance
Our results highlight a potential new utility of small molecule ligands for protein kinases. Small molecules may be capable of inducing changes to secondary, tertiary and even quaternary structure of protein kinases and protein kinase complexes in cells, which are not sampled by the protein normally. This could open up the possibility of regulating protein kinase function through inducing conformational changes in protein-protein or enzymatic domains outside of the kinase catalytic domain (Papa et al., 2003), providing new therapeutic modalities.
Methods
A detailed description of methods used for protein expression and purification, in vitro kinase assays, crystallization and structure determination, and chemical synthesis are described in the supplemental data.
Supplementary Material
Acknowledgments
We would like to thank Yi Liu for helpful discussions on design of potent DFG-out binding inhibitors. We thank Brianna Burden for assistance in synthesizing 3 and members of the Shokat laboratory for advice and insights. We thank Jimmy Blair and Chao Zhang, along with Pingda Ren, and Yi Liu for comments on this manuscript. Mass spectrometry was made possible by NIH shared resource grants NCRR RR015804 and NCRR RR001614. We thank Beamline Scientists, in particular Peter Zwart, at the Advance Light Source (ALS), Berkeley, beamlines 5.0.1 and 5.0.3, for technical assistance. We thank John Kuriyan and Markus Seeliger for expression plasmids and advice on crystallography. ACD is a recipient of a U.S. Department of Defense Breast Cancer Research Program Postdoctoral Award (W81XWH-06-1-0727).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angell RM, Angell TD, Bamborough P, Bamford MJ, Chung CW, Cockerill SG, Flack SS, Jones KL, Laine DI, Longstaff T, et al. Biphenyl amide p38 kinase inhibitors 4: DFG-in and DFG-out binding modes. Bioorg Med Chem Lett. 2008;18:4433–4437. doi: 10.1016/j.bmcl.2008.06.028. [DOI] [PubMed] [Google Scholar]
- Bishop AC, Kung CY, Shah K, Witucki L, Shokat KM, Liu Y. Generation of monospecific nanomolar tyrosine kinase inhibitors via a chemical genetic approach. Journal of the American Chemical Society. 1999;121:627–631. [Google Scholar]
- Bishop AC, Shah K, Liu Y, Witucki L, Kung C, Shokat KM. Design of allele-specific inhibitors to probe protein kinase signaling. Curr Biol. 1998;8:257–266. doi: 10.1016/s0960-9822(98)70198-8. [DOI] [PubMed] [Google Scholar]
- Blethrow J, Zhang C, Shokat KM, Weiss EL. Design and use of analog-sensitive protein kinases. Curr Protoc Mol Biol. 2004;Chapter 18(Unit 18):11. doi: 10.1002/0471142727.mb1811s66. [DOI] [PubMed] [Google Scholar]
- Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1:493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- Cumming JG, McKenzie CL, Bowden SG, Campbell D, Masters DJ, Breed J, Jewsbury PJ. Novel, potent and selective anilinoquinazoline and anilinopyrimidine inhibitors of p38 MAP kinase. Bioorg Med Chem Lett. 2004;14:5389–5394. doi: 10.1016/j.bmcl.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- Dimauro EF, Newcomb J, Nunes JJ, Bemis JE, Boucher C, Buchanan JL, Buckner WH, Cee VJ, Chai L, Deak HL, et al. Discovery of aminoquinazolines as potent, orally bioavailable inhibitors of Lck: Synthesis, SAR, and in vivo anti-inflammatory activity. Journal of Medicinal Chemistry. 2006;49:5671–5686. doi: 10.1021/jm0605482. [DOI] [PubMed] [Google Scholar]
- Fersht A. Structure and Mechanism in Protein Science: a guide to enzyme catalysis and protein folding. New York: W.H. Freeman and Co; 1999. [Google Scholar]
- Gill AL, Frederickson M, Cleasby A, Woodhead SJ, Carr MG, Woodhead AJ, Walker MT, Congreve MS, Devine LA, Tisi D, et al. Identification of novel p38alpha MAP kinase inhibitors using fragment-based lead generation. J Med Chem. 2005;48:414–426. doi: 10.1021/jm049575n. [DOI] [PubMed] [Google Scholar]
- Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, Lippke J, Saxena K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–178. doi: 10.1016/s1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Heron NM, Anderson M, Blowers DP, Breed J, Eden JM, Green S, Hill GB, Johnson T, Jung FH, McMiken HH, et al. SAR and inhibitor complex structure determination of a novel class of potent and specific Aurora kinase inhibitors. Bioorg Med Chem Lett. 2006;16:1320–1323. doi: 10.1016/j.bmcl.2005.11.053. [DOI] [PubMed] [Google Scholar]
- Hubbard SR, Wei L, Ellis L, Hendrickson WA. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- Levinson NM, Kuchment O, Shen K, Young MA, Koldobskiy M, Karplus M, Cole PA, Kuriyan J. A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol. 2006;4:e144. doi: 10.1371/journal.pbio.0040144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Bishop A, Witucki L, Kraybill B, Shimizu E, Tsien J, Ubersax J, Blethrow J, Morgan DO, Shokat KM. Structural basis for selective inhibition of Src family kinases by PP1. Chem Biol. 1999;6:671–678. doi: 10.1016/s1074-5521(99)80118-5. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC, Wilson KP. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279:31655–31663. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571) Cancer Research. 2002;62:4236–4243. [PubMed] [Google Scholar]
- Okram B, Nagle A, Adrian FJ, Lee C, Ren P, Wang X, Sim T, Xie Y, Wang X, Xia G, et al. A general strategy for creating "inactive-conformation" abl inhibitors. Chem Biol. 2006;13:779–786. doi: 10.1016/j.chembiol.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Papa FR, Zhang C, Shokat K, Walter P. Bypassing a kinase activity with an ATP-competitive drug. Science. 2003;302:1533–1537. doi: 10.1126/science.1090031. [DOI] [PubMed] [Google Scholar]
- Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, Regan J. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9:268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- Sawyers CL. Disabling Abl-perspectives on Abl kinase regulation and cancer therapeutics. Cancer Cell. 2002;1:13–15. doi: 10.1016/s1535-6108(02)00022-3. [DOI] [PubMed] [Google Scholar]
- Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- Schindler T, Sicheri F, Pico A, Gazit A, Levitzki A, Kuriyan J. Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol Cell. 1999;3:639–648. doi: 10.1016/s1097-2765(00)80357-3. [DOI] [PubMed] [Google Scholar]
- Seeliger MA, Nagar B, Frank F, Cao X, Henderson MN, Kuriyan J. c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure. 2007;15:299–311. doi: 10.1016/j.str.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- Vajpai N, Strauss A, Fendrich G, Cowan-Jacob SW, Manley PW, Grzesiek S, Jahnke W. Solution conformations and dynamics of ABL kinase-inhibitor complexes determined by NMR substantiate the different binding modes of imatinib/nilotinib and dasatinib. J Biol Chem. 2008;283:18292–18302. doi: 10.1074/jbc.M801337200. [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- Xu WQ, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.